
Virus-Targeted Transcriptomic Analyses Implicate Ranaviral Interaction with Host Interferon Response in Frog Virus 3-Infected Frog Tissues

 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Virus
2.2. Animal Infection and Tissue Sampling
2.3. DNA/RNA Extraction and qPCR FV3 Gene Copy Assays
2.4. Transcriptomic Assays (RNA-Seq)
2.5. Novel Viral Gene Prediction and Functional Analysis
2.6. Statistical Analysis
3. Results and Discussions
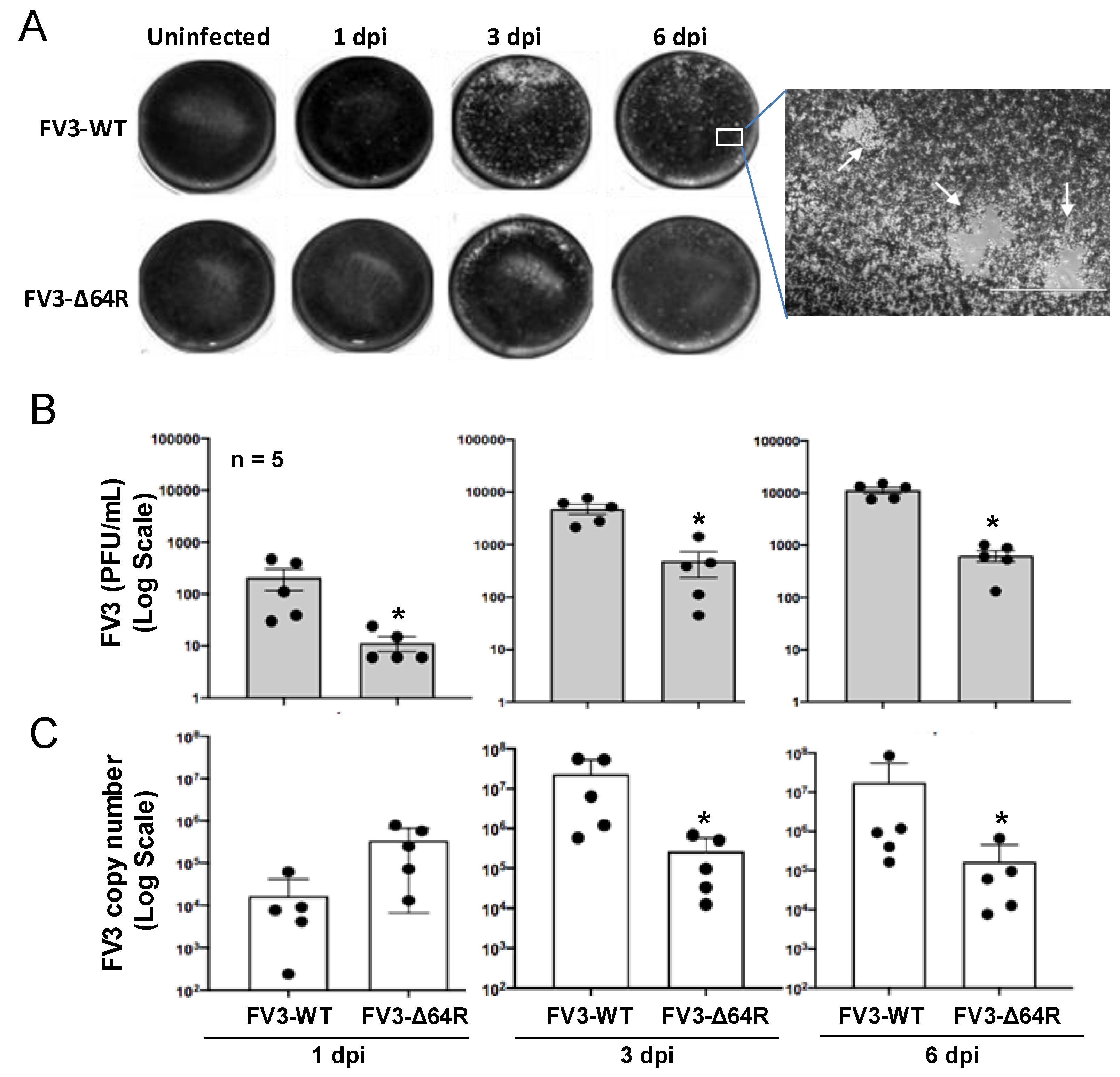
3.1. FV3 Infection and Comparative Viral Determination Between FV3-WT and FV3-∆64R Strains in the Kidneys of Adult Frogs
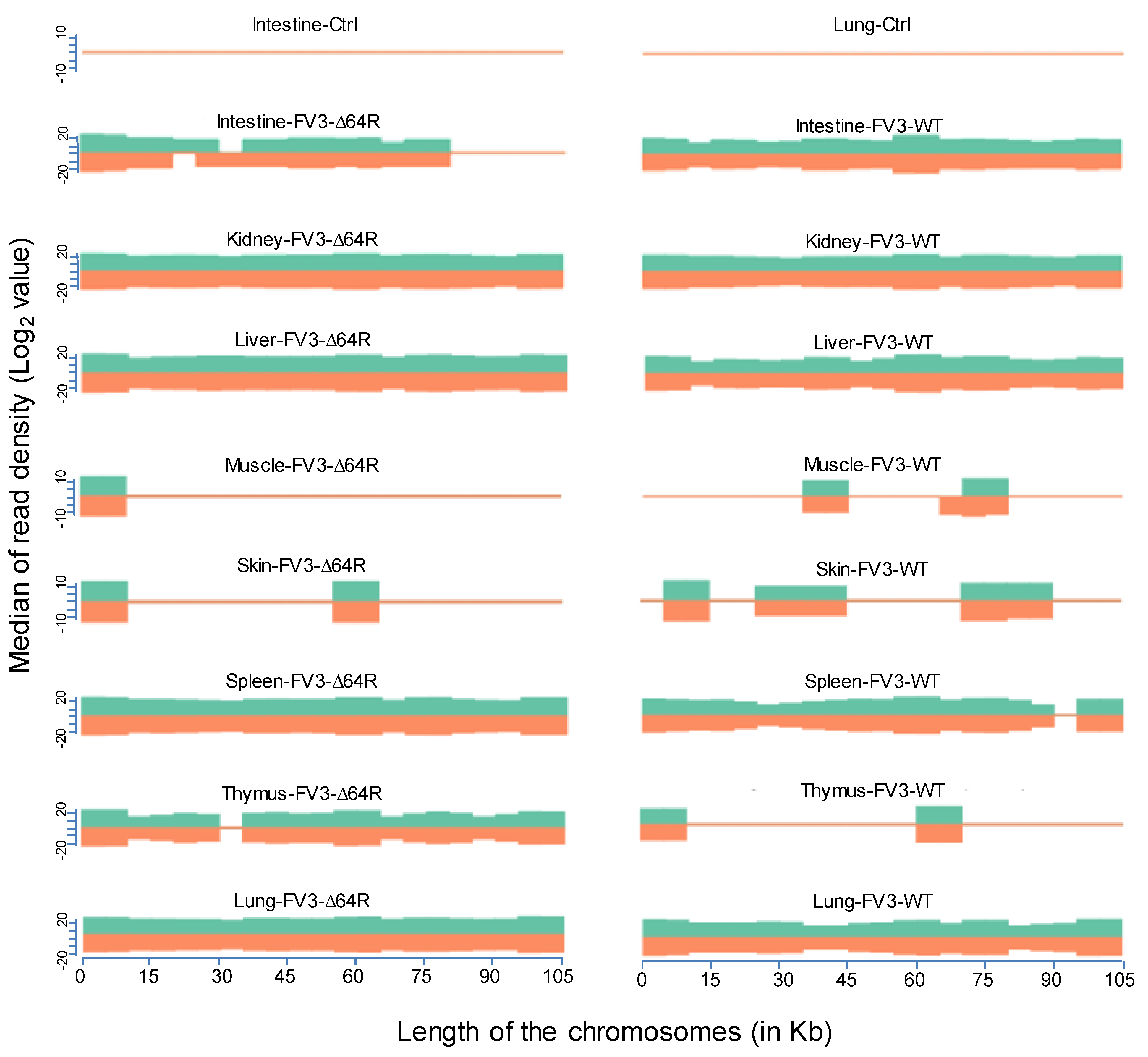
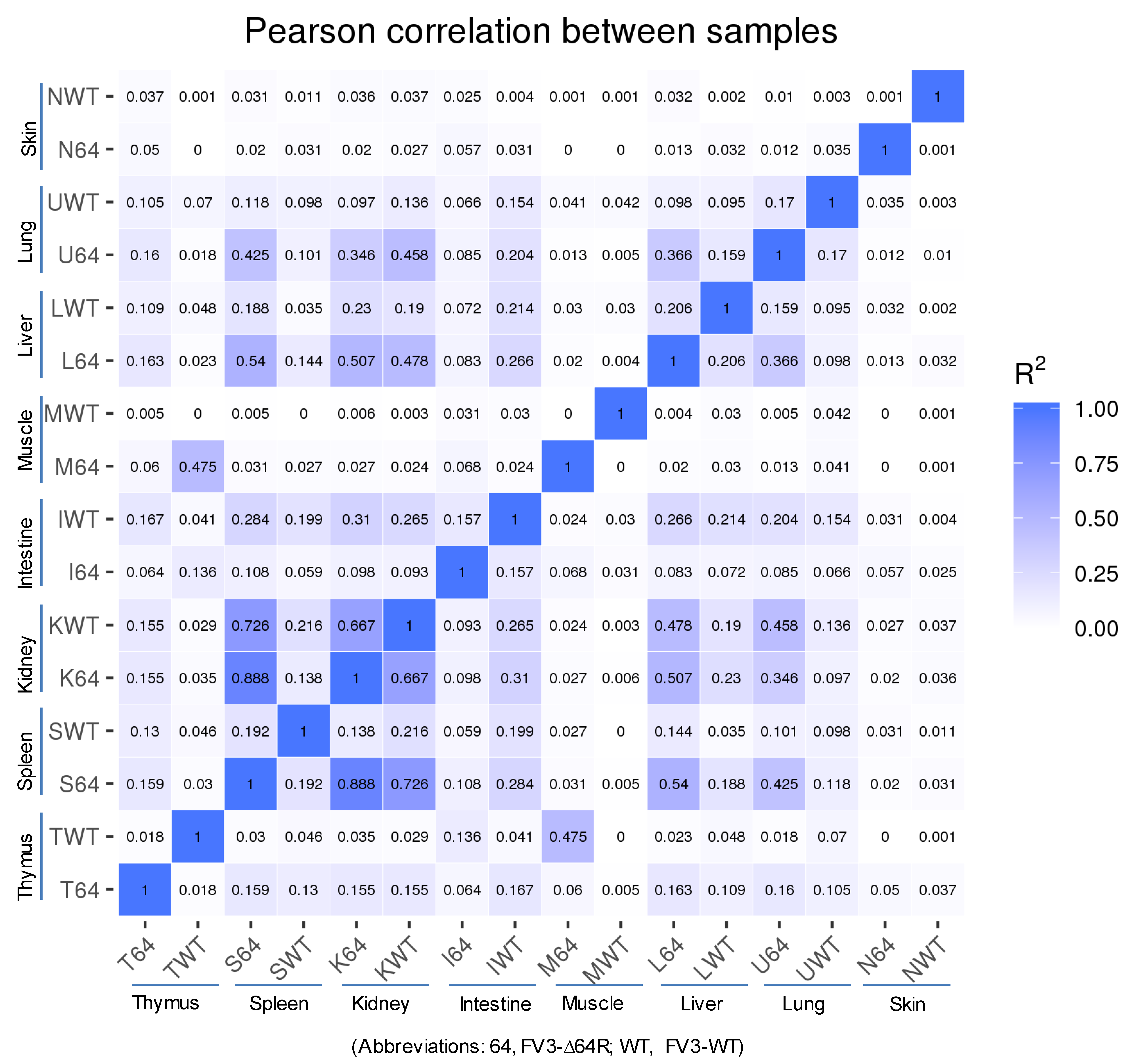
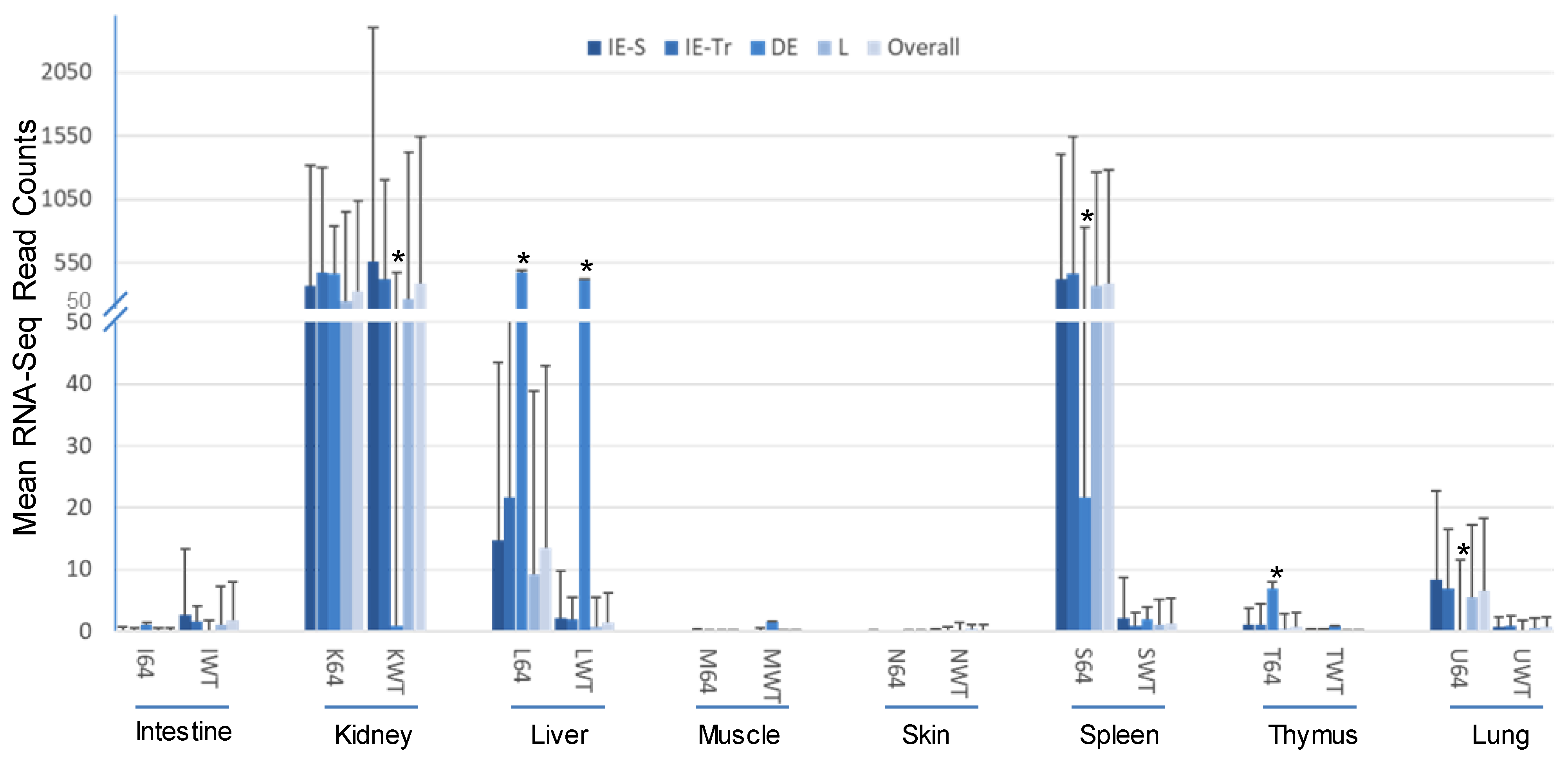
3.2. Virus-Targeting Transcriptome Analysis and Difference Dependent on Tissue Types and FV3 Strains
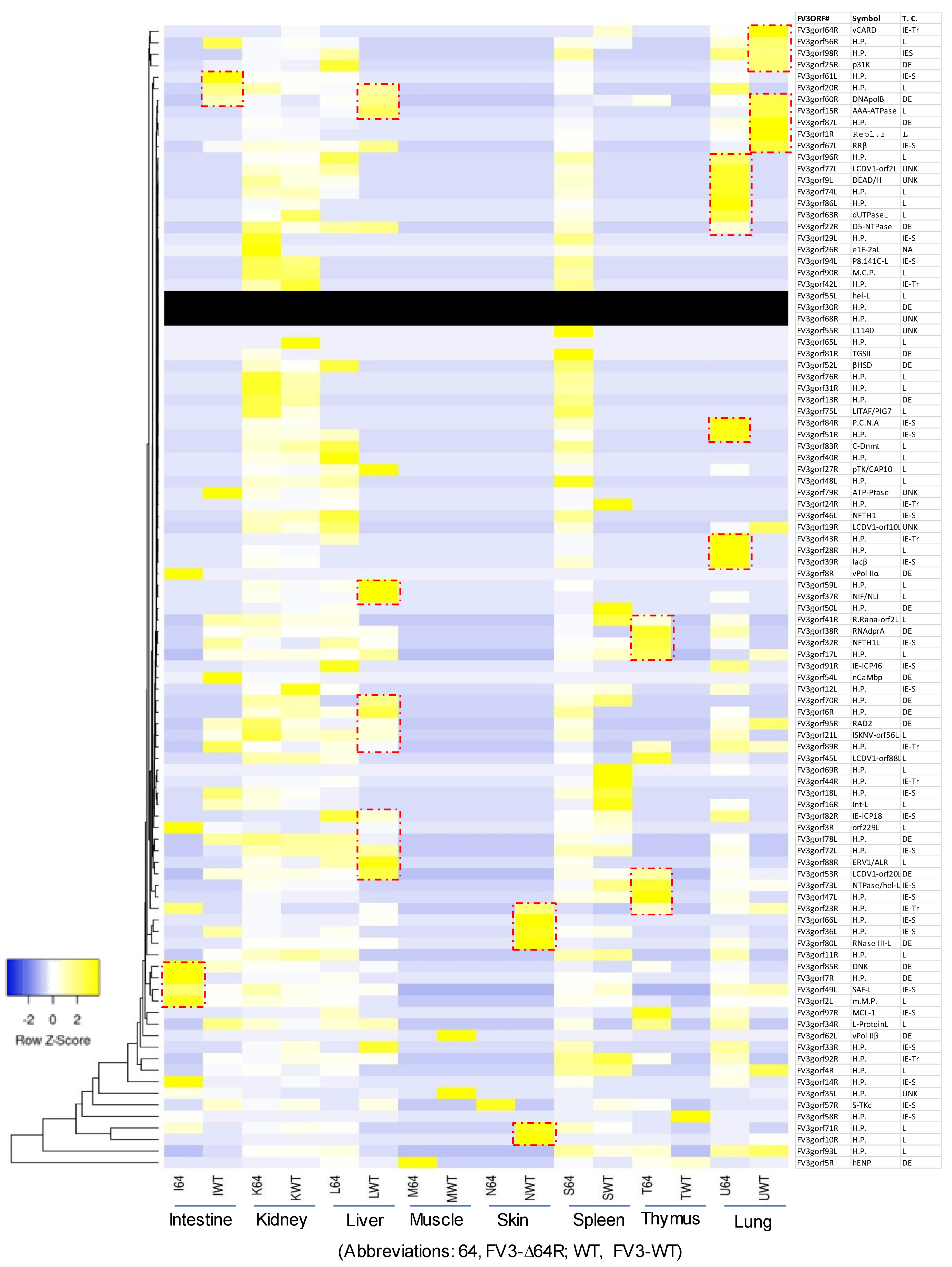
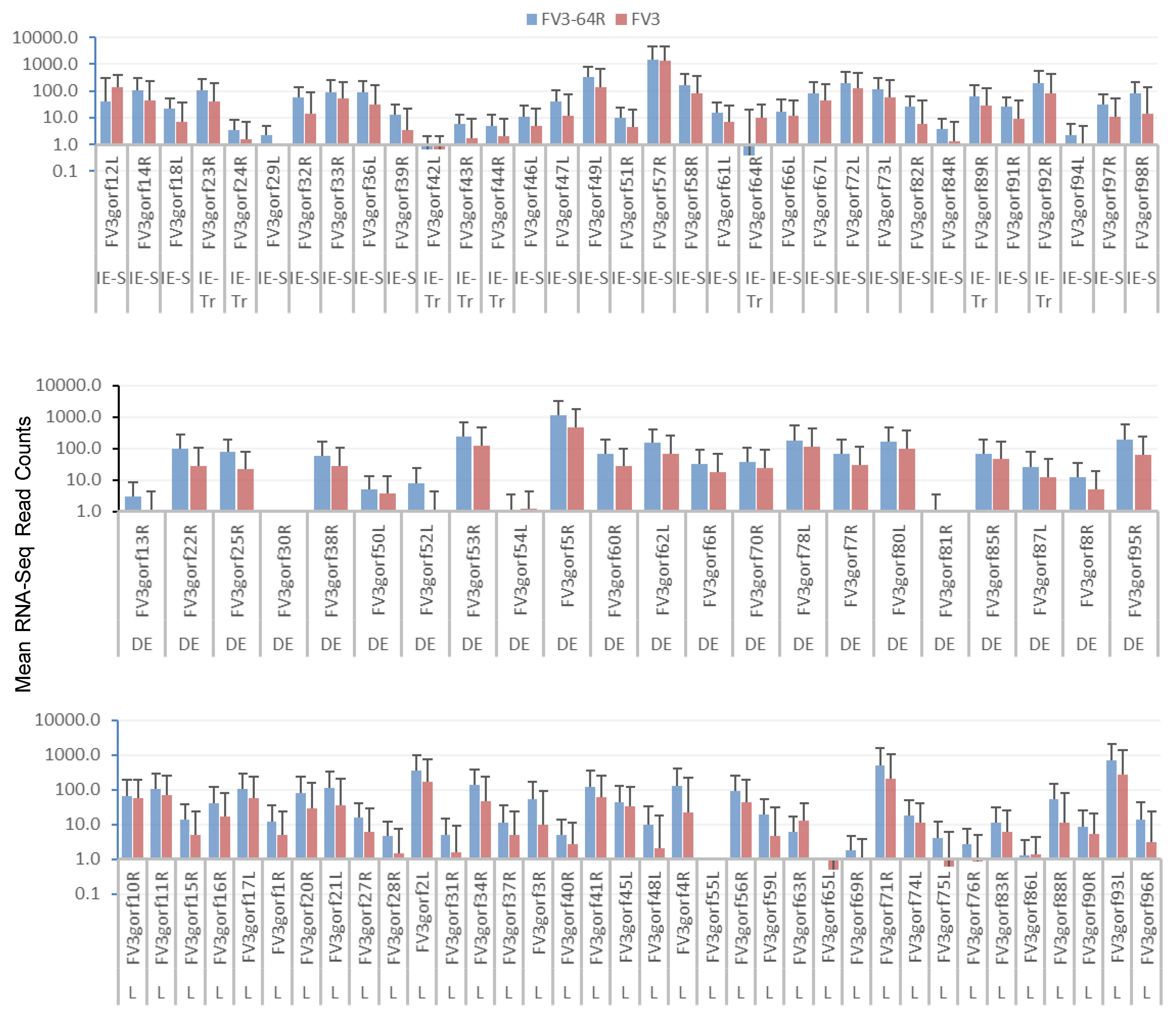
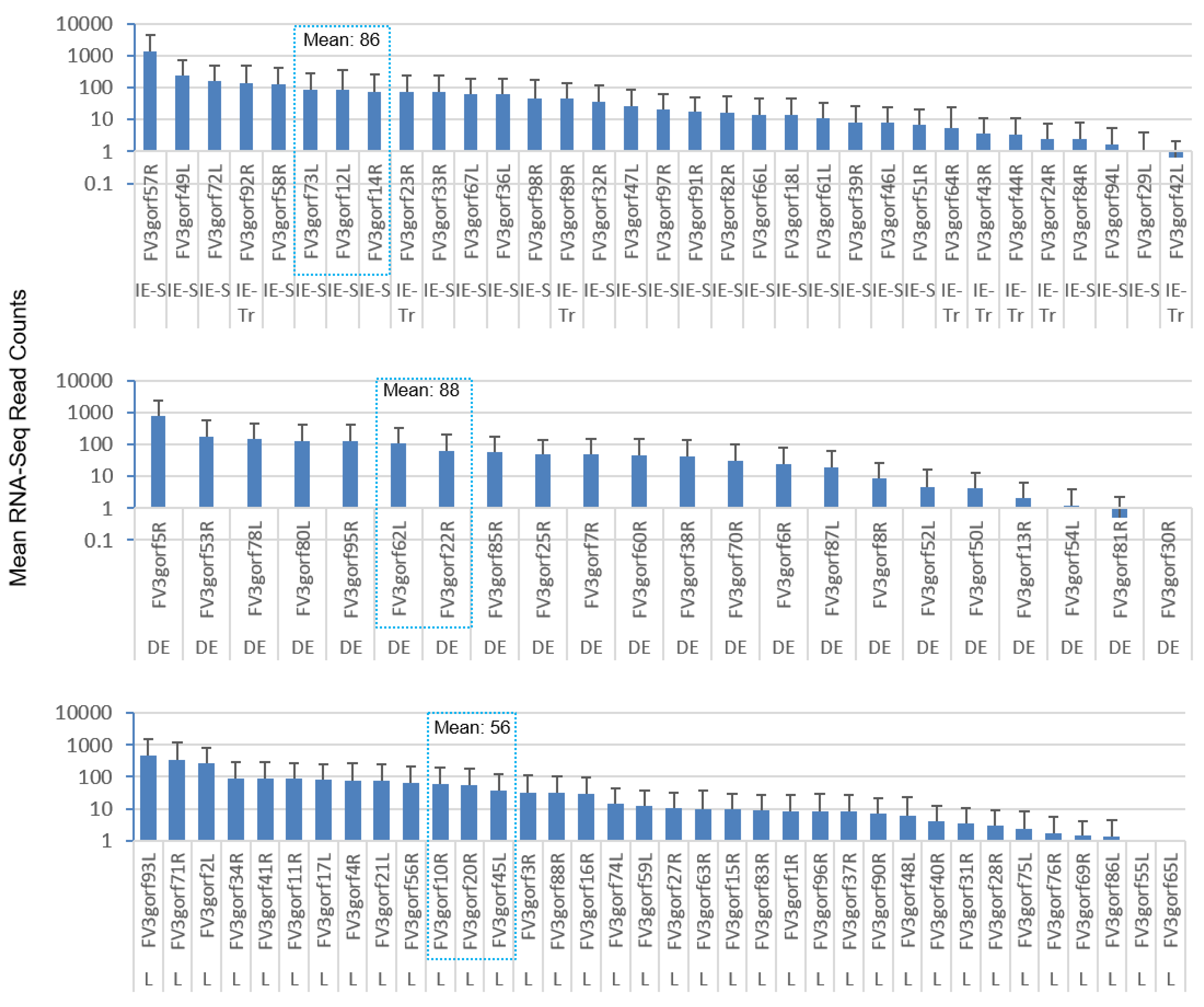
3.3. Genome-Wide Differential Expression Analysis of FV3 Coding Genes
3.4. Differential Expression Analyses According to the Temporal Class of FV3 Coding Genes
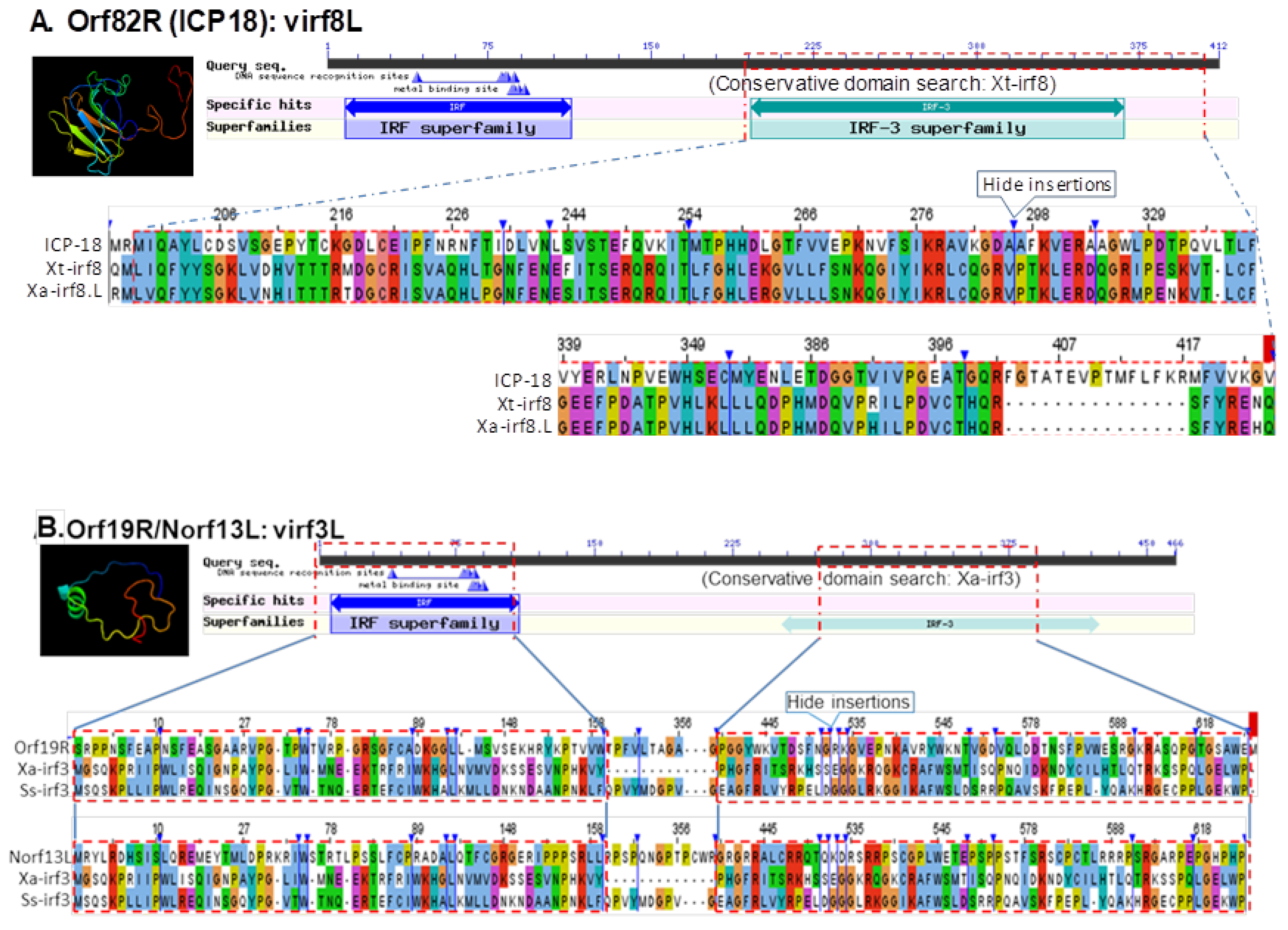
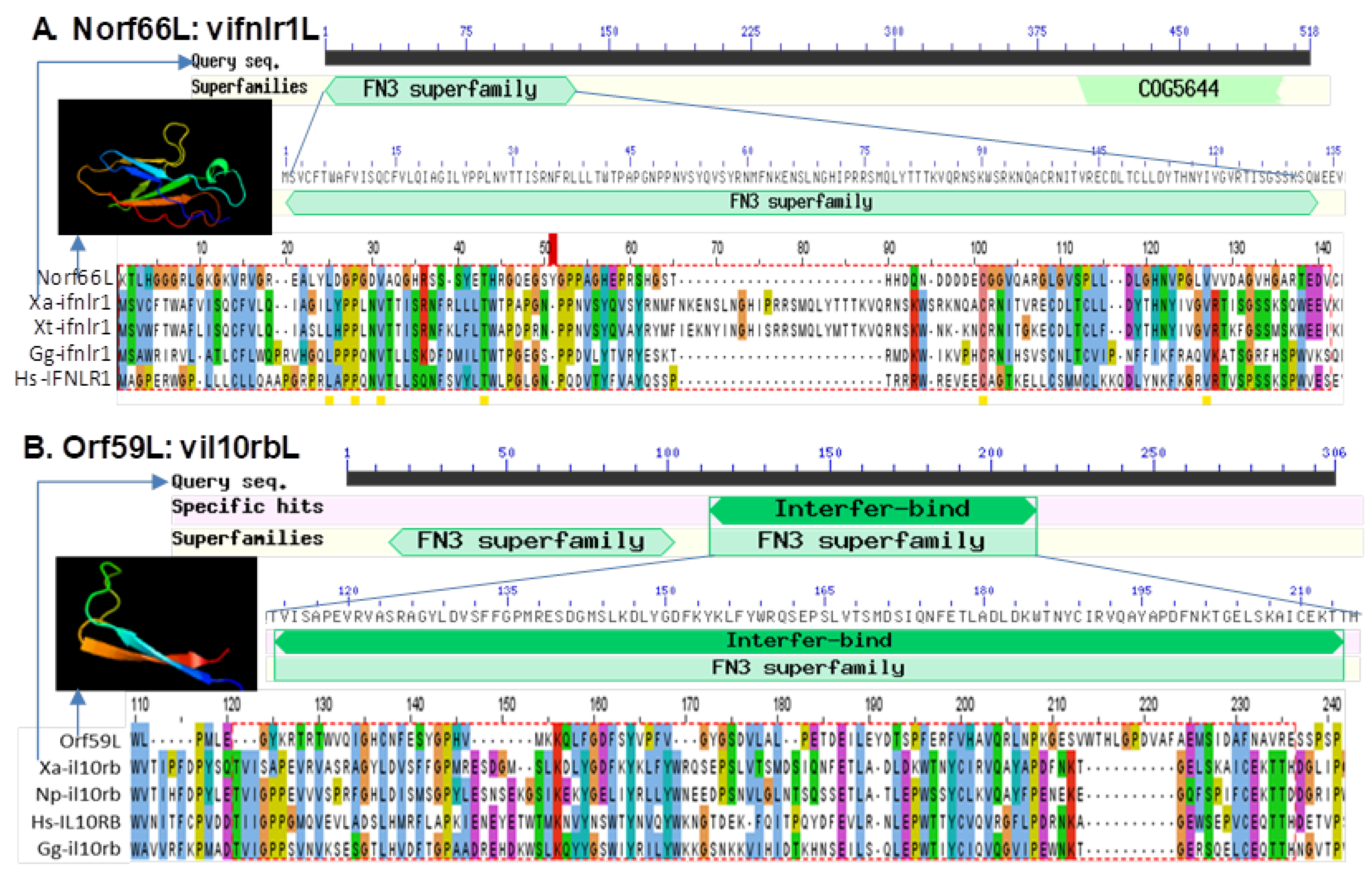
3.5. Detection of Interferon Regulatory Factor Domain or Fibronectin Type 3 Domain in Several FV3 Hypothetical Proteins
4. Conclusive Highlights
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chinchar, V.G. Ranaviruses (family Iridoviridae): Emerging cold-blooded killers. Arch. Virol. 2002, 147, 447–470. [Google Scholar] [CrossRef]
- Chinchar, V.G.; Yu, K.H.; Jancovich, J.K. The molecular biology of frog virus 3 and other iridoviruses infecting cold-blooded vertebrates. Viruses 2011, 3, 1959–1985. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Robert, J. Antiviral immunity in amphibians. Viruses 2011, 3, 2065–2086. [Google Scholar] [CrossRef]
- Grayfer, L.; De Jesús Andino, F.; Robert, J. The amphibian (Xenopus laevis) type I interferon response to frog virus 3: New insight into ranavirus pathogenicity. J. Virol. 2014, 88, 5766–5777. [Google Scholar] [CrossRef] [Green Version]
- Pounds, J.A.; Bustamante, M.R.; Coloma, L.A.; Consuegra, J.A.; Fogden, M.P.; Foster, P.N.; La Marca, E.; Masters, K.L.; Merino-Viteri, A.; Puschendorf, R.; et al. Widespread amphibian extinctions from epidemic disease driven by global warming. Nature 2006, 439, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Green, D.E.; Converse, K.A.; Schrader, A.K. Epizootiology of sixty-four amphibian morbidity and mortality events in the USA, 1996–2001. Ann. N. Y. Acad. Sci. 2002, 969, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.P. Amphibian decline and extinction: What we know and what we need to learn. Dis. Aquat. Organ. 2010, 92, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.; Gray, M.; Storfer, A. Ecopathology of ranaviruses infecting amphibians. Viruses 2011, 3, 2351–2373. [Google Scholar] [CrossRef]
- Vilaça, S.T.; Bienentreu, J.F.; Brunetti, C.R.; Lesbarrères, D.; Murray, D.L.; Kyle, C.J. Frog Virus 3 Genomes Reveal Prevalent Recombination between Ranavirus Lineages and Their Origins in Canada. J. Virol. 2019, 93, e00765-19. [Google Scholar] [CrossRef] [Green Version]
- Saucedo, B.; Garner, T.W.J.; Kruithof, N.; Allain, S.J.; Goodman, M.J.; Cranfield, R.J.; Sergeant, C.; Vergara, D.A.; Kik, M.J.; Forzán, M.J.; et al. Common midwife toad ranaviruses replicate first in the oral cavity of smooth newts (Lissotriton vulgaris) and show distinct strain-associated pathogenicity. Sci. Rep. 2019, 9, 4453. [Google Scholar] [CrossRef] [PubMed]
- Goorha, R. Frog virus 3 DNA replication occurs in two stages. J. Virol. 1982, 43, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Iyer, L.M.; Balaji, S.; Koonin, E.V.; Aravind, L. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res. 2006, 117, 156–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonin, E.V.; Yutin, N. Evolution of the Large Nucleocytoplasmic DNA Viruses of Eukaryotes and Convergent Origins of Viral Gigantism. Adv. Virus Res. 2019, 103, 167–202. [Google Scholar] [PubMed]
- Gallot-Lavallée, L.; Blanc, G. A Glimpse of Nucleo-Cytoplasmic Large DNA Virus Biodiversity through the Eukaryotic Genomics Window. Viruses 2017, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaján, G.L.; Doszpoly, A.; Tarján, Z.L.; Vidovszky, M.Z.; Papp, T. Virus-Host Coevolution with a Focus on Animal and Human DNA Viruses. J. Mol. Evol. 2020, 88, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Willis, D.B.; Goorha, R.; Miles, M.; Granoff, A. Macromolecular synthesis in cells infected by frog virus 3. VII. Transcriptional and post-transcriptional regulation of virus gene expression. J. Virol. 1977, 24, 326–342. [Google Scholar] [CrossRef] [Green Version]
- Goorha, R.; Willis, D.B.; Granoff, A. Macromolecular synthesis in cells infected by frog virus 3. XII. Viral regulatory proteins in transcriptional and post-transcriptional controls. J. Virol. 1979, 32, 442–448. [Google Scholar] [CrossRef] [Green Version]
- Majji, S.; Thodima, V.; Sample, R.; Whitley, D.; Deng, Y.; Mao, J.; Chinchar, V.G. Transcriptome analysis of Frog virus 3, the type species of the genus Ranavirus, family Iridoviridae. Virology 2009, 391, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Rebollar, E.A.; Antwis, R.E.; Becker, M.H.; Belden, L.K.; Bletz, M.C.; Brucker, R.M.; Harrison, X.A.; Hughey, M.C.; Kueneman, J.G.; Loudon, A.H.; et al. Using “Omics” and Integrated Multi-Omics Approaches to Guide Probiotic Selection to Mitigate Chytridiomycosis and Other Emerging Infectious Diseases. Front. Microbiol. 2016, 7, 68. [Google Scholar] [CrossRef]
- Russo, A.G.; Eden, J.S.; Enosi Tuipulotu, D.; Shi, M.; Selechnik, D.; Shine, R.; Rollins, L.A.; Holmes, E.C.; White, P.A. Viral Discovery in the Invasive Australian Cane Toad (Rhinella marina) Using Metatranscriptomic and Genomic Approaches. J. Virol. 2018, 92, e00768-18. [Google Scholar] [CrossRef] [Green Version]
- Andino Fde, J.; Grayfer, L.; Chen, G.; Chinchar, V.G.; Edholm, E.S.; Robert, J. Characterization of Frog Virus 3 knockout mutants lacking putative virulence genes. Virology 2015, 485, 162–170. [Google Scholar] [CrossRef]
- Jacques, R.; Edholm, E.S.; Jazz, S.; Odalys, T.L.; Francisco, J.A. Xenopus-FV3 host-pathogen interactions and immune evasion. Virology 2017, 511, 309–319. [Google Scholar] [CrossRef]
- Qi, Z.; Nie, P.; Secombes, C.J.; Zou, J. Intron-containing type I and type III IFN coexist in amphibians: Refuting the concept that a retroposition event gave rise to type I IFNs. J. Immunol. 2010, 184, 5038–5046. [Google Scholar] [CrossRef] [Green Version]
- Sang, Y.; Liu, Q.; Lee, J.; Ma, W.; McVey, D.S.; Blecha, F. Expansion of amphibian intronless interferons revises the paradigm for interferon evolution and functional diversity. Sci. Rep. 2016, 6, 29072. [Google Scholar] [PubMed] [Green Version]
- Tian, Y.; Jennings, J.; Gong, Y.; Sang, Y. Xenopus Interferon Complex: Inscribing the Amphibiotic Adaption and Species-Specific Pathogenic Pressure in Vertebrate Evolution? Cells 2019, 9, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, H.H.; Schneider, W.M.; Rice, C.M. Interferons and viruses: An evolutionary arms race of molecular interactions. Trends Immunol. 2015, 36, 124–138. [Google Scholar] [PubMed] [Green Version]
- Xenopus Laevis Research Resource for Immunology at the University of Rochester. Available online: http://www.urmc.rochester.edu/mbi/resources/xenopus-laevis/ (accessed on 7 July 2021).
- Grayfer, L.; De Jesús Andino, F.; Robert, J. Prominent amphibian (Xenopus laevis) tadpole type III interferon response to the frog virus 3 ranavirus. J. Virol. 2015, 89, 5072–5082. [Google Scholar] [PubMed] [Green Version]
- Sang, Y.; Brichalli, W.; Rowland, R.R.; Blecha, F. Genome-wide analysis of antiviral signature genes in porcine macrophages at different activation statuses. PLoS ONE 2014, 9, e87613. [Google Scholar] [CrossRef]
- Xenbase. Available online: http://ftp.xenbase.org/ (accessed on 7 July 2021).
- NCBI Genome Portal. Available online: ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF (accessed on 7 July 2021).
- NCBI Blast Portal. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 7 July 2021).
- Program fgenesV0 and fgenesV. Available online: http://www.softberry.com/ (accessed on 7 July 2021).
- HDOCK Server. Available online: http://hdock.phys.hust.edu.cn/ (accessed on 7 July 2021).
- Park, H.H.; Lo, Y.C.; Lin, S.C.; Wang, L.; Yang, J.K.; Wu, H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu. Rev. Immunol. 2007, 25, 561–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, K.E.; Gilmour, J.S.; Coutinho, A.E.; Savill, J.S.; Seckl, J.R. Beta-hydroxysteroid dehydrogenase type 1—A role in inflammation? Mol. Cell Endocrinol. 2006, 248, 3–8. [Google Scholar] [CrossRef]
- Sang, E.R.; Tian, Y.; Miller, L.C.; Sang, Y. Epigenetic Evolution of ACE2 and IL-6 Genes: Non-Canonical Interferon-Stimulated Genes Correlate to COVID-19 Susceptibility in Vertebrates. Genes 2021, 12, 154. [Google Scholar] [CrossRef]
- Barrat, F.J.; Crow, M.K.; Ivashkiv, L.B. Interferon target-gene expression and epigenomic signatures in health and disease. Nat. Immunol. 2019, 20, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Piehler, J.; Thomas, C.; Garcia, K.C.; Schreiber, G. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol. Rev. 2012, 250, 317–334. [Google Scholar] [PubMed] [Green Version]
- Jefferies, C.A. Regulating IRFs in IFN Driven Disease. Front. Immunol. 2019, 10, 325. [Google Scholar] [PubMed] [Green Version]
- Chiang, H.S.; Liu, H.M. The Molecular Basis of Viral Inhibition of IRF- and STAT-Dependent Immune Responses. Front. Immunol. 2019, 9, 3086. [Google Scholar] [PubMed] [Green Version]
- Alcami, A. Viral mimicry of cytokines, chemokines and their receptors. Nat. Rev. Immunol. 2003, 3, 36–50. [Google Scholar]
- McSharry, B.P.; Avdic, S.; Slobedman, B. Human cytomegalovirus encoded homologs of cytokines, chemokines and their receptors: Roles in immunomodulation. Viruses 2012, 4, 2448–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Qi, Z.T.; Xu, Z.; Nie, P. Global characterization of interferon regulatory factor (IRF) genes in vertebrates: Glimpse of the diversification in evolution. BMC Immunol. 2010, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Tailor, P.; Tamura, T.; Kong, H.J.; Kubota, T.; Kubota, M.; Borghi, P.; Gabriele, L.; Ozato, K. The feedback phase of type I interferon induction in dendritic cells requires interferon regulatory factor 8. Immunity 2007, 27, 228–239. [Google Scholar]
- Tian, Y.; Khwatenge, C.N.; Li, J.; De Jesus Andino, F.; Robert, J.; Sang, Y. Targeted Transcriptomics of Frog Virus 3 in Infected Frog Tissues Reveal Non-Coding Regulatory Elements and microRNAs in the Ranaviral Genome and Their Potential Interaction with Host Immune Response. Front. Immunol. 2021, 12, 705253. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Rivera, A.; Parker, D.; Durbin, J.E. Type III IFNs: Beyond antiviral protection. Semin. Immunol. 2019, 43, 101303. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, I.; Odendall, C. Editorial: Interferon-λs: New regulators of inflammatory processes. Front. Immunol. 2019, 10, 2117. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-λ orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location on FV3 Genome | Viral Gene (Temporal) Designation | ORF/Protein Size | Validation/Prediction Algorithm | Analogy to IFN-Interfering Domain (E < 0.5 and ~30% id.) |
|---|---|---|---|---|
| 14,685–15,092 (−) | Norf13L (UNK) | 408 nt/135 aa | FGEESV0 (Markov chain-based) | IRF domains in Xa-irf3 (NP_001079588) Ss-irf3 (NP_001165753) |
| 21,916–24,471 (+) | Orf19R (UNK) | 2556 nt/851 aa | NCBI Annotation/FGEESV0 | IRF domains in Xa-irf3 (NP_001079588) Ss-irf3 (NP_001165753) |
| 33,728–36,640 (+) | Orf27R (L) | 2913 nt/970 aa | NCBI Annotation/FGEESV0 | IRF domains in Dr-irf8 (NP_001002622) Xt-irf8 (XP_004913664) |
| 38,635–39,102 (−) | Norf42L (UNK) | 468 nt/155 aa | FGEESV0 | IRF domains in Dr-irf6 (NP_956892) Xt-irf6 (NP_001025493) |
| 46,691–50,188 (+) | Orf41R (L) | 3498 nt/1165 aa | NCBI Annotation/FGEESV0 | IRF domain in Dr-irf4a (NP_001116182) Xt-irf4 (XP_002936464) |
| 59,162–60,037 (−) | Norf66L (UNK) | 876 nt/291 aa | FGEESV0 | FN3 domain in Xa-ifnlr1 (XP_018097809) Xt-ifnlr1 (ACV32138) |
| 65,956–67,014 (−) | Orf59L (L) | 1059 nt/352 aa | NCBI Annotation/FGEESV0 | FN3 domain in Xa-il10rb (XP_018101420) Hs-il10rb (NP_000619) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; De Jesús Andino, F.; Khwatenge, C.N.; Li, J.; Robert, J.; Sang, Y. Virus-Targeted Transcriptomic Analyses Implicate Ranaviral Interaction with Host Interferon Response in Frog Virus 3-Infected Frog Tissues. Viruses 2021, 13, 1325. https://doi.org/10.3390/v13071325
Tian Y, De Jesús Andino F, Khwatenge CN, Li J, Robert J, Sang Y. Virus-Targeted Transcriptomic Analyses Implicate Ranaviral Interaction with Host Interferon Response in Frog Virus 3-Infected Frog Tissues. Viruses. 2021; 13(7):1325. https://doi.org/10.3390/v13071325
Chicago/Turabian StyleTian, Yun, Francisco De Jesús Andino, Collins N. Khwatenge, Jiuyi Li, Jacques Robert, and Yongming Sang. 2021. "Virus-Targeted Transcriptomic Analyses Implicate Ranaviral Interaction with Host Interferon Response in Frog Virus 3-Infected Frog Tissues" Viruses 13, no. 7: 1325. https://doi.org/10.3390/v13071325
APA StyleTian, Y., De Jesús Andino, F., Khwatenge, C. N., Li, J., Robert, J., & Sang, Y. (2021). Virus-Targeted Transcriptomic Analyses Implicate Ranaviral Interaction with Host Interferon Response in Frog Virus 3-Infected Frog Tissues. Viruses, 13(7), 1325. https://doi.org/10.3390/v13071325