

DExD/H Box Helicases DDX24 and DDX49 Inhibit Reactivation of Kaposi’s Sarcoma Associated Herpesvirus by Interacting with Viral mRNAs
 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Helicase siRNA Screen
2.3. Real-Time qPCR
2.4. Transfection by Electroporation for BCBL-1 Overexpression
2.5. Antibodies and Western Blot Analysis
2.6. Lentiviral Stable Transduction for BCBL-1 Knockdown
2.7. Lentiviral Transduction for TREx BCBL1-Rta Overexpression
2.8. RNA Immunoprecipitation
2.9. Next Generation Sequencing (NGS)
2.10. Statistical Analyses
2.11. Type I Interferon Detection by SEAP Assay and Transcript Measurement by RT-qPCR
3. Results
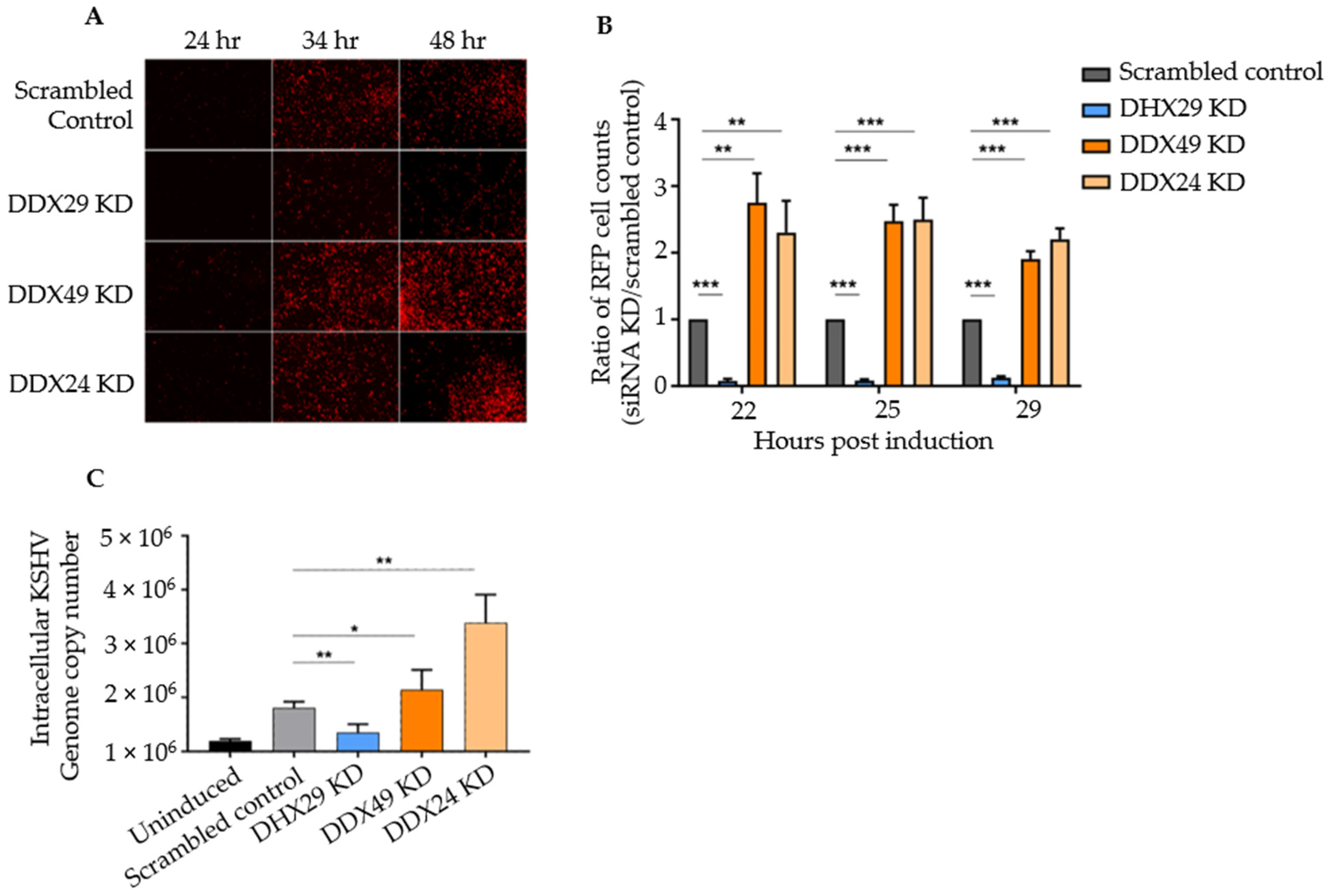
3.1. Knockdown of DexD/H Box Helicases to Screen for Effects on KSHV Reactivation
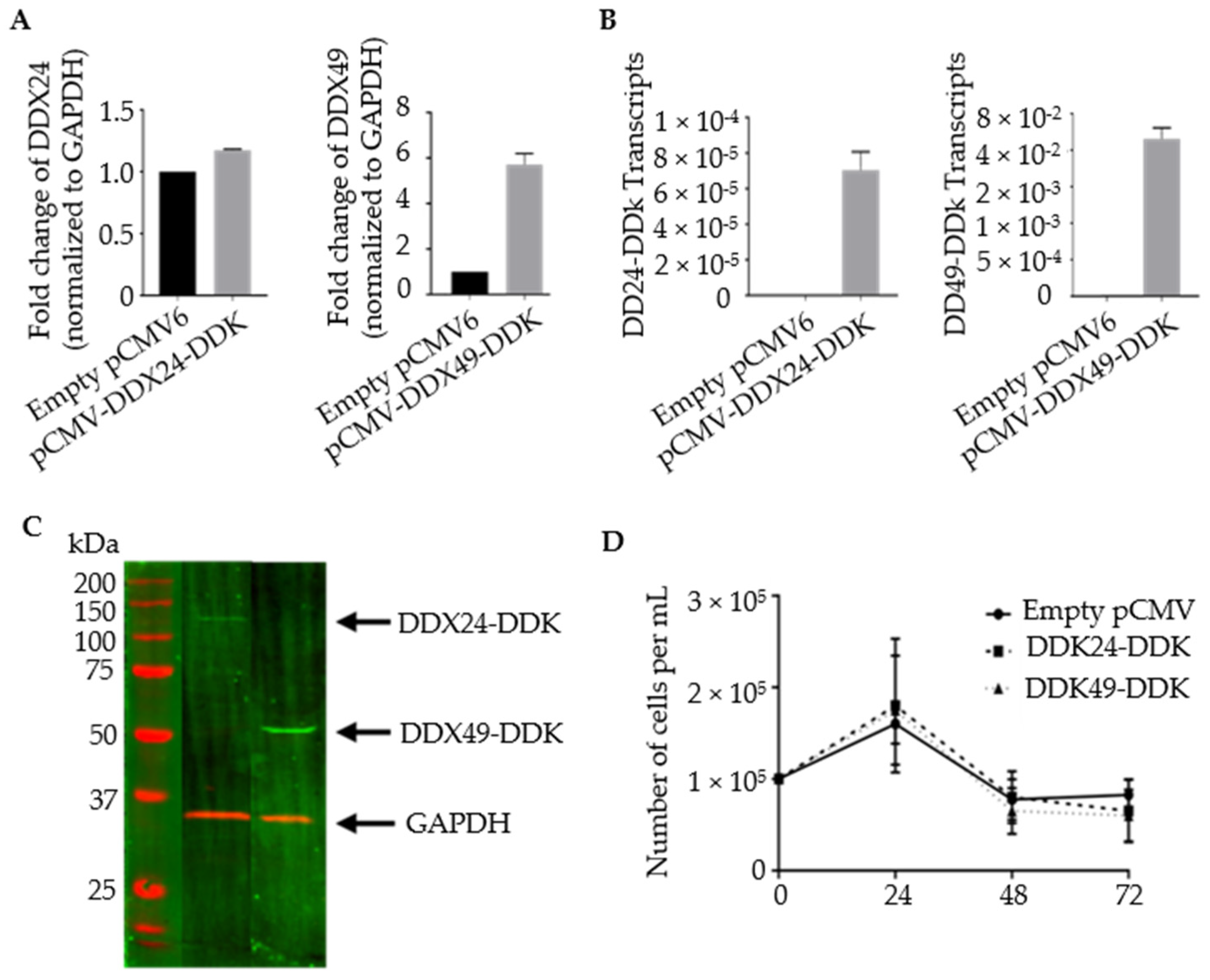
3.2. Construction of Stably Transfected BCBL-1 Cells Expressing DDX24-DDK and DDX49-DDK
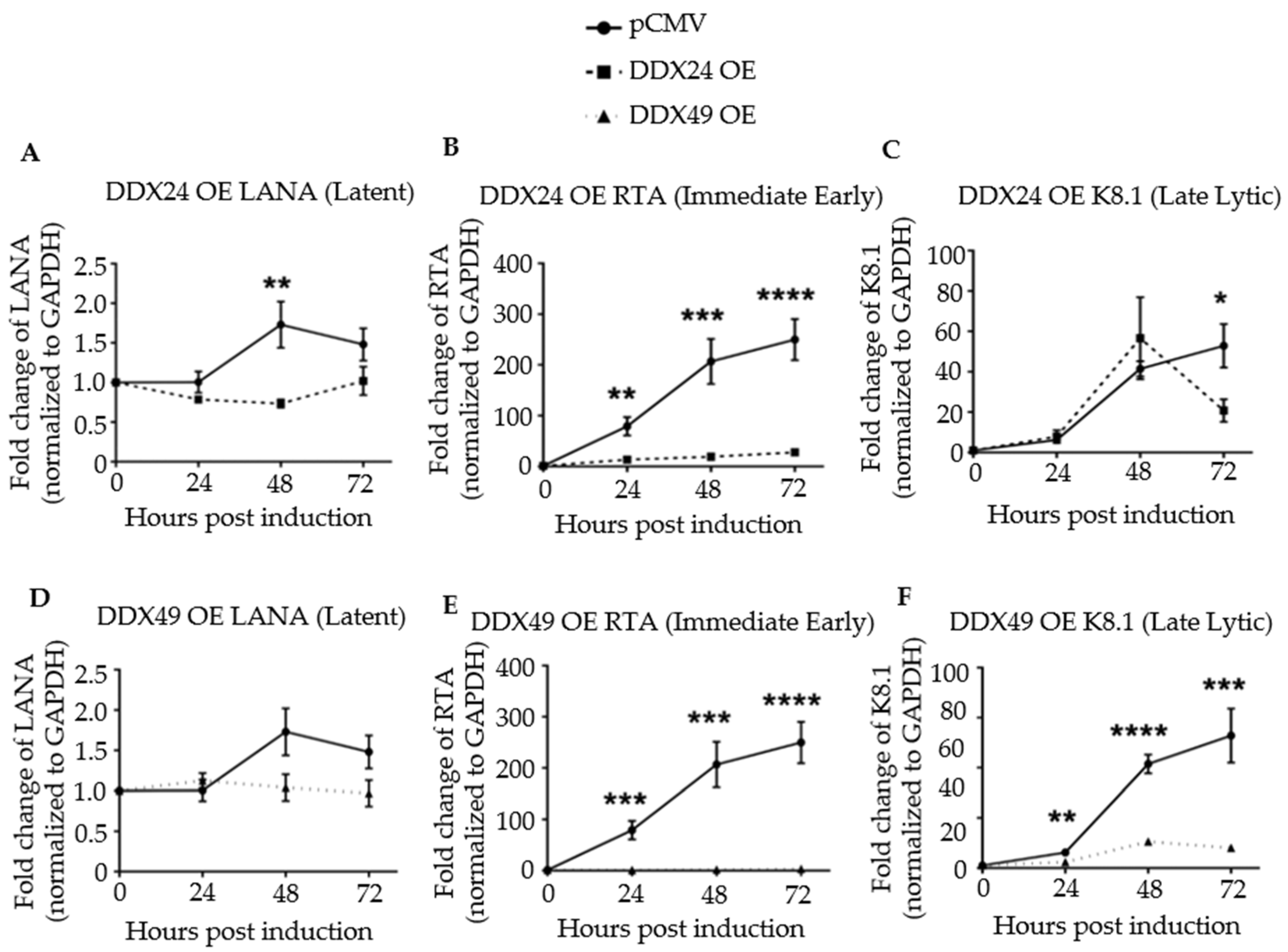
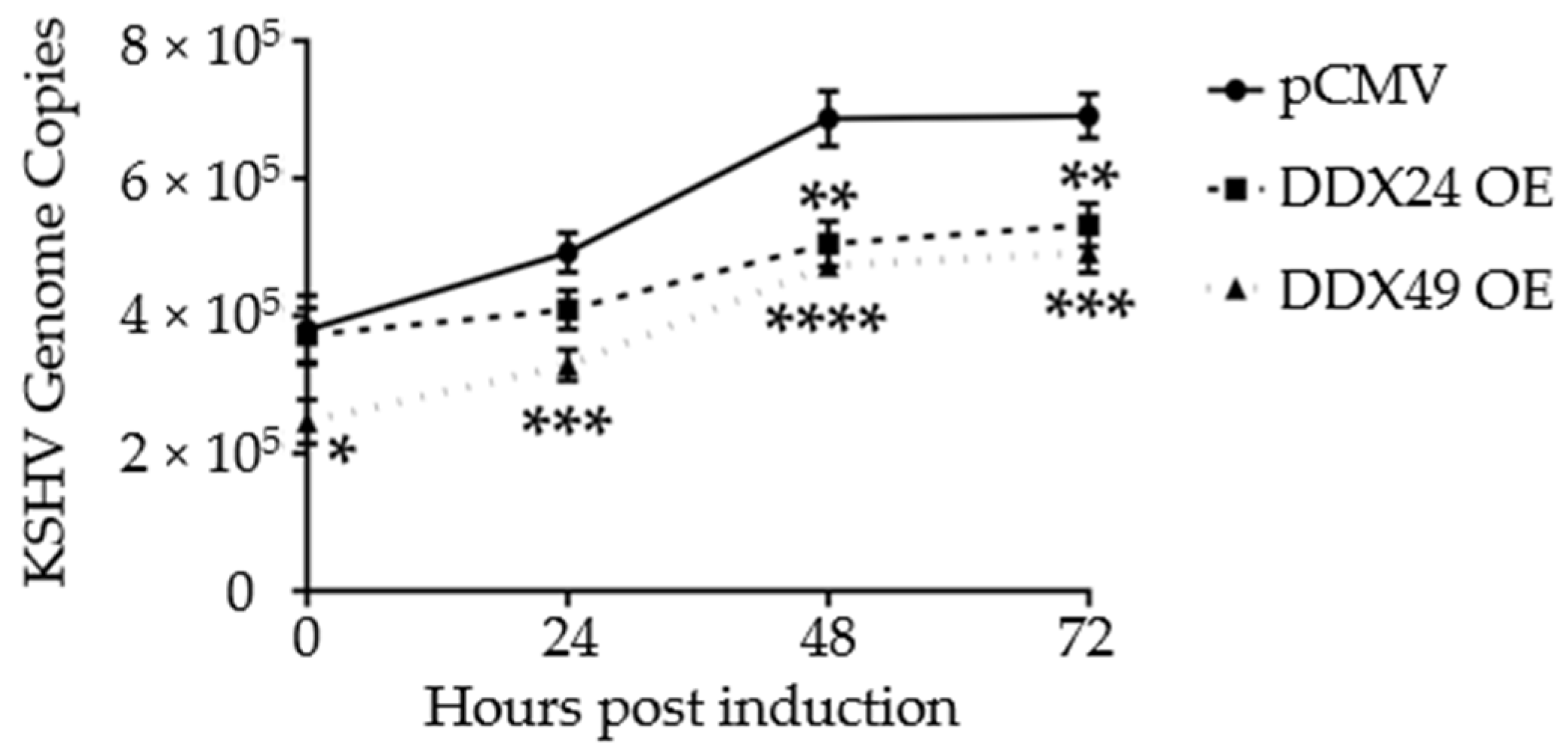
3.3. Effects of DDX24-DDK and DDX49-DDK on KSHV Gene Expression following Induction
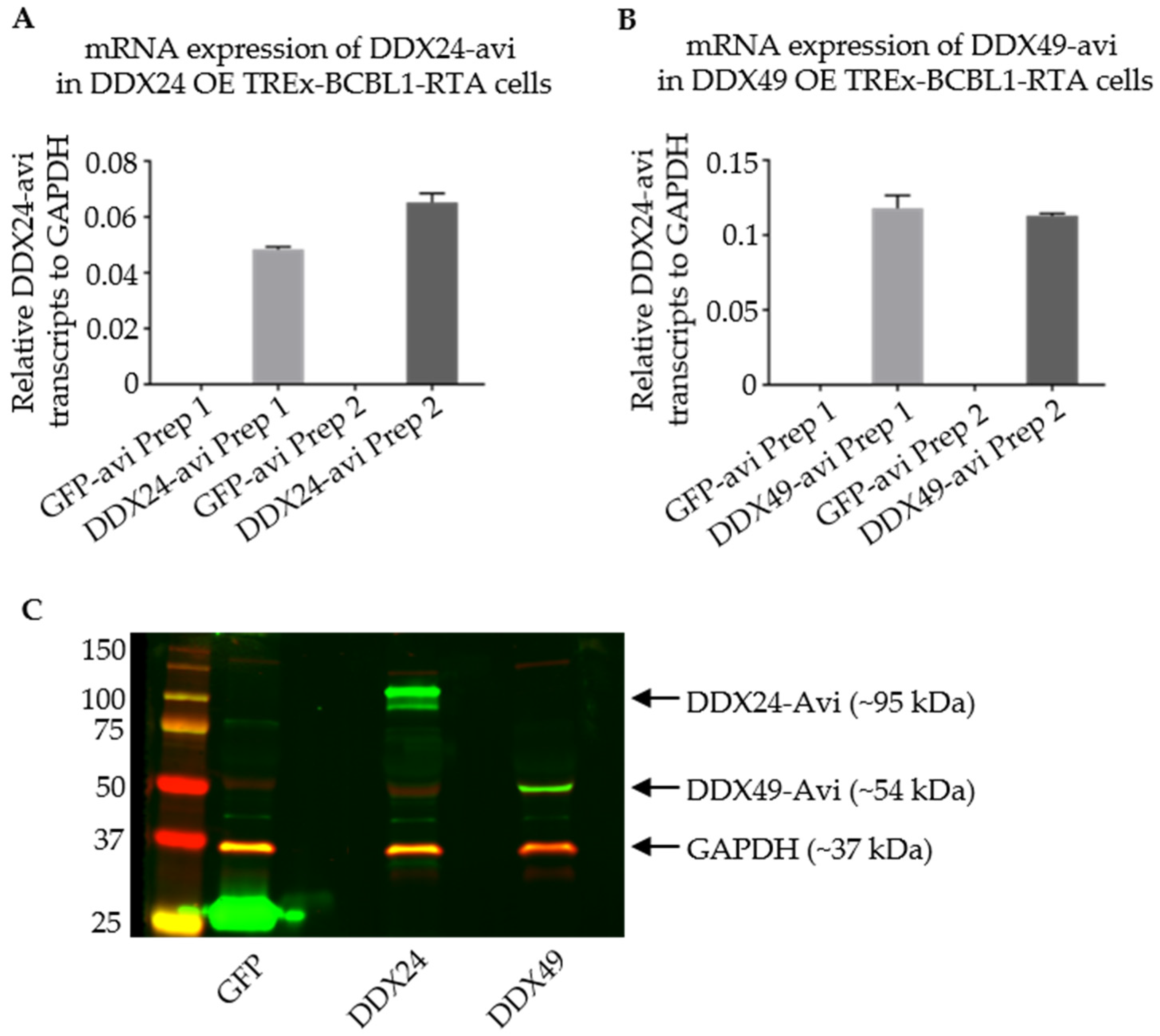
3.4. Stably Transduced TREx BCBL1-Rta Cells Expressing DDX24-Avi and DDX49-Avi
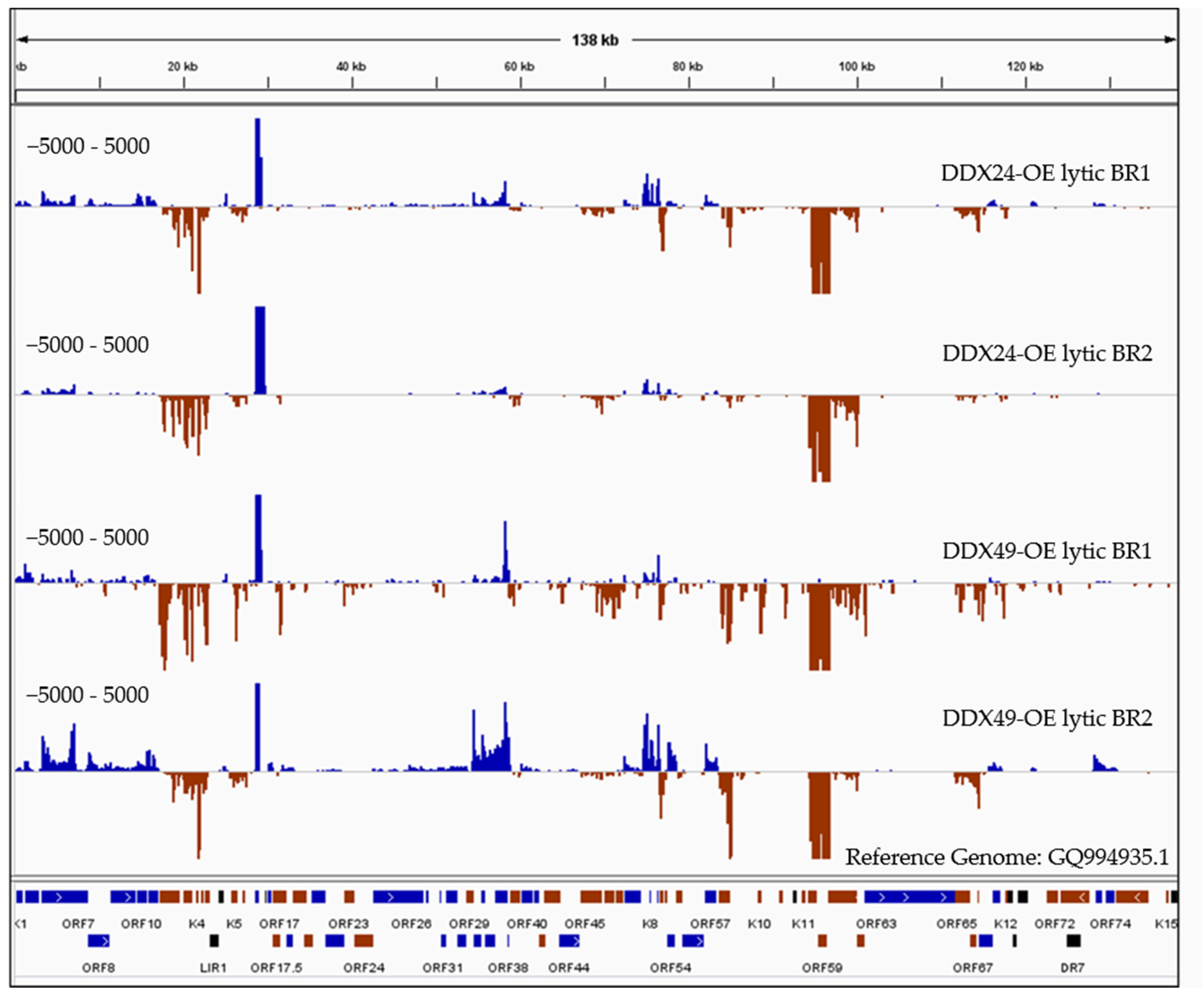
3.5. RNA Immunoprecipitation of TREx BCBL1-Rta Cells
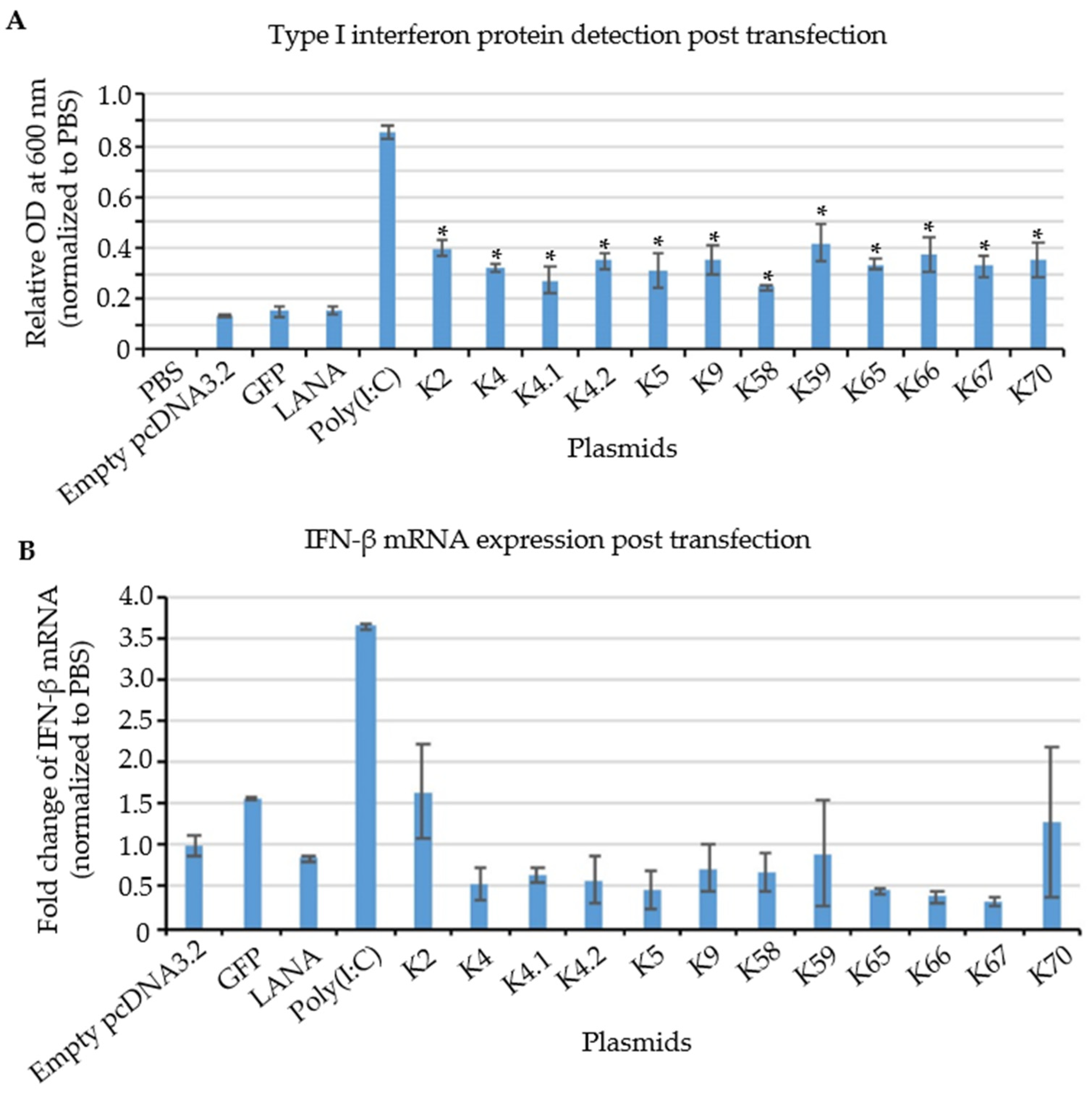
3.6. Testing of Cloned KSHV Genes for Their Ability to Induce a Type I Interferon Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harley, J.B.; Chen, X.; Pujato, M.; Miller, D.; Maddox, A.; Forney, C.; Magnusen, A.F.; de la Cruz-Lynch, A.; Chetal, K.; Yukawa, M.; et al. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet. 2018, 50, 699–707. [Google Scholar] [CrossRef]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef]
- National Cancer Institute DoEA. Investigation of the Transmission of Kaposi’s Sarcoma-Associated Herpesvirus 2017. Available online: https://deainfo.nci.nih.gov/advisory/joint/1117/07-Huppi.pdf (accessed on 15 September 2022).
- The American Cancer Society. Kaposi Sarcoma, Atlanta, GA, USA. Available online: https://www.cancer.org/cancer/kaposi-sarcoma/about/what-is-key-statistics.html (accessed on 15 September 2022).
- Jha, H.C.; Banerjee, S.; Robertson, E.S. The Role of Gammaherpesviruses in Cancer Pathogenesis. Pathogens 2016, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Renne, R.; Zhong, W.; Herndier, B.; Mcgrath, M.; Abbey, N.; Kedes, D.; Ganem, D. Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med. 1996, 2, 342–346. [Google Scholar] [CrossRef]
- Lukac, D.M.; Yuan, Y. Reactivation and lytic replication of KSHV. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Giffin, L.; West, J.A.; Damania, B. Kaposi’s Sarcoma-Associated Herpesvirus Interleukin-6 Modulates Endothelial Cell Movement by Upregulating Cellular Genes Involved in Migration. MBio 2015, 6, e01499-15. [Google Scholar] [CrossRef] [PubMed]
- Cuconati, A.; White, E. Viral homologs of BCL-2: Role of apoptosis in the regulation of virus infection. Genes Dev. 2002, 16, 2465–2478. [Google Scholar] [CrossRef]
- Gregory, S.M.; West, J.A.; Dillon, P.J.; Hilscher, C.; Dittmer, D.P.; Damania, B. Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. USA 2009, 106, 11725–11730. [Google Scholar] [CrossRef]
- West, J.A.; Wicks, M.; Gregory, S.M.; Chugh, P.; Jacobs, S.R.; Zhang, Z.; Host, K.M.; Dittmer, D.P.; Damania, B. An important role for mitochondrial antiviral signaling protein in the Kaposi’s sarcoma-associated herpesvirus life cycle. J. Virol. 2014, 88, 5778–5787. [Google Scholar] [CrossRef]
- Inn, K.S.; Lee, S.H.; Rathbun, J.Y.; Wong, L.Y.; Toth, Z.; Machida, K.; Ou, J.H.J.; Jung, J.U. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 2011, 85, 10899–10904. [Google Scholar] [CrossRef]
- Zhao, Y.; Ye, X.; Dunker, W.; Song, Y.; Karijolich, J. RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nat. Commun. 2018, 9, 4841. [Google Scholar] [CrossRef] [PubMed]
- Sathish, N.; Yuan, Y. Evasion and subversion of interferon-mediated antiviral immunity by Kaposi’s sarcoma-associated herpesvirus: An overview. J. Virol. 2011, 85, 10934–10944. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-R.; Choi, U.Y.; Hwang, S.-W.; Kim, S.; Jung, A.J.U. Viral Inhibition of PRR-Mediated Innate Immune Response: Learning from KSHV Evasion Strategies. Mol. Cells 2016, 39, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dittmer, D.P.; Mieczkowski, P.A.; Host, K.M.; Fusco, W.G.; Duncan, J.A.; Damania, B. RIG-I Detects Kaposi’s Sarcoma-Associated Herpesvirus Transcripts in a RNA Polymerase III-Independent Manner. mBio 2018, 9, e00823-18. [Google Scholar] [CrossRef] [PubMed]
- Zevini, A.; Olagnier, D.; Hiscott, J. Crosstalk between Cytoplasmic RIG-I and STING Sensing Pathways. Trends Immunol. 2017, 38, 194–205. [Google Scholar] [CrossRef]
- Wu, J.-J.; Li, W.; Shao, Y.; Avey, D.; Fu, B.; Gillen, J.; Hand, T.; Ma, S.; Liu, X.; Miley, W.; et al. Inhibition of cGAS DNA Sensing by a Herpesvirus Virion Protein. Cell Host Microbe 2015, 18, 333–344. [Google Scholar] [CrossRef]
- Bourgeois, C.; Mortreux, F.; Auboeuf, D. The multiple functions of RNA helicases as drivers and regulators of gene expression. Nat. Rev. Mol. Cell Biol. 2016, 17, 426–438. [Google Scholar] [CrossRef]
- Young, C.L.; Khoshnevis, S.; Karbstein, K. Cofactor-dependent specificity of a DEAD-box protein. Proc. Natl. Acad. Sci. USA 2013, 110, E2668–E2676. [Google Scholar] [CrossRef]
- Mallam, A.L.; Del Campo, M.; Gilman, B.; Sidote, D.J.; Lambowitz, A.M. Structural basis for RNA-duplex recognition and unwinding by the DEAD-box helicase Mss116p. Nature 2012, 490, 121–125. [Google Scholar] [CrossRef]
- Jankowsky, E.; Bowers, H. Remodeling of ribonucleoprotein complexes with DExH/D RNA helicases. Nucleic Acids Res. 2006, 34, 4181–4188. [Google Scholar] [CrossRef]
- Dempsey, A.; Bowie, A.G. Innate immune recognition of DNA: A recent history. Virology 2015, 479–480, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Fullam, A.; Schroder, M. DExD/H-box RNA helicases as mediators of anti-viral innate immunity and essential host factors for viral replication. Biochim. Biophys. Acta 2013, 1829, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Steimer, L.; Klostermeier, D. RNA helicases in infection and disease. RNA Biol. 2012, 9, 751–771. [Google Scholar] [CrossRef]
- Hull, C.M.; Bevilacqua, P.C. Discriminating Self and Non-Self by RNA: Roles for RNA Structure, Misfolding, and Modification in Regulating the Innate Immune Sensor PKR. Accounts Chem. Res. 2016, 49, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Tanner, N.; Linder, P. DExD/H Box RNA Helicases: From Generic Motors to Specific Dissociation Functions. Mol. Cell 2001, 8, 251–262. [Google Scholar] [CrossRef]
- Jarmoskaite, I.; Russell, R. DEAD-box proteins as RNA helicases and chaperones. Wiley Interdiscip. Rev. RNA 2011, 2, 135–152. [Google Scholar] [CrossRef]
- Linder, P.; Jankowsky, E. From unwinding to clamping—The DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef]
- Fairman-Williams, M.E.; Guenther, U.-P.; Jankowsky, E. SF1 and SF2 helicases: Family matters. Curr. Opin. Struct. Biol. 2010, 20, 313–324. [Google Scholar] [CrossRef]
- Putnam, A.A.; Jankowsky, E. DEAD-box helicases as integrators of RNA, nucleotide and protein binding. Biochim. Biophys. Acta 2013, 1829, 884–893. [Google Scholar] [CrossRef]
- Li, C.; Ge, L.-L.; Li, P.-P.; Wang, Y.; Dai, J.-J.; Sun, M.-X.; Huang, L.; Shen, Z.-Q.; Hu, X.-C.; Ishag, H.; et al. Cellular DDX3 regulates Japanese encephalitis virus replication by interacting with viral un-translated regions. Virology 2014, 449, 70–81. [Google Scholar] [CrossRef]
- Wang, X.; Wang, R.; Luo, M.; Li, C.; Wang, H.X.; Huan, C.C.; Qu, Y.R.; Liao, Y.; Mao, X. (DEAD)-box RNA helicase 3 modulates NF-kappaB signal pathway by controlling the phosphorylation of PP2A-C subunit. Oncotarget 2017, 8, 33197–33213. [Google Scholar] [CrossRef] [PubMed]
- Valiente-Echeverría, F.; Hermoso, M.A.; Rifo, R.S. RNA helicase DDX3: At the crossroad of viral replication and antiviral immunity. Rev. Med Virol. 2015, 25, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhu, Y.; Liu, Z.-J.; Ouyang, S. The emerging roles of the DDX41 protein in immunity and diseases. Protein Cell 2017, 8, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Moore, R.; Xu, X.; Barber, G.N. DDX24 Negatively Regulates Cytosolic RNA-Mediated Innate Immune Signaling. PLoS Pathog. 2013, 9, e1003721. [Google Scholar] [CrossRef]
- Vieira, J.; O’Hearn, P.M. Use of the red fluorescent protein as a marker of Kaposi’s sarcoma-associated herpesvirus lytic gene expression. Virology 2004, 325, 225–240. [Google Scholar] [CrossRef]
- Nakamura, H.; Lu, M.; Gwack, Y.; Souvlis, J.; Zeichner, S.L.; Jung, J.U. Global changes in Kaposi’s sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J. Virol. 2003, 77, 4205–4220. [Google Scholar] [CrossRef]
- Rahman, M.M.; Bagdassarian, E.; Ali, M.A.M.; McFadden, G. Identification of host DEAD-box RNA helicases that regulate cellular tropism of oncolytic Myxoma virus in human cancer cells. Sci. Rep. 2017, 7, 15710. [Google Scholar] [CrossRef]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 479–480, 110–121. [Google Scholar] [CrossRef]
- Gack, M.U. Mechanisms of RIG-I-Like Receptor Activation and Manipulation by Viral Pathogens. J. Virol. 2014, 88, 5213–5216. [Google Scholar] [CrossRef]
- Beckett, D.; Kovaleva, E.; Schatz, P.J. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999, 8, 921–929. [Google Scholar] [CrossRef]
- Purushothaman, P.; Uppal, T.; Verma, S.C. Molecular Biology of KSHV Lytic Reactivation. Viruses 2015, 7, 116–153. [Google Scholar] [CrossRef] [PubMed]
- Brulois, K.; Toth, Z.; Wong, L.Y.; Feng, P.; Gao, S.J.; Ensser, A.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus K3 and K5 ubiquitin E3 ligases have stage-specific immune evasion roles during lytic replication. J. Virol. 2014, 88, 9335–9349. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Chauhan, D.; Teoh, G.; Raje, N.; Treon, S.P.; Tai, Y.T.; Shima, Y.; Anderson, K.C. Characterization of signaling cascades triggered by human interleukin-6 versus Kaposi’s sarcoma-associated herpes virus-encoded viral interleukin 6. Clin. Cancer Res. 2000, 6, 1180–1189. [Google Scholar] [PubMed]
- Majerciak, V.; Yamanegi, K.; Zheng, Z.M. Gene structure and expression of Kaposi’s sarcoma-associated herpesvirus ORF56, ORF57, ORF58, and ORF59. J. Virol. 2006, 80, 11968–11981. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Gregory, S.M.; West, J.A.; Wollish, A.C.; Bennett, C.L.; Blackbourn, D.J.; Heise, M.T.; Damania, B. The viral interferon regulatory factors of kaposi’s sarcoma-associated herpesvirus differ in their inhibition of interferon activation mediated by toll-like receptor 3. J. Virol. 2013, 87, 798–806. [Google Scholar] [CrossRef]
- Burýsek, L.; Yeow, W.S.; Lubyová, B.; Kellum, M.; Schafer, S.L.; Huang, Y.Q.; Pitha, P.M. Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J. Virol. 1999, 73, 7334–7342. [Google Scholar] [CrossRef]
- Lin, R.; Genin, P.; Mamane, Y.; Sgarbanti, M.; Battistini, A., Jr.; Harrington, W.J.; Barber, G.N.; Hiscott, J. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene 2001, 20, 800–811. [Google Scholar] [CrossRef]
- Nishimura, M.; Watanabe, T.; Yagi, S.; Yamanaka, T.; Fujimuro, M. Kaposi’s sarcoma-associated herpesvirus ORF34 is essential for late gene expression and virus production. Sci. Rep. 2017, 7, 329. [Google Scholar] [CrossRef]
- Weidle, U.H.; Weissmann, C. The 5′-flanking region of a human IFN-α gene mediates viral induction of transcription. Nature 1983, 303, 442–446. [Google Scholar] [CrossRef]
- Ma, J.; Rong, L.; Zhou, Y.; Roy, B.B.; Lu, J.; Abrahamyan, L.; Mouland, A.J.; Pan, Q.; Liang, C. The requirement of the DEAD-box protein DDX24 for the packaging of human immunodeficiency virus type 1 RNA. Virology 2008, 375, 253–264. [Google Scholar] [CrossRef]
- Yedavalli, V.S.; Neuveut, C.; Chi, Y.-H.; Kleiman, L.; Jeang, K.-T. Requirement of DDX3 DEAD Box RNA Helicase for HIV-1 Rev-RRE Export Function. Cell 2004, 119, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Gringhuis, S.I.; Hertoghs, N.; Kaptein, T.M.; Zijlstra-Willems, E.M.; Sarrami-Forooshani, R.; Sprokholt, J.K.; van Teijlingen, N.H.; Kootstra, N.A.; Booiman, T.; van Dort, K.A.; et al. HIV-1 blocks the signaling adaptor MAVS to evade antiviral host defense after sensing of abortive HIV-1 RNA by the host helicase DDX3. Nat. Immunol. 2017, 18, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Nishiyama, M.; Moroishi, T.; Yumimoto, K.; Nakayama, K.I. MDM2 Mediates Nonproteolytic Polyubiquitylation of the DEAD-Box RNA Helicase DDX24. Mol. Cell. Biol. 2014, 34, 3321–3340. [Google Scholar] [CrossRef] [PubMed]
- Mocarski, E.S.; Upton, J.; Kaiser, W.J. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat. Rev. Immunol. 2011, 12, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Gay, L.A.; Sethuraman, S.; Thomas, M.; Turner, P.C.; Renne, R. Modified Cross-Linking, Ligation, and Sequencing of Hybrids (qCLASH) Identifies Kaposi’s Sarcoma-Associated Herpesvirus MicroRNA Targets in Endothelial Cells. J. Virol. 2018, 92, e02138-17. [Google Scholar] [CrossRef]
- Bullard, W.L.; Kara, M.; Gay, L.A.; Sethuraman, S.; Wang, Y.; Nirmalan, S.; Esemenli, A.; Feswick, A.; Hoffman, B.A.; Renne, R.; et al. Identification of murine gammaherpesvirus 68 miRNA-mRNA hybrids reveals miRNA target conservation among gammaherpesviruses including host translation and protein modification machinery. PLoS Pathog. 2019, 15, e1007843. [Google Scholar] [CrossRef]
- Ungerleider, N.; Bullard, W.; Kara, M.; Wang, X.; Roberts, C.; Renne, R.; Tibbetts, S.; Flemington, E.K. EBV miRNAs are potent effectors of tumor cell transcriptome remodeling in promoting immune escape. PLoS Pathog. 2021, 17, e1009217. [Google Scholar] [CrossRef]
- Awasthi, S.; Verma, M.; Mahesh, A.; Khan, M.I.K.; Govindaraju, G.; Rajavelu, A.; Chavali, P.L.; Chavali, S.; Dhayalan, A. DDX49 is an RNA helicase that affects translation by regulating mRNA export and the levels of pre-ribosomal RNA. Nucleic Acids Res. 2018, 46, 6304–6317. [Google Scholar] [CrossRef]
- Schumann, S.; Jackson, B.R.; Baquero-Perez, B.; Whitehouse, A. Kaposi’s Sarcoma-Associated Herpesvirus ORF57 Protein: Exploiting All Stages of Viral mRNA Processing. Viruses 2013, 5, 1901–1923. [Google Scholar] [CrossRef]
- Gong, D.; Kim, Y.H.; Xiao, Y.; Du, Y.; Xie, Y.; Lee, K.K.; Feng, J.; Farhat, N.; Zhao, D.; Shu, S.; et al. A Herpesvirus Protein Selectively Inhibits Cellular mRNA Nuclear Export. Cell Host Microbe 2016, 20, 642–653. [Google Scholar] [CrossRef]
- Haecker, I.; Gay, L.A.; Yang, Y.; Hu, J.; Morse, A.M.; McIntyre, L.M.; Renne, R. Ago HITS-CLIP expands understanding of Kaposi’s sarcoma-associated herpesvirus miRNA function in primary effusion lymphomas. PLoS Pathog. 2012, 8, e1002884. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Huang, Y.; Li, W.; Zhu, Y.; Jung, J.U.; Lu, C.; Gao, S.J. Genomewide mapping and screening of Kaposi’s sarcoma-associated herpesvirus (KSHV) 3’ untranslated regions identify bicistronic and polycistronic viral transcripts as frequent targets of KSHV microRNAs. J. Virol. 2014, 88, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ni, G.; Damania, B. ADAR1 Facilitates KSHV Lytic Reactivation by Modulating the RLR-Dependent Signaling Pathway. Cell Rep. 2020, 31, 107564. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KSHV Gene | Phase | Expressed from Spliced mRNAs | KSHV Protein | Function | Reference |
|---|---|---|---|---|---|
| K2 | Immediate-Early | Yes | viral interleukin-6 (v-IL6) | activates JAK/STAT, MAPK, and Akt signaling pathways to regulate B-cell proliferation and KSHV reactivation | [44,46] |
| ORF70/K4/K4.1/K4.2 | Immediate-Early/Early | No | v-CCL3 (v-MIP-III), v-CCL3 (v-MIP-II) | ORF70: Govern the transition of the KSHV genome from latent to lytic phase. K4/K4.1: Homologs of cellular chemokines | [44] |
| K5 | Immediate-Early | Yes | Modulator of Immune Recognition (MIR2) | encodes for a ubiquitin E3 ligase that can degrade MHC-I molecules | [45] |
| ORF58 | Early | No | EBV BMRF2 homologue | A component of the tegument that interacts with the envelope | [47] |
| ORF59 | Early | Yes | DNA polymerase processivity factor, Cytomegalovirus (CMV) and EBV homolog | A viral DNA polymerase processivity factor required for lytic DNA replication | [47] |
| K9 (only in DDX49) | Early | No | vIRF1 | Homolog of cellular interferon regulatory factor | [48,49,50] |
| ORF65/66/67 (only in DDX49) | Early/Late | No | EBV BFRF2 | Required for virion production | [51] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serfecz, J.C.; Hong, Y.; Gay, L.A.; Shekhar, R.; Turner, P.C.; Renne, R. DExD/H Box Helicases DDX24 and DDX49 Inhibit Reactivation of Kaposi’s Sarcoma Associated Herpesvirus by Interacting with Viral mRNAs. Viruses 2022, 14, 2083. https://doi.org/10.3390/v14102083
Serfecz JC, Hong Y, Gay LA, Shekhar R, Turner PC, Renne R. DExD/H Box Helicases DDX24 and DDX49 Inhibit Reactivation of Kaposi’s Sarcoma Associated Herpesvirus by Interacting with Viral mRNAs. Viruses. 2022; 14(10):2083. https://doi.org/10.3390/v14102083
Chicago/Turabian StyleSerfecz, Jacquelyn C., Yuan Hong, Lauren A. Gay, Ritu Shekhar, Peter C. Turner, and Rolf Renne. 2022. "DExD/H Box Helicases DDX24 and DDX49 Inhibit Reactivation of Kaposi’s Sarcoma Associated Herpesvirus by Interacting with Viral mRNAs" Viruses 14, no. 10: 2083. https://doi.org/10.3390/v14102083
APA StyleSerfecz, J. C., Hong, Y., Gay, L. A., Shekhar, R., Turner, P. C., & Renne, R. (2022). DExD/H Box Helicases DDX24 and DDX49 Inhibit Reactivation of Kaposi’s Sarcoma Associated Herpesvirus by Interacting with Viral mRNAs. Viruses, 14(10), 2083. https://doi.org/10.3390/v14102083