
Viral Agents as Potential Drivers of Diffuse Large B-Cell Lymphoma Tumorigenesis

 ,
,  ,
,  , and
, and
Abstract
1. Introduction: Diffuse Large B-Cell Lymphoma
2. Viruses in DLBCL
2.1. Epstein–Barr Virus

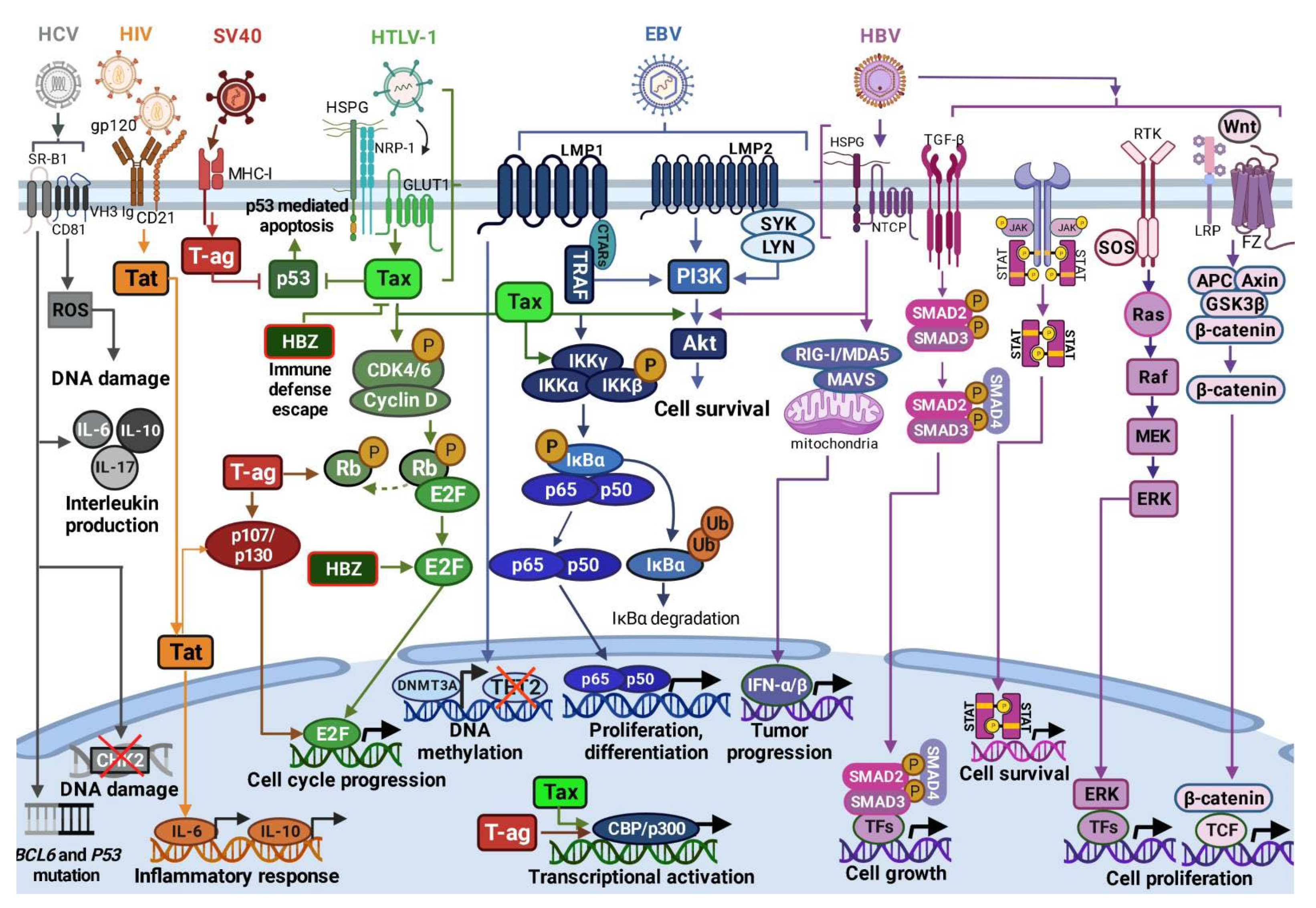{kind=link}
| Virus/Parameter | Genome | Mechanism(s) of Infection | Infection Outcomes in DLBCL | Therapeutic Approach in DLBCL |
|---|---|---|---|---|
| EBV | ds DNA [77] | Latency III Expression of nuclear antigens EBNA 1, 2, 3A, 3B, 3C, and LP. Latent membrane proteins (LMP) 1, 2A and 2B and EBER expression. Latency II: Expression of EBNA1, LMP1/2A and EBER. Latency I: Expression of EBNA1 and EBER [60,61,62]. | LMP1 and LMP2A mimic the B-cell receptor and CD40 signaling pathways, constitutively activating NF-κB signaling [6,54,79,81]. | R-CHOP treatment [87,88] |
| HTLV-1 | ss RNA [59,60] | Cell-to-cell manner through GLUT1 receptor. Infected cells form virological synapses with uninfected cells [95,96] | Inhibition of p53; activation of PI3K/Akt signaling [62,63,97]; constitutive activation of NF-κB signaling [64,65]; Stimulation of CCND2 gene expression and phosphorylation of Rb protein [97,98,99] | CHOP and R-CHOP treatment [48,90] |
| HIV | ss linear RNA [66] | Direct mechanisms: Interaction with B-lymphocyte surface molecules: viral glycoprotein gp120, membrane immunoglobulins, C-type lectin receptors [100,101]; CD1 and CD21. Indirect mechanisms: Regulation of cytokine secretion deregulating B-cell differentiation and activation, leading to DNA modifications [102] Low CD21 levels and low B-cell response to antigenic stimuli; Induction of B-cell hyperactivation by increasing expression of activation-related molecules [103]. | Direct cell cycle control by Tat through Rb2/p130 protein interaction [66,67] | Antiretroviral therapy (HAART) [46,104]; R-CHOP and R-EPOCH [105] |
| SV40 | ds circular DNA [106] | Viral entry/endocytosis by interaction with major histocompatibility complex class I (MHC-I) molecules [107,108]. | T-ag-mediated inhibition of tumor suppressors p-Rb, p53, p107 and p130 [68,69,109]; T-ag- induced dissociation of E2F from p-RB [68,69,70,106,110]; Induction of cell growth, cell transformation and resistance to apoptosis by increased levels of the CBP/p300 protein [69,111,112]. | - |
| HBV | ds circular DNA [113,114] | Cell membrane interaction with glypican 5 or heparan sulfate proteoglycans, following clathrin-mediated endocytosis coordinated by EGFR into the endosomal network. HBV transition into the nucleus [115,116] | Activation of RIG-I, NF-κB, Wnt/β-catenin, TGF-β, GAS-STING, PI3K/AKT, JAK/STAT, RAS/MEK/ERK signaling pathways; Increased production IFN-α/-β, IL-1 and IL-6; Inhibition of TLR signaling, reduced production of TNF-α [117]. | Rituximab [118] |
| HCV | ss RNA [119] | Viral uptake and internalization by claudin-1, CD81, and SR-BI receptors [120] | BCL6 and p53 gene mutations [121]; Increased IL-6, IL-10, IL-17, and TGF-β production [122,123,124]; NS3 protein overexpression induces ROS leading to DNA damage [122,125,126] | Rituximab [127]; AVT [128] |
2.2. Human T-Cell Leukemia Virus Type 1
2.3. Human Immunodeficiency Virus
2.4. Simian Virus 40
2.5. Hepatitis B Virus
2.6. Hepatitis C Virus
2.7. Other Viruses
3. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Padala, S.A.; Kallam, A. Diffuse Large B Cell Lymphoma; StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Li, S.; Young, K.H.; Medeiros, L.J. Diffuse Large B-Cell Lymphoma. Pathology 2018, 50, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.H.; Salles, G. Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2021, 384, 842–858. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, L.; Young, K.H. New Agents and Regimens for Diffuse Large B Cell Lymphoma. J. Hematol. Oncol. 2020, 13, 175. [Google Scholar] [CrossRef]
- Harrington, F.; Greenslade, M.; Talaulikar, D.; Corboy, G. Genomic Characterisation of Diffuse Large B-Cell Lymphoma. Pathology 2021, 53, 367–376. [Google Scholar] [CrossRef]
- Crombie, J.L.; Armand, P. Diffuse Large B-Cell Lymphoma and High-Grade B-Cell Lymphoma. Hematol. Oncol. Clin. N. Am. 2019, 33, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Kesavan, M.; Eyre, T.A.; Collins, G.P. Front-Line Treatment of High Grade B Cell Non-Hodgkin Lymphoma. Curr. Hematol. Malig. Rep. 2019, 14, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Chiappella, A.; Castellino, A.; Nicolosi, M.; Santambrogio, E.; Vitolo, U. Diffuse Large B-Cell Lymphoma in the Elderly: Standard Treatment and New Perspectives. Expert Rev. Hematol. 2017, 10, 289–297. [Google Scholar] [CrossRef]
- Epperla, N.; Hamadani, M. Hematopoietic Cell Transplantation for Diffuse Large B-Cell and Follicular Lymphoma: Current Controversies and Advances. Hematol. Oncol. Stem Cell Ther. 2017, 10, 277–284. [Google Scholar] [CrossRef]
- Cioroianu, A.I.; Stinga, P.I.; Sticlaru, L.; Cioplea, M.D.; Nichita, L.; Popp, C.; Staniceanu, F. Tumor Microenvironment in Diffuse Large B-Cell Lymphoma: Role and Prognosis. Anal. Cell. Pathol. 2019, 2019, 8586354. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct Types of Diffuse Large B-Cell Lymphoma Identified by Gene Expression Profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Lenz, G.; Davis, R.E.; Ngo, V.N.; Lam, L.; George, T.C.; Wright, G.W.; Dave, S.S.; Zhao, H.; Xu, W.; Rosenwald, A.; et al. Oncogenic CARD11 Mutations in Human Diffuse Large B Cell Lymphoma. Science 2008, 319, 1676–1679. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Compagno, M.; Houldsworth, J.; Monti, S.; Grunn, A.; Nandula, S.V.; Aster, J.C.; Murty, V.V.; Shipp, M.A.; Dalla-Favera, R. Inactivation of the PRDM1/BLIMP1 Gene in Diffuse Large B Cell Lymphoma. J. Exp. Med. 2006, 203, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G. Insights into the Molecular Pathogenesis of Activated B-Cell-like Diffuse Large B-Cell Lymphoma and Its Therapeutic Implications. Cancers 2015, 7, 811–822. [Google Scholar] [CrossRef]
- Staudt, L.M. Oncogenic Activation of NF-KappaB. Cold Spring Harb. Perspect. Biol. 2010, 2, a000109. [Google Scholar] [CrossRef] [PubMed]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of Multiple Genes Cause Deregulation of NF-ΚB in Diffuse Large B-Cell Lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Nagel, I.; Siebert, R.; Roschke, A.V.; Sanger, W.; Wright, G.W.; Dave, S.S.; Tan, B.; Zhao, H.; Rosenwald, A.; et al. Aberrant Immunoglobulin Class Switch Recombination and Switch Translocations in Activated B Cell–like Diffuse Large B Cell Lymphoma. J. Exp. Med. 2007, 204, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic Active B-Cell-Receptor Signalling in Diffuse Large B-Cell Lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically Active MYD88 Mutations in Human Lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef]
- Iqbal, J.; Greiner, T.C.; Patel, K.; Dave, B.J.; Smith, L.; Ji, J.; Wright, G.; Sanger, W.G.; Pickering, D.L.; Jain, S.; et al. Distinctive Patterns of BCL6 Molecular Alterations and Their Functional Consequences in Different Subgroups of Diffuse Large B-Cell Lymphoma. Leukemia 2007, 21, 2332–2343. [Google Scholar] [CrossRef]
- Zhang, B.; Calado, D.P.; Wang, Z.; Fröhler, S.; Köchert, K.; Qian, Y.; Koralov, S.B.; Schmidt-Supprian, M.; Sasaki, Y.; Unitt, C.; et al. An Oncogenic Role for Alternative NF-ΚB Signaling in DLBCL Revealed upon Deregulated BCL6 Expression. Cell Rep. 2015, 11, 715–726. [Google Scholar] [CrossRef]
- Frick, M.; Dörken, B.; Lenz, G. The Molecular Biology of Diffuse Large B-Cell Lymphoma. Ther. Adv. Hematol. 2011, 2, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Wright, G.W.; Emre, N.C.T.; Kohlhammer, H.; Dave, S.S.; Davis, R.E.; Carty, S.; Lam, L.T.; Shaffer, A.L.; Xiao, W.; et al. Molecular Subtypes of Diffuse Large B-Cell Lymphoma Arise by Distinct Genetic Pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 13520–13525. [Google Scholar] [CrossRef] [PubMed]
- Bea, S. Diffuse Large B-Cell Lymphoma Subgroups Have Distinct Genetic Profiles That Influence Tumor Biology and Improve Gene-Expression-Based Survival Prediction. Blood 2005, 106, 3183–3190. [Google Scholar] [CrossRef]
- Weber, A.N.R.; Cardona Gloria, Y.; Çınar, Ö.; Reinhardt, H.C.; Pezzutto, A.; Wolz, O.-O. Oncogenic MYD88 Mutations in Lymphoma: Novel Insights and Therapeutic Possibilities. Cancer Immunol. Immunother. 2018, 67, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Basso, K.; Klein, U.; Niu, H.; Stolovitzky, G.A.; Tu, Y.; Califano, A.; Cattoretti, G.; Dalla-Favera, R. Tracking CD40 Signaling during Germinal Center Development. Blood 2004, 104, 4088–4096. [Google Scholar] [CrossRef] [PubMed]
- Lossos, I.S. Molecular Pathogenesis of Diffuse Large B-Cell Lymphoma. JCO 2005, 23, 6351–6357. [Google Scholar] [CrossRef]
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The Use of Molecular Profiling to Predict Survival after Chemotherapy for Diffuse Large-B-Cell Lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947. [Google Scholar] [CrossRef]
- Frontzek, F.; Lenz, G. Novel Insights into the Pathogenesis of Molecular Subtypes of Diffuse Large B-Cell Lymphoma and Their Clinical Implications. Expert Rev. Clin. Pharmacol. 2019, 12, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Dal Bo, M.; Bomben, R.; Hernández, L.; Gattei, V. The MYC/MiR-17-92 Axis in Lymphoproliferative Disorders: A Common Pathway with Therapeutic Potential. Oncotarget 2015, 6, 19381–19392. [Google Scholar] [CrossRef]
- Béguelin, W.; Popovic, R.; Teater, M.; Jiang, Y.; Bunting, K.L.; Rosen, M.; Shen, H.; Yang, S.N.; Wang, L.; Ezponda, T.; et al. EZH2 Is Required for Germinal Center Formation and Somatic EZH2 Mutations Promote Lymphoid Transformation. Cancer Cell 2013, 23, 677–692. [Google Scholar] [CrossRef]
- Liu, Y.; Barta, S.K. Diffuse Large B-cell Lymphoma: 2019 Update on Diagnosis, Risk Stratification, and Treatment. Am. J. Hematol. 2019, 94, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Högfeldt, T.; Jaing, C.; Loughlin, K.M.; Thissen, J.; Gardner, S.; Bahnassy, A.A.; Gharizadeh, B.; Lundahl, J.; Österborg, A.; Porwit, A.; et al. Differential Expression of Viral Agents in Lymphoma Tissues of Patients with ABC Diffuse Large B-Cell Lymphoma from High and Low Endemic Infectious Disease Regions. Oncol. Lett. 2016, 12, 2782–2788. [Google Scholar] [CrossRef]
- Jarrett, R.F. Viruses and Lymphoma/Leukaemia. J. Pathol. 2006, 208, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Chang, Y. Why Do Viruses Cause Cancer? Highlights of the First Century of Human Tumour Virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef]
- Ringelhan, M.; McKeating, J.A.; Protzer, U. Viral Hepatitis and Liver Cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160274. [Google Scholar] [CrossRef] [PubMed]
- Wardak, S. Human Papillomavirus (HPV) and Cervical Cancer. Med. Dosw. Mikrobiol. 2016, 68, 73–84. [Google Scholar]
- Mahmutović, L.; Bilajac, E.; Hromić-Jahjefendić, A. Meet the Insidious Players: Review of Viral Infections in Head and Neck Cancer Etiology with an Update on Clinical Trials. Microorganisms 2021, 9, 1001. [Google Scholar] [CrossRef]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global Burden of Cancer Attributable to Infections in 2018: A Worldwide Incidence Analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Kellogg, C.; Kouznetsova, V.L.; Tsigelny, I.F. Implications of Viral Infection in Cancer Development. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188622. [Google Scholar] [CrossRef]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human Viral Oncogenesis: A Cancer Hallmarks Analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef]
- Akram, N.; Imran, M.; Noreen, M.; Ahmed, F.; Atif, M.; Fatima, Z.; Bilal Waqar, A. Oncogenic Role of Tumor Viruses in Humans. Viral Immunol. 2017, 30, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Desfarges, S.; Ciuffi, A. Viral Integration and Consequences on Host Gene Expression. In Viruses: Essential Agents of Life; Witzany, G., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 147–175. ISBN 978-94-007-4898-9. [Google Scholar]
- Wang, Y.; Wang, H.; Pan, S.; Hu, T.; Shen, J.; Zheng, H.; Xie, S.; Xie, Y.; Lu, R.; Guo, L. Capable Infection of Hepatitis B Virus in Diffuse Large B-Cell Lymphoma. J. Cancer 2018, 9, 1575–1581. [Google Scholar] [CrossRef] [PubMed]
- Shibusawa, M.; Kidoguchi, K.; Tanimoto, T. Epstein-Barr Virus-Positive Diffuse Large B Cell Lymphoma. In Lymphoma; Gallamini, A., Juweid, M., Eds.; Exon Publications: Brisbane, Australia, 2021; pp. 27–46. ISBN 978-0-645-33200-1. [Google Scholar]
- Besson, C.; Lancar, R.; Prevot, S.; Algarte-Genin, M.; Delobel, P.; Bonnet, F.; Meyohas, M.-C.; Partisani, M.; Oberic, L.; Gabarre, J.; et al. Outcomes for HIV-Associated Diffuse Large B-Cell Lymphoma in the Modern Combined Antiretroviral Therapy Era. AIDS 2017, 31, 2493–2501. [Google Scholar] [CrossRef] [PubMed]
- Visco, C.; Finotto, S. Hepatitis C Virus and Diffuse Large B-Cell Lymphoma: Pathogenesis, Behavior and Treatment. World J. Gastroenterol. 2014, 20, 11054–11061. [Google Scholar] [CrossRef]
- Beltran, B.E.; Quiñones, P.; Morales, D.; Revilla, J.C.; Alva, J.C.; Castillo, J.J. Diffuse Large B-Cell Lymphoma in Human T-Lymphotropic Virus Type 1 Carriers. Leuk. Res. Treat. 2012, 2012, 262363. [Google Scholar] [CrossRef]
- Amara, K.; Trimeche, M.; Ziadi, S.; Laatiri, A.; Hachana, M.; Sriha, B.; Mokni, M.; Korbi, S. Presence of Simian Virus 40 DNA Sequences in Diffuse Large B-Cell Lymphomas in Tunisia Correlates with Aberrant Promoter Hypermethylation of Multiple Tumor Suppressor Genes. Int. J. Cancer 2007, 121, 2693–2702. [Google Scholar] [CrossRef]
- Amara, K.; Trimeche, M.; Ziadi, S.; Laatiri, A.; Mestiri, S.; Sriha, B.; Mokni, M.; Korbi, S. Presence of Simian Virus 40 in Diffuse Large B-Cell Lymphomas in Tunisia Correlates with Germinal Center B-Cell Immunophenotype, t(14;18) Translocation, and P53 Accumulation. Mod. Pathol. 2008, 21, 282–296. [Google Scholar] [CrossRef]
- Sreehari, S. High Risk HPV, HSIL and Primary Diffuse Large B Cell Lymphoma of Cervix: An Unsual Case. IJCSMB 2019, 5, 555666. [Google Scholar] [CrossRef]
- Van, J.; Dave, A.A.; Schwartz, M.; Mitchell, J. S2443 A Case of HHV-8 Diffuse Large B-Cell Lymphoma—Not Otherwise Specified With Liver Infiltration. Am. J. Gastroenterol. 2020, 115, S1294. [Google Scholar] [CrossRef]
- Schiller, J.T.; Lowy, D.R. An Introduction to Virus Infections and Human Cancer. In Viruses and Human Cancer; Wu, T.-C., Chang, M.-H., Jeang, K.-T., Eds.; Recent Results in Cancer Research; Springer International Publishing: Cham, Swizherland, 2021; Volume 217, pp. 1–11. ISBN 978-3-030-57361-4. [Google Scholar]
- Montes-Moreno, S.; Odqvist, L.; Diaz-Perez, J.A.; Lopez, A.B.; de Villambrosía, S.G.; Mazorra, F.; Castillo, M.E.; Lopez, M.; Pajares, R.; García, J.F.; et al. EBV-Positive Diffuse Large B-Cell Lymphoma of the Elderly Is an Aggressive Post-Germinal Center B-Cell Neoplasm Characterized by Prominent Nuclear Factor-KB Activation. Mod. Pathol. 2012, 25, 968–982. [Google Scholar] [CrossRef]
- Mancao, C.; Hammerschmidt, W. Epstein-Barr Virus Latent Membrane Protein 2A Is a B-Cell Receptor Mimic and Essential for B-Cell Survival. Blood 2007, 110, 3715–3721. [Google Scholar] [CrossRef] [PubMed]
- Mosialos, G.; Birkenbach, M.; Yalamanchili, R.; VanArsdale, T.; Ware, C.; Kieff, E. The Epstein-Barr Virus Transforming Protein LMP1 Engages Signaling Proteins for the Tumor Necrosis Factor Receptor Family. Cell 1995, 80, 389–399. [Google Scholar] [CrossRef]
- Dawson, C.W.; Tramountanis, G.; Eliopoulos, A.G.; Young, L.S. Epstein-Barr Virus Latent Membrane Protein 1 (LMP1) Activates the Phosphatidylinositol 3-Kinase/Akt Pathway to Promote Cell Survival and Induce Actin Filament Remodeling. J. Biol. Chem. 2003, 278, 3694–3704. [Google Scholar] [CrossRef] [PubMed]
- Miura, M.; Naito, T.; Saito, M. Current Perspectives in Human T-Cell Leukemia Virus Type 1 Infection and Its Associated Diseases. Front. Med. 2022, 9, 867478. [Google Scholar] [CrossRef]
- Panfil, A.R.; Martinez, M.P.; Ratner, L.; Green, P.L. Human T-Cell Leukemia Virus-Associated Malignancy. Curr. Opin. Virol. 2016, 20, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi Ghezeldasht, S.; Shirdel, A.; Assarehzadegan, M.A.; Hassannia, T.; Rahimi, H.; Miri, R.; Rezaee, S.A.R. Human T Lymphotropic Virus Type I (HTLV-I) Oncogenesis: Molecular Aspects of Virus and Host Interactions in Pathogenesis of Adult T Cell Leukemia/Lymphoma (ATL). Iran J. Basic Med. Sci. 2013, 16, 179–195. [Google Scholar] [PubMed]
- Mota, T.M.; Jones, R.B. HTLV-1 as a Model for Virus and Host Coordinated Immunoediting. Front. Immunol. 2019, 10, 2259. [Google Scholar] [CrossRef]
- Jeong, S.-J.; Pise-Masison, C.A.; Radonovich, M.F.; Park, H.U.; Brady, J.N. Activated AKT Regulates NF-KappaB Activation, P53 Inhibition and Cell Survival in HTLV-1-Transformed Cells. Oncogene 2005, 24, 6719–6728. [Google Scholar] [CrossRef]
- Peloponese, J.-M.; Jeang, K.-T. Role for Akt/Protein Kinase B and Activator Protein-1 in Cellular Proliferation Induced by the Human T-Cell Leukemia Virus Type 1 Tax Oncoprotein. J. Biol. Chem. 2006, 281, 8927–8938. [Google Scholar] [CrossRef]
- Sun, S.-C.; Yamaoka, S. Activation of NF-ΚB by HTLV-I and Implications for Cell Transformation. Oncogene 2005, 24, 5952–5964. [Google Scholar] [CrossRef]
- Iha, H.; Kibler, K.V.; Yedavalli, V.R.K.; Peloponese, J.-M.; Haller, K.; Miyazato, A.; Kasai, T.; Jeang, K.-T. Segregation of NF-KappaB Activation through NEMO/IKKgamma by Tax and TNFalpha: Implications for Stimulus-Specific Interruption of Oncogenic Signaling. Oncogene 2003, 22, 8912–8923. [Google Scholar] [CrossRef] [PubMed]
- Grogg, K.L.; Miller, R.F.; Dogan, A. HIV Infection and Lymphoma. J. Clin. Pathol. 2007, 60, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Bellan, C.; Lazzi, S.; De Falco, G.; Nyongo, A.; Giordano, A.; Leoncini, L. Burkitt’s Lymphoma: New Insights into Molecular Pathogenesis. J. Clin. Pathol. 2003, 56, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Butel, J.S.; Lednicky, J.A. Cell and Molecular Biology of Simian Virus 40: Implications for Human Infections and Disease. JNCI J. Natl. Cancer Inst. 1999, 91, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Sáenz-Robles, M.T.; Sullivan, C.S.; Pipas, J.M. Transforming Functions of Simian Virus 40. Oncogene 2001, 20, 7899–7907. [Google Scholar] [CrossRef]
- Wang, J.Y.; Knudsen, E.S.; Welch, P.J. The Retinoblastoma Tumor Suppressor Protein. Adv. Cancer Res. 1994, 64, 25–85. [Google Scholar] [CrossRef]
- Ayee, R.; Ofori, M.E.; Wright, E.; Quaye, O. Epstein Barr Virus Associated Lymphomas and Epithelia Cancers in Humans. J. Cancer 2020, 11, 1737–1750. [Google Scholar] [CrossRef]
- Nowalk, A.; Green, M. Epstein-Barr Virus. Microbiol. Spectr. 2016, 4, 47. [Google Scholar] [CrossRef]
- Bakkalci, D.; Jia, Y.; Winter, J.R.; Lewis, J.E.; Taylor, G.S.; Stagg, H.R. Risk Factors for Epstein Barr Virus-Associated Cancers: A Systematic Review, Critical Appraisal, and Mapping of the Epidemiological Evidence. J. Glob. Health 2020, 10, 010405. [Google Scholar] [CrossRef]
- Bauer, M.; Jasinski-Bergner, S.; Mandelboim, O.; Wickenhauser, C.; Seliger, B. Epstein-Barr Virus-Associated Malignancies and Immune Escape: The Role of the Tumor Microenvironment and Tumor Cell Evasion Strategies. Cancers 2021, 13, 5189. [Google Scholar] [CrossRef]
- Cohen, J.I. Epstein-Barr Virus Infection. N. Engl. J. Med. 2000, 343, 481–492. [Google Scholar] [CrossRef]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr Virus: More than 50 Years Old and Still Providing Surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Santpere, G.; Darre, F.; Blanco, S.; Alcami, A.; Villoslada, P.; Mar Albà, M.; Navarro, A. Genome-Wide Analysis of Wild-Type Epstein-Barr Virus Genomes Derived from Healthy Individuals of the 1000 Genomes Project. Genome Biol. Evol. 2014, 6, 846–860. [Google Scholar] [CrossRef]
- Oyama, T.; Yamamoto, K.; Asano, N.; Oshiro, A.; Suzuki, R.; Kagami, Y.; Morishima, Y.; Takeuchi, K.; Izumo, T.; Mori, S.; et al. Age-Related EBV-Associated B-Cell Lymphoproliferative Disorders Constitute a Distinct Clinicopathologic Group: A Study of 96 Patients. Clin. Cancer Res. 2007, 13, 5124–5132. [Google Scholar] [CrossRef] [PubMed]
- Ok, C.Y.; Papathomas, T.G.; Medeiros, L.J.; Young, K.H. EBV-Positive Diffuse Large B-Cell Lymphoma of the Elderly. Blood 2013, 122, 328–340. [Google Scholar] [CrossRef]
- Nicolae, A.; Pittaluga, S.; Abdullah, S.; Steinberg, S.M.; Pham, T.A.; Davies-Hill, T.; Xi, L.; Raffeld, M.; Jaffe, E.S. EBV-Positive Large B-Cell Lymphomas in Young Patients: A Nodal Lymphoma with Evidence for a Tolerogenic Immune Environment. Blood 2015, 126, 863–872. [Google Scholar] [CrossRef]
- Hofscheier, A.; Ponciano, A.; Bonzheim, I.; Adam, P.; Lome-Maldonado, C.; Vela, T.; Cortes, E.; Ortiz-Hidalgo, C.; Fend, F.; Quintanilla-Martinez, L. Geographic Variation in the Prevalence of Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma of the Elderly: A Comparative Analysis of a Mexican and a German Population. Mod. Pathol. 2011, 24, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Park, S.; Ju, H.; Ha, S.Y.; Sohn, I.; Jo, J.; Do, I.-G.; Min, S.; Kim, S.J.; Kim, W.S.; et al. Integrated Copy Number and Gene Expression Profiling Analysis of Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma. Genes Chromosomes Cancer 2015, 54, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Karube, K.; Yamamoto, K.; Takizawa, J.; Tsuzuki, S.; Yatabe, Y.; Kanda, T.; Katayama, M.; Ozawa, Y.; Ishitsuka, K.; et al. Gene Expression Profiling of Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma of the Elderly Reveals Alterations of Characteristic Oncogenetic Pathways. Cancer Sci. 2014, 105, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Miyoshi, H.; Sakata, S.; Dobashi, A.; Couronné, L.; Kogure, Y.; Sato, Y.; Nishida, K.; Gion, Y.; Shiraishi, Y.; et al. Frequent Structural Variations Involving Programmed Death Ligands in Epstein-Barr Virus-Associated Lymphomas. Leukemia 2019, 33, 1687–1699. [Google Scholar] [CrossRef]
- Gebauer, N.; Gebauer, J.; Hardel, T.T.; Bernard, V.; Biersack, H.; Lehnert, H.; Rades, D.; Feller, A.C.; Thorns, C. Prevalence of Targetable Oncogenic Mutations and Genomic Alterations in Epstein-Barr Virus-Associated Diffuse Large B-Cell Lymphoma of the Elderly. Leuk. Lymphoma 2015, 56, 1100–1106. [Google Scholar] [CrossRef]
- Castillo, J.J.; Beltran, B.E.; Miranda, R.N.; Paydas, S.; Winer, E.S.; Butera, J.N. Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma of the Elderly: What We Know so Far. Oncologist 2011, 16, 87–96. [Google Scholar] [CrossRef]
- Lu, T.-X.; Liang, J.-H.; Miao, Y.; Fan, L.; Wang, L.; Qu, X.-Y.; Cao, L.; Gong, Q.-X.; Wang, Z.; Zhang, Z.-H.; et al. Epstein-Barr Virus Positive Diffuse Large B-Cell Lymphoma Predict Poor Outcome, Regardless of the Age. Sci. Rep. 2015, 5, 12168. [Google Scholar] [CrossRef]
- Murthy, S.L.; Hitchcock, M.A.; Endicott-Yazdani, T.R.; Watson, J.T.; Krause, J.R. Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma. Bayl. Univ. Med. Cent. Proc. 2017, 30, 443–444. [Google Scholar] [CrossRef]
- Bourbon, E.; Maucort-Boulch, D.; Fontaine, J.; Mauduit, C.; Sesques, P.; Safar, V.; Ferrant, E.; Golfier, C.; Ghergus, D.; Karlin, L.; et al. Clinicopathological Features and Survival in EBV-Positive Diffuse Large B-Cell Lymphoma Not Otherwise Specified. Blood Adv. 2021, 5, 3227–3239. [Google Scholar] [CrossRef]
- Beltran, B.E.; Castro, D.; Paredes, S.; Miranda, R.N.; Castillo, J.J. EBV-Positive Diffuse Large B-Cell Lymphoma, Not Otherwise Specified: 2020 Update on Diagnosis, Risk-Stratification and Management. Am. J. Hematol. 2020, 95, 435–445. [Google Scholar] [CrossRef]
- Long, H.M.; Taylor, G.S.; Rickinson, A.B. Immune Defence against EBV and EBV-Associated Disease. Curr. Opin. Immunol. 2011, 23, 258–264. [Google Scholar] [CrossRef]
- Rooney, C.M.; Smith, C.A.; Ng, C.Y.; Loftin, S.; Li, C.; Krance, R.A.; Brenner, M.K.; Heslop, H.E. Use of Gene-Modified Virus-Specific T Lymphocytes to Control Epstein-Barr-Virus-Related Lymphoproliferation. Lancet 1995, 345, 9–13. [Google Scholar] [CrossRef]
- Savoldo, B.; Goss, J.A.; Hammer, M.M.; Zhang, L.; Lopez, T.; Gee, A.P.; Lin, Y.-F.; Quiros-Tejeira, R.E.; Reinke, P.; Schubert, S.; et al. Treatment of Solid Organ Transplant Recipients with Autologous Epstein Barr Virus-Specific Cytotoxic T Lymphocytes (CTLs). Blood 2006, 108, 2942–2949. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-Associated B7-H1 Promotes T-Cell Apoptosis: A Potential Mechanism of Immune Evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Igakura, T.; Stinchcombe, J.C.; Goon, P.K.C.; Taylor, G.P.; Weber, J.N.; Griffiths, G.M.; Tanaka, Y.; Osame, M.; Bangham, C.R.M. Spread of HTLV-I between Lymphocytes by Virus-Induced Polarization of the Cytoskeleton. Science 2003, 299, 1713–1716. [Google Scholar] [CrossRef]
- Manel, N.; Kim, F.J.; Kinet, S.; Taylor, N.; Sitbon, M.; Battini, J.-L. The Ubiquitous Glucose Transporter GLUT-1 Is a Receptor for HTLV. Cell 2003, 115, 449–459. [Google Scholar] [CrossRef]
- Matsuoka, M.; Jeang, K.-T. Human T-Cell Leukaemia Virus Type 1 (HTLV-1) Infectivity and Cellular Transformation. Nat. Rev. Cancer 2007, 7, 270–280. [Google Scholar] [CrossRef]
- Iwanaga, R.; Ohtani, K.; Hayashi, T.; Nakamura, M. Molecular Mechanism of Cell Cycle Progression Induced by the Oncogene Product Tax of Human T-Cell Leukemia Virus Type I. Oncogene 2001, 20, 2055–2067. [Google Scholar] [CrossRef]
- Haller, K.; Wu, Y.; Derow, E.; Schmitt, I.; Jeang, K.-T.; Grassmann, R. Physical Interaction of Human T-Cell Leukemia Virus Type 1 Tax with Cyclin-Dependent Kinase 4 Stimulates the Phosphorylation of Retinoblastoma Protein. Mol. Cell Biol. 2002, 22, 3327–3338. [Google Scholar] [CrossRef]
- Cagigi, A.; Du, L.; Dang, L.V.P.; Grutzmeier, S.; Atlas, A.; Chiodi, F.; Pan-Hammarström, Q.; Nilsson, A. CD27(-) B-Cells Produce Class Switched and Somatically Hyper-Mutated Antibodies during Chronic HIV-1 Infection. PLoS ONE 2009, 4, e5427. [Google Scholar] [CrossRef]
- He, B.; Qiao, X.; Klasse, P.J.; Chiu, A.; Chadburn, A.; Knowles, D.M.; Moore, J.P.; Cerutti, A. HIV-1 Envelope Triggers Polyclonal Ig Class Switch Recombination through a CD40-Independent Mechanism Involving BAFF and C-Type Lectin Receptors. J. Immunol. 2006, 176, 3931–3941. [Google Scholar] [CrossRef]
- Ho, J.; Moir, S.; Kulik, L.; Malaspina, A.; Donoghue, E.T.; Miller, N.J.; Wang, W.; Chun, T.-W.; Fauci, A.S.; Holers, V.M. Role for CD21 in the Establishment of an Extracellular HIV Reservoir in Lymphoid Tissues. J. Immunol. 2007, 178, 6968–6974. [Google Scholar] [CrossRef]
- De Carvalho, P.S.; Leal, F.E.; Soares, M.A. Clinical and Molecular Properties of Human Immunodeficiency Virus-Related Diffuse Large B-Cell Lymphoma. Front. Oncol. 2021, 11, 675353. [Google Scholar] [CrossRef]
- Grulich, A.E.; Li, Y.; McDonald, A.M.; Correll, P.K.; Law, M.G.; Kaldor, J.M. Decreasing Rates of Kaposi’s Sarcoma and Non-Hodgkin’s Lymphoma in the Era of Potent Combination Anti-Retroviral Therapy. AIDS 2001, 15, 629–633. [Google Scholar] [CrossRef]
- Vandenhende, M.-A.; Roussillon, C.; Henard, S.; Morlat, P.; Oksenhendler, E.; Aumaitre, H.; Georget, A.; May, T.; Rosenthal, E.; Salmon, D.; et al. Cancer-Related Causes of Death among HIV-Infected Patients in France in 2010: Evolution since 2000. PLoS ONE 2015, 10, e0129550. [Google Scholar] [CrossRef]
- Vilchez, R.A.; Butel, J.S. SV40 in Human Brain Cancers and Non-Hodgkin’s Lymphoma. Oncogene 2003, 22, 5164–5172. [Google Scholar] [CrossRef] [PubMed]
- Stang, E.; Kartenbeck, J.; Parton, R.G. Major Histocompatibility Complex Class I Molecules Mediate Association of SV40 with Caveolae. MBoC 1997, 8, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Toscano, M.G.; de Haan, P. How Simian Virus 40 Hijacks the Intracellular Protein Trafficking Pathway to Its Own Benefit … and Ours. Front. Immunol. 2018, 9, 1160. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, J.C.; Mazzoni, E.; Bononi, I.; Tognon, M.; Martini, F. Association Between Simian Virus 40 and Human Tumors. Front. Oncol. 2019, 9, 670. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.S.; Pipas, J.M. T Antigens of Simian Virus 40: Molecular Chaperones for Viral Replication and Tumorigenesis. Microbiol. Mol. Biol. Rev. 2002, 66, 179–202. [Google Scholar] [CrossRef]
- Bocchetta, M.; Eliasz, S.; De Marco, M.A.; Rudzinski, J.; Zhang, L.; Carbone, M. The SV40 Large T Antigen-P53 Complexes Bind and Activate the Insulin-like Growth Factor-I Promoter Stimulating Cell Growth. Cancer Res. 2008, 68, 1022–1029. [Google Scholar] [CrossRef]
- Atkin, S.J.L.; Griffin, B.E.; Dilworth, S.M. Polyoma Virus and Simian Virus 40 as Cancer Models: History and Perspectives. Semin. Cancer Biol. 2009, 19, 211–217. [Google Scholar] [CrossRef]
- Tsai, K.-N.; Kuo, C.-F.; Ou, J.-H.J. Mechanisms of Hepatitis B Virus Persistence. Trends Microbiol. 2018, 26, 33–42. [Google Scholar] [CrossRef]
- Tong, S.; Revill, P. Overview of Hepatitis B Viral Replication and Genetic Variability. J. Hepatol. 2016, 64, S4–S16. [Google Scholar] [CrossRef]
- Herrscher, C.; Roingeard, P.; Blanchard, E. Hepatitis B Virus Entry into Cells. Cells 2020, 9, 1486. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Hildt, E. Intracellular Trafficking of HBV Particles. Cells 2020, 9, 2023. [Google Scholar] [CrossRef] [PubMed]
- Vincent, I.E.; Zannetti, C.; Lucifora, J.; Norder, H.; Protzer, U.; Hainaut, P.; Zoulim, F.; Tommasino, M.; Trépo, C.; Hasan, U.; et al. Hepatitis B Virus Impairs TLR9 Expression and Function in Plasmacytoid Dendritic Cells. PLoS ONE 2011, 6, e26315. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, Y.; Yamamoto, Y.; Ito, S.; Ohigashi, H.; Shiratori, S.; Naruse, H.; Teshima, T. Hepatitis B Virus Reactivation with a Rituximab-Containing Regimen. World J. Hepatol. 2015, 7, 2344–2351. [Google Scholar] [CrossRef]
- Douam, F.; Lavillette, D.; Cosset, F.-L. The Mechanism of HCV Entry into Host Cells. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2015; Volume 129, pp. 63–107. ISBN 978-0-12-802461-4. [Google Scholar]
- Burlone, M.E.; Budkowska, A. Hepatitis C Virus Cell Entry: Role of Lipoproteins and Cellular Receptors. J. Gen. Virol. 2009, 90, 1055–1070. [Google Scholar] [CrossRef]
- Machida, K.; Cheng, K.T.-N.; Sung, V.M.-H.; Shimodaira, S.; Lindsay, K.L.; Levine, A.M.; Lai, M.-Y.; Lai, M.M.C. Hepatitis C Virus Induces a Mutator Phenotype: Enhanced Mutations of Immunoglobulin and Protooncogenes. Proc. Natl. Acad. Sci. USA 2004, 101, 4262–4267. [Google Scholar] [CrossRef]
- Couronné, L.; Bachy, E.; Roulland, S.; Nadel, B.; Davi, F.; Armand, M.; Canioni, D.; Michot, J.M.; Visco, C.; Arcaini, L.; et al. From Hepatitis C Virus Infection to B-Cell Lymphoma. Ann. Oncol. 2018, 29, 92–100. [Google Scholar] [CrossRef]
- Machida, K.; Cheng, K.T.-H.; Pavio, N.; Sung, V.M.-H.; Lai, M.M.C. Hepatitis C Virus E2-CD81 Interaction Induces Hypermutation of the Immunoglobulin Gene in B Cells. J. Virol. 2005, 79, 8079–8089. [Google Scholar] [CrossRef]
- Ishikawa, T.; Shibuya, K.; Yasui, K.; Mitamura, K.; Ueda, S. Expression of Hepatitis C Virus Core Protein Associated with Malignant Lymphoma in Transgenic Mice. Comp. Immunol. Microbiol. Infect. Dis. 2003, 26, 115–124. [Google Scholar] [CrossRef]
- Dai, B.; Chen, A.Y.; Corkum, C.P.; Peroutka, R.J.; Landon, A.; Houng, S.; Muniandy, P.A.; Zhang, Y.; Lehrmann, E.; Mazan-Mamczarz, K.; et al. Hepatitis C Virus Upregulates B-Cell Receptor Signaling: A Novel Mechanism for HCV-Associated B-Cell Lymphoproliferative Disorders. Oncogene 2016, 35, 2979–2990. [Google Scholar] [CrossRef]
- Machida, K.; Cheng, K.T.-H.; Sung, V.M.-H.; Lee, K.J.; Levine, A.M.; Lai, M.M.C. Hepatitis C Virus Infection Activates the Immunologic (Type II) Isoform of Nitric Oxide Synthase and Thereby Enhances DNA Damage and Mutations of Cellular Genes. J. Virol. 2004, 78, 8835–8843. [Google Scholar] [CrossRef] [PubMed]
- Hosry, J.; Mahale, P.; Turturro, F.; Miranda, R.N.; Economides, M.P.; Granwehr, B.P.; Torres, H.A. Antiviral Therapy Improves Overall Survival in Hepatitis C Virus-Infected Patients Who Develop Diffuse Large B-Cell Lymphoma. Int. J. Cancer 2016, 139, 2519–2528. [Google Scholar] [CrossRef] [PubMed]
- Michot, J.-M.; Canioni, D.; Driss, H.; Alric, L.; Cacoub, P.; Suarez, F.; Sibon, D.; Thieblemont, C.; Dupuis, J.; Terrier, B.; et al. Antiviral Therapy Is Associated with a Better Survival in Patients with Hepatitis C Virus and B-Cell Non-Hodgkin Lymphomas, ANRS HC-13 Lympho-C Study. Am. J. Hematol. 2015, 90, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Valcarcel, B.; Ampuero, G.S.; de la Cruz-Ku, G.; Enriquez, D.J.; Malpica, L. Outcomes of HTLV-1 Carriers with Diffuse Large B-Cell Lymphoma: A Single-Center Retrospective Matched Cohort Study. Clin. Lymphoma. Myeloma. Leuk. 2022, 22, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Kasinathan, G.; Sathar, J. Peripheral Lymphocytosis Presenting as EBV/HTLV-1 Co-Infection Adult T-Cell Leukemia. Hematol. Transfus. Cell Ther. 2022, 44, 279–283. [Google Scholar] [CrossRef]
- Proietti, F.A.; Carneiro-Proietti, A.B.F.; Catalan-Soares, B.C.; Murphy, E.L. Global Epidemiology of HTLV-I Infection and Associated Diseases. Oncogene 2005, 24, 6058–6068. [Google Scholar] [CrossRef]
- Gonçalves, D.U.; Proietti, F.A.; Ribas, J.G.R.; Araújo, M.G.; Pinheiro, S.R.; Guedes, A.C.; Carneiro-Proietti, A.B.F. Epidemiology, Treatment, and Prevention of Human T-Cell Leukemia Virus Type 1-Associated Diseases. Clin. Microbiol. Rev. 2010, 23, 577–589. [Google Scholar] [CrossRef]
- Zhao, T. The Role of HBZ in HTLV-1-Induced Oncogenesis. Viruses 2016, 8, 34. [Google Scholar] [CrossRef]
- Zhao, T.; Matsuoka, M. HBZ and Its Roles in HTLV-1 Oncogenesis. Front. Microbiol. 2012, 3, 247. [Google Scholar] [CrossRef]
- Suefuji, H.; Ohshima, K.; Hayabuchi, N.; Nakamura, K.; Kikuchi, M. HTLV-1 Carriers with B-Cell Lymphoma of Localized Stage Head and Neck: Prognosis, Clinical and Immunopathological Features. Br. J. Haematol. 2003, 123, 606–612. [Google Scholar] [CrossRef]
- Valcarcel, B.; Enriquez, D.J.; Sandival-Ampuero, G.; Aviles-Perez, U.; Haro, J.C.; Alcarraz, C.; Quintana, S.; Villena, M.; Dueñas, D.; Casavilca, S.; et al. Clinical Characteristics and Outcome of Diffuse Large B Cell Lymphoma Among HTLV-1 Carriers in Peru: A Matched Cohort Study. Blood 2020, 136, 38–39. [Google Scholar] [CrossRef]
- Isaguliants, M.; Bayurova, E.; Avdoshina, D.; Kondrashova, A.; Chiodi, F.; Palefsky, J.M. Oncogenic Effects of HIV-1 Proteins, Mechanisms Behind. Cancers 2021, 13, 305. [Google Scholar] [CrossRef] [PubMed]
- Laurence, J.; Astrin, S.M. Human Immunodeficiency Virus Induction of Malignant Transformation in Human B Lymphocytes. Proc. Natl. Acad. Sci. USA 1991, 88, 7635–7639. [Google Scholar] [CrossRef] [PubMed]
- Planès, R.; Serrero, M.; Leghmari, K.; BenMohamed, L.; Bahraoui, E. HIV-1 Envelope Glycoproteins Induce the Production of TNF-α and IL-10 in Human Monocytes by Activating Calcium Pathway. Sci. Rep. 2018, 8, 17215. [Google Scholar] [CrossRef] [PubMed]
- Malaspina, A.; Moir, S.; Kottilil, S.; Hallahan, C.W.; Ehler, L.A.; Liu, S.; Planta, M.A.; Chun, T.-W.; Fauci, A.S. Deleterious Effect of HIV-1 Plasma Viremia on B Cell Costimulatory Function. J. Immunol. 2003, 170, 5965–5972. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, S.; Sun, J.; Xu, J.; Zhang, X. Irreversible Phenotypic Perturbation and Functional Impairment of B Cells during HIV-1 Infection. Front. Med. 2017, 11, 536–547. [Google Scholar] [CrossRef]
- Wu, J.; Miao, Y.; Qian, C.; Tao, P.; Wang, X.; Dong, X.; Li, X.; Lou, J.; Liang, J.; Xu, W.; et al. Clinical Characteristics and Outcomes in HIV-Associated Diffuse Large B-Cell Lymphoma in China: A Retrospective Single-Center Study. J. Cancer 2021, 12, 2903–2911. [Google Scholar] [CrossRef]
- Diamond, C.; Taylor, T.H.; Aboumrad, T.; Anton-Culver, H. Changes in Acquired Immunodeficiency Syndrome-Related Non-Hodgkin Lymphoma in the Era of Highly Active Antiretroviral Therapy: Incidence, Presentation, Treatment, and Survival. Cancer 2006, 106, 128–135. [Google Scholar] [CrossRef]
- Hoffmann, C.; Wolf, E.; Fätkenheuer, G.; Buhk, T.; Stoehr, A.; Plettenberg, A.; Stellbrink, H.-J.; Jaeger, H.; Siebert, U.; Horst, H.-A. Response to Highly Active Antiretroviral Therapy Strongly Predicts Outcome in Patients with AIDS-Related Lymphoma. AIDS 2003, 17, 1521–1529. [Google Scholar] [CrossRef]
- Tsuyama, N.; Sakata, S.; Baba, S.; Mishima, Y.; Nishimura, N.; Ueda, K.; Yokoyama, M.; Terui, Y.; Hatake, K.; Kitagawa, M.; et al. BCL2 Expression in DLBCL: Reappraisal of Immunohistochemistry with New Criteria for Therapeutic Biomarker Evaluation. Blood 2017, 130, 489–500. [Google Scholar] [CrossRef]
- Philippe, L.; Lancar, R.; Laurent, C.; Algarte-Genin, M.; Chassagne-Clément, C.; Fabiani, B.; Pierre Chenard, M.; Lazure, T.; Parrens, M.; Charlotte, F.; et al. In Situ BCL2 Expression Is an Independent Prognostic Factor in HIV-Associated DLBCL, a LYMPHOVIR Cohort Study. Br. J. Haematol. 2020, 188, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Stratton, K.; Almario, D.A.; McCormick, M.C. (Eds.) Immunization Safety Review: SV40 Contamination of Polio Vaccine and Cancer; National Academies Press: Washington, DC, USA, 2002; p. 10534. ISBN 978-0-309-08610-3. [Google Scholar]
- McNees, A.L.; Butel, J.S. Simian Virus 40. In Encyclopedia of Virology; Elsevier: Amsterdam, The Netherlands, 2008; pp. 630–638. ISBN 978-0-12-374410-4. [Google Scholar]
- Tognon, M.; Luppi, M.; Corallini, A.; Taronna, A.; Barozzi, P.; Rotondo, J.C.; Comar, M.; Casali, M.V.; Bovenzi, M.; D’Agostino, A.; et al. Immunologic Evidence of a Strong Association between Non-Hodgkin Lymphoma and Simian Virus 40: NHL and SV40. Cancer 2015, 121, 2618–2626. [Google Scholar] [CrossRef]
- Jasani, B.; Cristaudo, A.; Emri, S.A.; Gazdar, A.F.; Gibbs, A.; Krynska, B.; Miller, C.; Mutti, L.; Radu, C.; Tognon, M.; et al. Association of SV40 with Human Tumours. Semin. Cancer Biol. 2001, 11, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Heinsohn, S.; Scholz, R.; Kabisch, H. SV40 and P53 as Team Players in Childhood Lymphoproliferative Disorders. Int. J. Oncol. 2011, 38, 1307–1317. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pipas, J.M.; Levine, A.J. Role of T Antigen Interactions with P53 in Tumorigenesis. Semin. Cancer Biol. 2001, 11, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, R.A.; Madden, C.R.; Kozinetz, C.A.; Halvorson, S.J.; White, Z.S.; Jorgensen, J.L.; Finch, C.J.; Butel, J.S. Association between Simian Virus 40 and Non-Hodgkin Lymphoma. Lancet 2002, 359, 817–823. [Google Scholar] [CrossRef]
- Zekri, A.-R.; Mohamed, W.; Bahnassy, A.; Refat, L.; Khaled, M.; Shalaby, S.; Hafez, M. Detection of Simian Virus 40 DNA Sequences in Egyptian Patients with Different Hematological Malignancies. Leuk. Lymphoma. 2007, 48, 1828–1834. [Google Scholar] [CrossRef]
- MacKenzie, J.; Wilson, K.S.; Perry, J.; Gallagher, A.; Jarrett, R.F. Association Between Simian Virus 40 DNA and Lymphoma in the United Kingdom. JNCI J. Natl. Cancer Inst. 2003, 95, 1001–1003. [Google Scholar] [CrossRef]
- Rollison, D.E.; Helzlsouer, K.J.; Halsey, N.A.; Shah, K.V.; Viscidi, R.P. Markers of Past Infection with Simian Virus 40 (SV40) and Risk of Incident Non-Hodgkin Lymphoma in a Maryland Cohort. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1448–1452. [Google Scholar] [CrossRef]
- Dinantia, N.; Anggorowati, N. Expression of Simian Virus 40 Large T-Antigen: A Case Control Study of Non-Hodgkin Lymphoma. Asian Pac. J. Cancer Biol. 2021, 6, 21–25. [Google Scholar] [CrossRef]
- Lamontagne, R.J.; Bagga, S.; Bouchard, M.J. Hepatitis B Virus Molecular Biology and Pathogenesis. Hepatoma Res. 2016, 2, 163. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Gripon, P.; Urban, S. Hepatitis B Virus Infection Initiates with a Large Surface Protein-Dependent Binding to Heparan Sulfate Proteoglycans. Hepatology 2007, 46, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Saso, W.; Sugiyama, R.; Ishii, K.; Ohki, M.; Nagamori, S.; Suzuki, R.; Aizaki, H.; Ryo, A.; Yun, J.-H.; et al. Epidermal Growth Factor Receptor Is a Host-Entry Cofactor Triggering Hepatitis B Virus Internalization. Proc. Natl. Acad. Sci. USA 2019, 116, 8487–8492. [Google Scholar] [CrossRef]
- Trebicka, J.; Bork, P.; Krag, A.; Arumugam, M. Utilizing the Gut Microbiome in Decompensated Cirrhosis and Acute-on-Chronic Liver Failure. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 167–180. [Google Scholar] [CrossRef]
- Ren, W.; Ye, X.; Su, H.; Li, W.; Liu, D.; Pirmoradian, M.; Wang, X.; Zhang, B.; Zhang, Q.; Chen, L.; et al. Genetic Landscape of Hepatitis B Virus–Associated Diffuse Large B-Cell Lymphoma. Blood 2018, 131, 2670–2681. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic Analysis of the Host Response to Hepatitis B Virus Infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674. [Google Scholar] [CrossRef]
- Luangsay, S.; Gruffaz, M.; Isorce, N.; Testoni, B.; Michelet, M.; Faure-Dupuy, S.; Maadadi, S.; Ait-Goughoulte, M.; Parent, R.; Rivoire, M.; et al. Early Inhibition of Hepatocyte Innate Responses by Hepatitis B Virus. J. Hepatol. 2015, 63, 1314–1322. [Google Scholar] [CrossRef]
- Evens, A.M.; Jovanovic, B.D.; Su, Y.-C.; Raisch, D.W.; Ganger, D.; Belknap, S.M.; Dai, M.-S.; Chiu, B.-C.C.; Fintel, B.; Cheng, Y.; et al. Rituximab-Associated Hepatitis B Virus (HBV) Reactivation in Lymphoproliferative Diseases: Meta-Analysis and Examination of FDA Safety Reports. Ann. Oncol. 2011, 22, 1170–1180. [Google Scholar] [CrossRef]
- Cabrerizo, M.; Bartolomé, J.; Caramelo, C.; Barril, G.; Carreno, V. Molecular Analysis of Hepatitis B Virus DNA in Serum and Peripheral Blood Mononuclear Cells from Hepatitis B Surface Antigen-Negative Cases. Hepatology 2000, 32, 116–123. [Google Scholar] [CrossRef]
- Loggi, E.; Gamal, N.; Bihl, F.; Bernardi, M.; Andreone, P. Adaptive Response in Hepatitis B Virus Infection. J. Viral. Hepat. 2014, 21, 305–313. [Google Scholar] [CrossRef]
- Deng, L.; Song, Y.; Young, K.H.; Hu, S.; Ding, N.; Song, W.; Li, X.; Shi, Y.; Huang, H.; Liu, W.; et al. Hepatitis B Virus-Associated Diffuse Large B-Cell Lymphoma: Unique Clinical Features, Poor Outcome, and Hepatitis B Surface Antigen-Driven Origin. Oncotarget 2015, 6, 25061–25073. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-Y.; Gao, F.; Zhao, Y.-W.; Ni, B.-W.; Huang, H.-H.; Hou, J. Inferior Survival and Frequent Hepatic Dysfunction in Non-Hodgkin’s Lymphoma Patients with HBV Infection: A Systematic Review and Meta-Analysis. Hematology 2022, 27, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Dalia, S.; Chavez, J.; Castillo, J.J.; Sokol, L. Hepatitis B Infection Increases the Risk of Non-Hodgkin Lymphoma: A Meta-Analysis of Observational Studies. Leuk. Res. 2013, 37, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Perrotti, D.; Caligiuri, M.A. Understanding the Molecular Basis of Imatinib Mesylate Therapy in Chronic Myelogenous Leukemia and the Related Mechanisms of Resistance: Commentary Re: A. N. Mohamed et al., The Effect of Imatinib Mesylate on Patients with Philadelphia Chromosome-Positive Chronic Myeloid Leukemia with Secondary Chromosomal Aberrations. Clin. Cancer Res., 9: 1333–1337, 2003. Clin. Cancer Res. 2003, 9, 1248–1252. [Google Scholar]
- Yan, X.; Zhou, M.; Lou, Z.; Mu, Q.; Sheng, L.; Zhang, P.; Wang, Y.; Ouyang, G. Diffuse Large B-Cell Lymphoma with Concurrent Hepatitis B Virus Infection in the MabThera Era: Unique Clinical Features and Worse Outcomes. J. Cancer Res. Ther. 2018, 14, S248–S253. [Google Scholar] [CrossRef]
- Pfreundschuh, M.; Ho, A.D.; Cavallin-Stahl, E.; Wolf, M.; Pettengell, R.; Vasova, I.; Belch, A.; Walewski, J.; Zinzani, P.-L.; Mingrone, W.; et al. Prognostic Significance of Maximum Tumour (Bulk) Diameter in Young Patients with Good-Prognosis Diffuse Large-B-Cell Lymphoma Treated with CHOP-like Chemotherapy with or without Rituximab: An Exploratory Analysis of the MabThera International Trial Group (MInT) Study. Lancet Oncol. 2008, 9, 435–444. [Google Scholar] [CrossRef]
- Mozessohn, L.; Chan, K.K.W.; Feld, J.J.; Hicks, L.K. Hepatitis B Reactivation in HBsAg-Negative/HBcAb-Positive Patients Receiving Rituximab for Lymphoma: A Meta-Analysis. J. Viral. Hepat. 2015, 22, 842–849. [Google Scholar] [CrossRef]
- Penin, F.; Dubuisson, J.; Rey, F.A.; Moradpour, D.; Pawlotsky, J.-M. Structural Biology of Hepatitis C Virus. Hepatology 2004, 39, 5–19. [Google Scholar] [CrossRef]
- Moradpour, D.; Penin, F. Hepatitis C Virus Proteins: From Structure to Function. In Hepatitis C Virus: From Molecular Virology to Antiviral Therapy; Bartenschlager, R., Ed.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2013; Volume 369, pp. 113–142. ISBN 978-3-642-27339-1. [Google Scholar]
- Spera, A.M. Safety of Direct Acting Antiviral Treatment for Hepatitis C in Oncologic Setting: A Clinical Experience and a Literature Review. World J. Hepatol. 2022, 14, 525–534. [Google Scholar] [CrossRef]
- Sène, D.; Limal, N.; Ghillani-Dalbin, P.; Saadoun, D.; Piette, J.-C.; Cacoub, P. Hepatitis C Virus-Associated B-Cell Proliferation--the Role of Serum B Lymphocyte Stimulator (BLyS/BAFF). Rheumatology 2007, 46, 65–69. [Google Scholar] [CrossRef][Green Version]
- Giordano, T.P.; Henderson, L.; Landgren, O.; Chiao, E.Y.; Kramer, J.R.; El-Serag, H.; Engels, E.A. Risk of Non-Hodgkin Lymphoma and Lymphoproliferative Precursor Diseases in US Veterans With Hepatitis C Virus. JAMA 2007, 297, 2010. [Google Scholar] [CrossRef] [PubMed]
- Fiorino, S. Possible Association between Hepatitis C Virus and Malignancies Different from Hepatocellular Carcinoma: A Systematic Review. World J. Gastroenterol. 2015, 21, 12896. [Google Scholar] [CrossRef]
- Nieters, A.; Kallinowski, B.; Brennan, P.; Ott, M.; Maynadié, M.; Benavente, Y.; Foretova, L.; Cocco, P.L.; Staines, A.; Vornanen, M.; et al. Hepatitis C and Risk of Lymphoma: Results of the European Multicenter Case-Control Study EPILYMPH. Gastroenterology 2006, 131, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- De Sanjose, S.; Benavente, Y.; Vajdic, C.M.; Engels, E.A.; Morton, L.M.; Bracci, P.M.; Spinelli, J.J.; Zheng, T.; Zhang, Y.; Franceschi, S.; et al. Hepatitis C and Non-Hodgkin Lymphoma Among 4784 Cases and 6269 Controls From the International Lymphoma Epidemiology Consortium. Clin. Gastroenterol. Hepatol. 2008, 6, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Pozzato, G.; Mazzaro, C.; Gattei, V. Hepatitis C Virus–Associated Non-Hodgkin Lymphomas. Clin. Liver Dis. 2017, 21, 499–515. [Google Scholar] [CrossRef]
- Pioltelli, P.; Zehender, G.; Monti, G.; Monteverde, A.; Galli, M. HCV and Non-Hodgkin Lymphoma. Lancet 1996, 347, 624–625. [Google Scholar] [CrossRef]
- Mele, A.; Pulsoni, A.; Bianco, E.; Musto, P.; Szklo, A.; Sanpaolo, M.G.; Iannitto, E.; De Renzo, A.; Martino, B.; Liso, V.; et al. Hepatitis C Virus and B-Cell Non-Hodgkin Lymphomas: An Italian Multicenter Case-Control Study. Blood 2003, 102, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, T.; Rowe, J. Pathogenesis and Prognostication in Acute Lymphoblastic Leukemia. F1000Prime Rep. 2014, 6, 59. [Google Scholar] [CrossRef]
- Zuckerman, E.; Zuckerman, T.; Sahar, D.; Streichman, S.; Attias, D.; Sabo, E.; Yeshurun, D.; Rowe, J.M. The Effect of Antiviral Therapy on t(14;18) Translocation and Immunoglobulin Gene Rearrangement in Patients with Chronic Hepatitis C Virus Infection. Blood 2001, 97, 1555–1559. [Google Scholar] [CrossRef]
- Giannelli, F.; Moscarella, S.; Giannini, C.; Caini, P.; Monti, M.; Gragnani, L.; Romanelli, R.G.; Solazzo, V.; Laffi, G.; La Villa, G.; et al. Effect of Antiviral Treatment in Patients with Chronic HCV Infection and t(14;18) Translocation. Blood 2003, 102, 1196–1201. [Google Scholar] [CrossRef]
- Ferri, C.; Caracciolo, F.; Zignego, A.L.; Civita, L.L.; Monti, M.; Longombardo, G.; Lombardini, F.; Greco, F.; Capochiani, E.; Mazzoni, A.; et al. Hepatitis C Virus Infection in Patients with Non-Hodgkin’s Lymphoma. Br. J. Haematol. 1994, 88, 392–394. [Google Scholar] [CrossRef] [PubMed]
- Galati, G.; Rampa, L.; Vespasiani-Gentilucci, U.; Marino, M.; Pisani, F.; Cota, C.; Guidi, A.; Picardi, A. Hepatitis C and Double-Hit B Cell Lymphoma Successfully Treated by Antiviral Therapy. World J. Hepatol. 2016, 8, 1244. [Google Scholar] [CrossRef] [PubMed]
- Duberg, A.-S.; Nordström, M.; Törner, A.; Reichard, O.; Strauss, R.; Janzon, R.; Bäck, E.; Ekdahl, K. Non-Hodgkin’s Lymphoma and Other Nonhepatic Malignancies in Swedish Patients with Hepatitis C Virus Infection. Hepatology 2005, 41, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Luppi, M.; Longo, G.; Ferrari, M.G.; Barozzi, P.; Marasca, R.; Morselli, M.; Valenti, C.; Mascia, T.; Vandelli, L.; Vallisa, D.; et al. Clinico-Pathological Characterization of Hepatitis C Virus-Related B-Cell Non-Hodgkin’s Lymphomas without Symptomatic Cryoglobulinemia. Ann. Oncol. 1998, 9, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Visco, C.; Arcaini, L.; Brusamolino, E.; Burcheri, S.; Ambrosetti, A.; Merli, M.; Bonoldi, E.; Chilosi, M.; Viglio, A.; Lazzarino, M.; et al. Distinctive Natural History in Hepatitis C Virus Positive Diffuse Large B-Cell Lymphoma: Analysis of 156 Patients from Northern Italy. Ann. Oncol. 2006, 17, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Besson, C.; Canioni, D.; Lepage, E.; Pol, S.; Morel, P.; Lederlin, P.; Van Hoof, A.; Tilly, H.; Gaulard, P.; Coiffier, B.; et al. Characteristics and Outcome of Diffuse Large B-Cell Lymphoma in Hepatitis C Virus-Positive Patients in LNH 93 and LNH 98 Groupe d’Etude Des Lymphomes de l’Adulte Programs. J. Clin. Oncol. 2006, 24, 953–960. [Google Scholar] [CrossRef] [PubMed]
- De Renzo, A.; Perna, F.; Persico, M.; Notaro, R.; Mainolfi, C.; de Sio, I.; Ciancia, G.; Picardi, M.; Del Vecchio, L.; Pane, F.; et al. Excellent Prognosis and Prevalence of HCV Infection of Primary Hepatic and Splenic Non-Hodgkin’s Lymphoma. Eur. J. Haematol. 2008, 81, 51–57. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, Y.; Ren, Y.; Tong, Y.; Hua, X.; Zhu, F.; Huang, L.; Liu, Y.; Luo, Y.; Lu, W.; et al. Hepatitis C Virus Protects Human B Lymphocytes from Fas-Mediated Apoptosis via E2-CD81 Engagement. PLoS ONE 2011, 6, e18933. [Google Scholar] [CrossRef]
- Stamataki, Z.; Shannon-Lowe, C.; Shaw, J.; Mutimer, D.; Rickinson, A.B.; Gordon, J.; Adams, D.H.; Balfe, P.; McKeating, J.A. Hepatitis C Virus Association with Peripheral Blood B Lymphocytes Potentiates Viral Infection of Liver-Derived Hepatoma Cells. Blood 2009, 113, 585–593. [Google Scholar] [CrossRef]
- Peveling-Oberhag, J.; Arcaini, L.; Hansmann, M.-L.; Zeuzem, S. Hepatitis C-Associated B-Cell Non-Hodgkin Lymphomas. Epidemiology, Molecular Signature and Clinical Management. J. Hepatol. 2013, 59, 169–177. [Google Scholar] [CrossRef]
- Bachy, E.; Besson, C.; Suarez, F.; Hermine, O. Hepatitis C Virus Infection and Lymphoma. Mediterr. J. Hematol. Infect. Dis. 2010, 2, e2010004. [Google Scholar] [CrossRef] [PubMed]
- Merli, M.; Visco, C.; Spina, M.; Luminari, S.; Ferretti, V.V.; Gotti, M.; Rattotti, S.; Fiaccadori, V.; Rusconi, C.; Targhetta, C.; et al. Outcome Prediction of Diffuse Large B-Cell Lymphomas Associated with Hepatitis C Virus Infection: A Study on Behalf of the Fondazione Italiana Linfomi. Haematologica 2014, 99, 489–496. [Google Scholar] [CrossRef]
- Xuan, C.; Steward, K.K.; Timmerman, J.M.; Morrison, S.L. Targeted Delivery of Interferon-Alpha via Fusion to Anti-CD20 Results in Potent Antitumor Activity against B-Cell Lymphoma. Blood 2010, 115, 2864–2871. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.-M.; Baker, C.C. Papillomavirus Genome Structure, Expression, and Post-Transcriptional Regulation. Front. Biosci. 2006, 11, 2286–2302. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Cheng, Y.; Wu, S.; Zeng, X.; Shi, X.; Ling, Q.; Li, Z.; Liang, Z.; Wang, B. Primary Non-Hodgkin Lymphoma of the Tongue Base: The Clinicopathology of Seven Cases and Evaluation of HPV and EBV Status. Diagn. Pathol. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Heawchaiyaphum, C.; Ekalaksananan, T.; Patarapadungkit, N.; Vatanasapt, P.; Pientong, C. Association of Human Papillomavirus and Epstein-Barr Virus Infection with Tonsil Cancer in Northeastern Thailand. Asian Pac. J. Cancer Prev. 2022, 23, 781–787. [Google Scholar] [CrossRef]
- Gillson, M.L.; Richard, A.F. Human Herpesvirus-8. Curr. Opin. Oncol. 1997, 9, 440–449. [Google Scholar] [CrossRef]
- Zhao, Y.; Maule, J.; Li, Y.; Neff, J.; McCall, C.M.; Hao, T.; Yang, W.; Rehder, C.; Yang, L.-H.; Wang, E. Sequential Development of Human Herpes Virus 8-Positive Diffuse Large B-Cell Lymphoma and Chronic Myelomonocytic Leukemia in a 59 Year Old Female Patient with Hemoglobin SC Disease. Pathol. Res. Pract. 2019, 215, 152704. [Google Scholar] [CrossRef]
- Tanière, P.; Manai, A.; Charpentier, R.; Terdjman, P.; Boucheron, S.; Cordier, J.F.; Berger, F. Pyothorax-Associated Lymphoma: Relationship with Epstein-Barr Virus, Human Herpes Virus-8 and Body Cavity-Based High Grade Lymphomas. Eur. Respir. J. 1998, 11, 779–783. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilajac, E.; Mahmutović, L.; Lundstrom, K.; Glamočlija, U.; Šutković, J.; Sezer, A.; Hromić-Jahjefendić, A. Viral Agents as Potential Drivers of Diffuse Large B-Cell Lymphoma Tumorigenesis. Viruses 2022, 14, 2105. https://doi.org/10.3390/v14102105
Bilajac E, Mahmutović L, Lundstrom K, Glamočlija U, Šutković J, Sezer A, Hromić-Jahjefendić A. Viral Agents as Potential Drivers of Diffuse Large B-Cell Lymphoma Tumorigenesis. Viruses. 2022; 14(10):2105. https://doi.org/10.3390/v14102105
Chicago/Turabian StyleBilajac, Esma, Lejla Mahmutović, Kenneth Lundstrom, Una Glamočlija, Jasmin Šutković, Abas Sezer, and Altijana Hromić-Jahjefendić. 2022. "Viral Agents as Potential Drivers of Diffuse Large B-Cell Lymphoma Tumorigenesis" Viruses 14, no. 10: 2105. https://doi.org/10.3390/v14102105
APA StyleBilajac, E., Mahmutović, L., Lundstrom, K., Glamočlija, U., Šutković, J., Sezer, A., & Hromić-Jahjefendić, A. (2022). Viral Agents as Potential Drivers of Diffuse Large B-Cell Lymphoma Tumorigenesis. Viruses, 14(10), 2105. https://doi.org/10.3390/v14102105