
Multiple Mammarenaviruses Circulating in Angolan Rodents


Abstract
1. Introduction
2. Materials and Methods
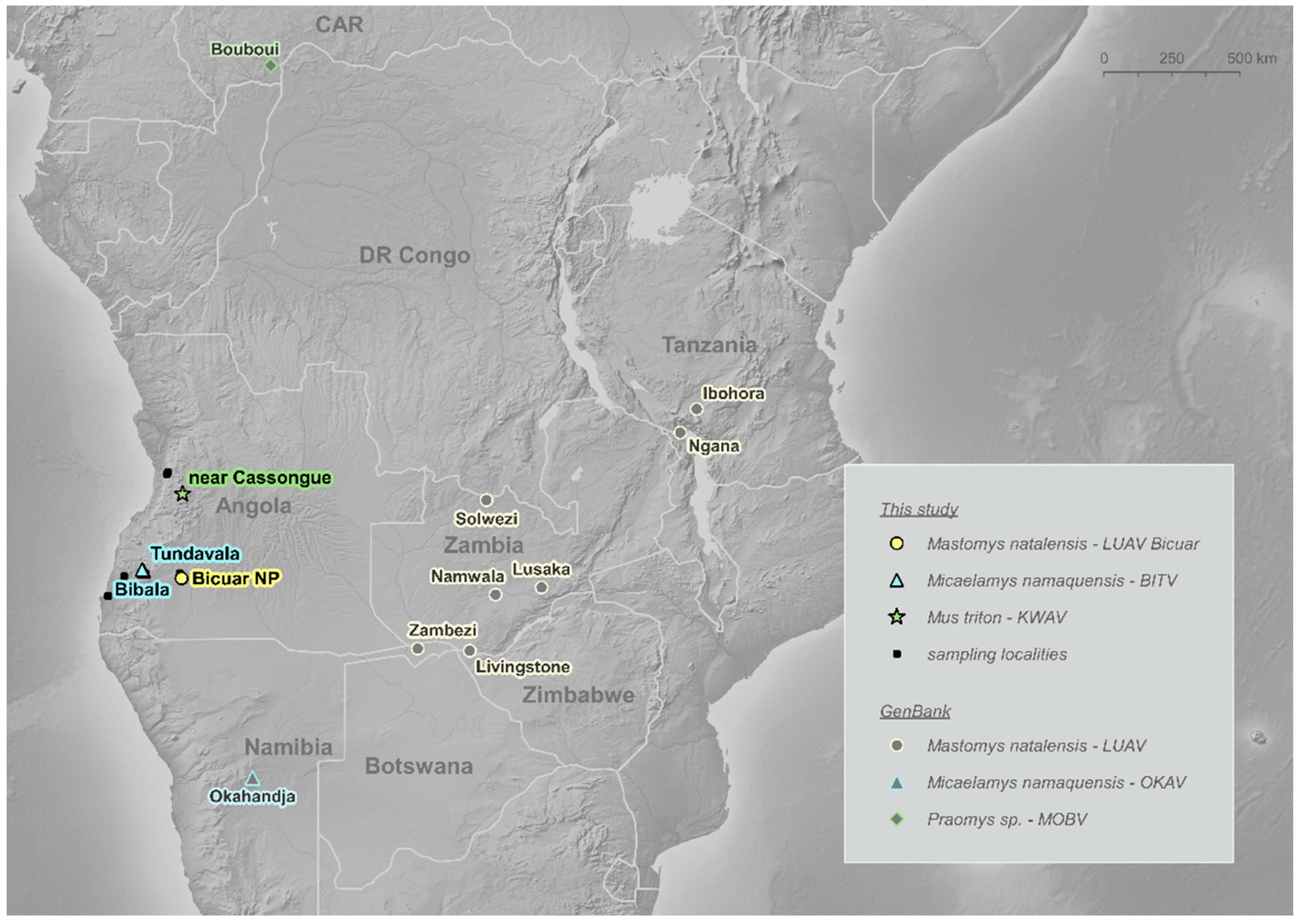
2.1. Sample Collection
2.2. Genetic Characterisation of Hosts
2.3. Molecular Screening of Viral RNA
2.4. Whole Genome Sequencing and Assembly of Mammarenavirus Genomes
2.5. Genetic Analyses of Mammarenaviruses
3. Results
3.1. Virus Detection and Prevalence
3.2. Characterisation of the Full Viral Genomes
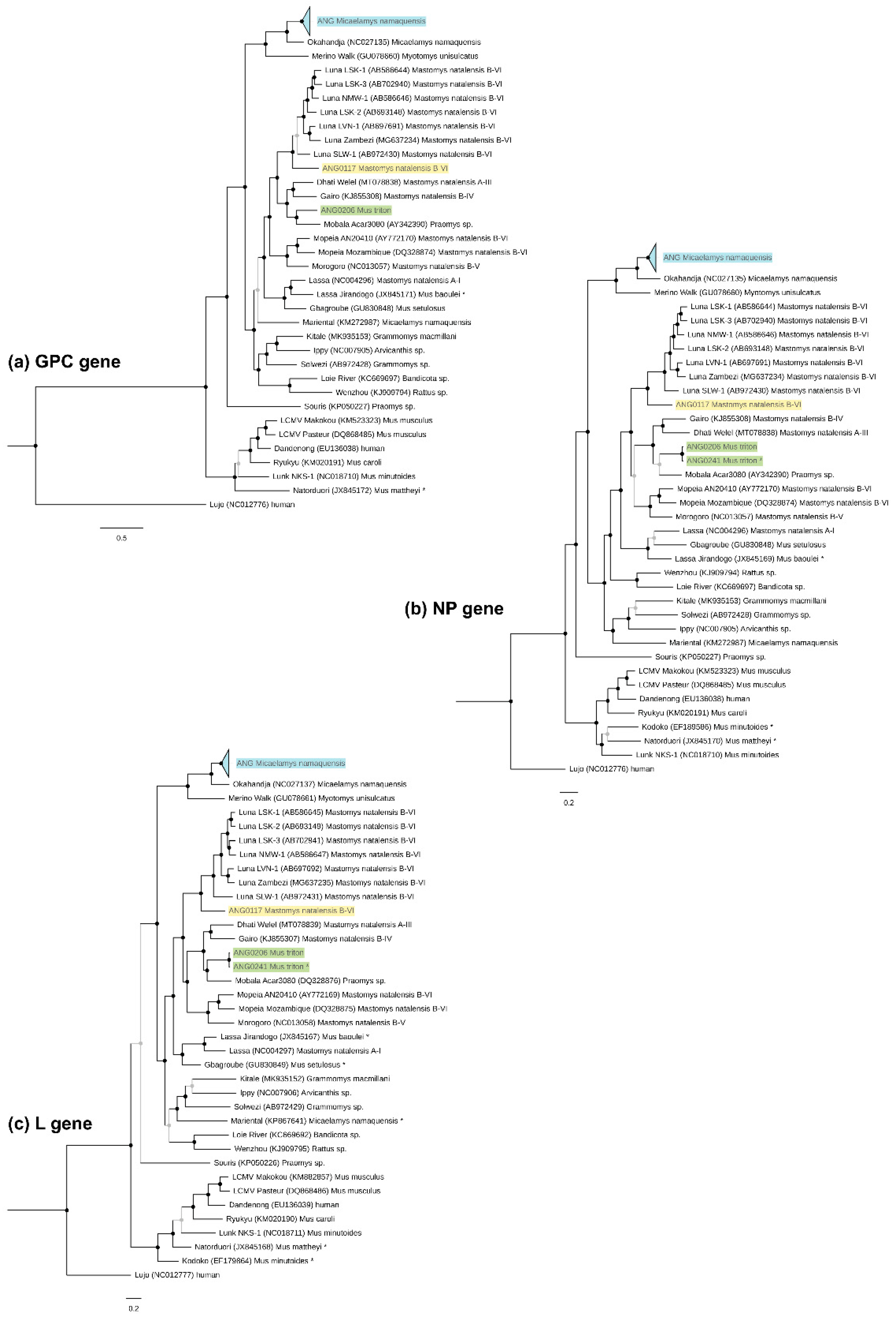
3.3. Phylogenetic Characterisation and Molecular Divergence
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, M.; Lin, X.D.; Chen, X.; Tian, J.H.; Chen, L.J.; Li, K.; Wang, W.; Eden, J.S.; Shen, J.J.; Liu, L.; et al. Author Correction: The evolutionary history of vertebrate RNA viruses. Nat. Cell Biol. 2018, 561, E6. [Google Scholar] [CrossRef] [PubMed]
- Mordecai, G.J.; Miller, K.M.; Di Cicco, E.; Schulze, A.D.; Kaukinen, K.H.; Ming, T.J.; Li, S.; Tabata, A.; Teffer, A.; A Patterson, D.; et al. Endangered wild salmon infected by newly discovered viruses. eLife 2019, 8, 47615. [Google Scholar] [CrossRef] [PubMed]
- Dietz, J.; Kolesnik, E.; Heckers, K.O.; Klingberg, M.N.; Marschang, R.E. Detection of an arenavirus in a group of captive wagler’s pit vipers (tropidolaemus wagleri). J. Zoo Wildl. Med. 2020, 51, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Downs, W.G.; Anderson, C.R.; Spence, L.; Aitken, T.H.G.; Greenhall, A.H. Tacaribe Virus, a New Agent Isolated from Artibeus Bats and Mosquitoes in Trinidad, West Indies. Am. J. Trop. Med. Hyg. 1963, 12, 640–646. [Google Scholar] [CrossRef]
- Wu, Z.; Du, J.; Lu, L.; Yang, L.; Dong, J.; Sun, L.; Zhu, Y.; Liu, Q.; Jin, Q. Detection of Hantaviruses and Arenaviruzses in three-toed jerboas from the Inner Mongolia Autonomous Region, China. Emerg. Microbes Infect. 2018, 7, 1–3. [Google Scholar] [CrossRef]
- Radoshitzky, S.R.; Buchmeier, M.J.; Charrel, R.N.; Clegg, J.C.S.; Gonzalez, J.-P.J.; Günther, S.; Hepojoki, J.; Kuhn, J.H.; Lukashevich, I.S.; Romanowski, V.; et al. ICTV Virus Taxonomy Profile: Arenaviridae. J. Gen. Virol. 2019, 100, 1200–1201. [Google Scholar] [CrossRef]
- Frame, J.D.; Baldwin, J.M.; Gocke, D.J.; Troup, J.M. Lassa Fever, a New Virus Disease of Man from West Africa. I. Clinical description and pathological findings. Am. J. Trop. Med. Hyg. 1970, 19, 670–676. [Google Scholar] [CrossRef]
- Briese, T.; Paweska, J.T.; McMullan, L.; Hutchison, S.K.; Street, C.; Palacios, G.; Khristova, M.L.; Weyer, J.; Swanepoel, R.; Egholm, M.; et al. Genetic Detection and Characterization of Lujo Virus, a New Hemorrhagic Fever–Associated Arenavirus from Southern Africa. PLoS Pathog. 2009, 5, e1000455. [Google Scholar] [CrossRef]
- Kronmann, K.C.; Nimo-Paintsil, S.; Guirguis, F.; Kronmann, L.C.; Bonney, K.; Obiri-Danso, K.; Ampofo, W.; Fichet-Calvet, E. Two Novel Arenaviruses Detected in Pygmy Mice, Ghana. Emerg. Infect. Dis. 2013, 19, 1832–1835. [Google Scholar] [CrossRef]
- Olayemi, A.; Cadar, D.; Magassouba, N.; Obadare, A.; Kourouma, F.; Oyeyiola, A.; Fasogbon, S.; Igbokwe, J.; Rieger, T.; Bockholt, S.; et al. New Hosts of The Lassa Virus. Sci. Rep. 2016, 6, 25280. [Google Scholar] [CrossRef]
- Meheretu, Y.; Čížková, D.; Těšíková, J.; Welegerima, K.; Tomas, Z.; Kidane, D.; Girmay, K.; Schmidt-Chanasit, J.; Bryja, J.; Günther, S.; et al. High Diversity of RNA Viruses in Rodents, Ethiopia. Emerg. Infect. Dis. 2012, 18, 2047–2050. [Google Scholar] [CrossRef]
- Goüy de Bellocq, J.; Borremans, B.; Katakweba, A.; Makundi, R.; Baird, S.J.; Becker-Ziaja, B.; Günther, S.; Leirs, H. Sympatric Occurrence of 3 Arenaviruses, Tanzania. Emerg. Infect. Dis. 2010, 16, 692–695. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; Savji, N.; Hui, J.; Da Rosa, A.T.; Popov, V.; Briese, T.; Tesh, R.; Lipkin, W.I. Genomic and phylogenetic characterization of Merino Walk virus, a novel arenavirus isolated in South Africa. J. Gen. Virol. 2010, 91, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, P.T.; Kallies, R.; Hoveka, J.; Auste, B.; Ithete, N.L.; Šoltys, K.; Szemes, T.; Drosten, C.; Preiser, W.; Klempa, B.; et al. Novel Arenavirus Isolates from Namaqua Rock Mice, Namibia, Southern Africa. Emerg. Infect. Dis. 2015, 21, 1213–1216. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.P.; McCormick, J.B.; Saluzzo, J.F.; Herve, J.P.; Georges, A.J.; Johnson, K.M. An Arenavirus Isolated from Wild-Caught Rodents (Praomys Species) in the Central African Republic. Intervirology 1983, 19, 105–112. [Google Scholar] [CrossRef]
- N′Dilimabaka, N.; Berthet, N.; Rougeron, V.; Mangombi, J.B.; Durand, P.; Maganga, G.D.; Bouchier, C.; Schneider, B.S.; Fair, J.; Renaud, F.; et al. Evidence of Lymphocytic Choriomeningitis Virus (LCMV) in Domestic Mice in Gabon: Risk of Emergence of LCMV Encephalitis in Central Africa. J. Virol. 2014, 89, 1456–1460. [Google Scholar] [CrossRef]
- Krásová, J.; Mikula, O.; Bryja, J.; Baptista, N.L.; António, T.; Aghová, T.; Šumbera, R. Biogeography of Angolan rodents: The first glimpse based on phylogenetic evidence. Divers Distrib. 2021. under review. [Google Scholar]
- Linder, H.P.; de Klerk, H.M.; Born, J.; Burgess, N.D.; Fjeldså, J.; Rahbek, C. The partitioning of Africa: Statistically defined bio-geographical regions in sub-Saharan Africa. J. Biogeogr. 2012, 39, 1189–1205. [Google Scholar] [CrossRef]
- Colangelo, P.; Verheyen, E.; Leirs, H.; Tatard, C.; Denys, C.; Dobigny, G.; Duplantier, J.-M.; Brouat, C.; Granjon, L.; Lecompte, E. A mitochondrial phylogeographic scenario for the most widespread African rodent, Mastomys natalensis. Biol. J. Linn. Soc. 2013, 108, 901–916. [Google Scholar] [CrossRef]
- Ishii, A.; Thomas, Y.; Moonga, L.; Nakamura, I.; Ohnuma, A.; Hang’Ombe, B.M.; Takada, A.S.; Mweene, A.; Sawa, H. Novel Arenavirus, Zambia. Emerg. Infect. Dis. 2011, 17, 1921–1924. [Google Scholar] [CrossRef]
- Cuypers, L.N.; Baird, S.J.E.; Hánová, A.; Locus, T.; Katakweba, A.S.; Gryseels, S.; Bryja, J.; Leirs, H.; Goüy de Bellocq, J. Three arenaviruses in three subspecific natal multimammate mouse taxa in Tanzania: Same host specificity, but different spatial genetic structure? Virus Evol. 2020, 6. [Google Scholar] [CrossRef]
- Wulff, H.; McIntosh, B.M.; Hamner, D.B.; Johnson, K.M. Isolation of an arenavirus closely related to Lassa virus from Masto-mys natalensis in south-east Africa. Bull. World Health Organ. 1977, 55, 441–444. [Google Scholar] [PubMed]
- Johnson, K.M.; Taylor, P.; Elliott, L.H.; Tomori, O. Recovery of a Lassa-Related Arenavirus in Zimbabwe. Am. J. Trop. Med. Hyg. 1981, 30, 1291–1293. [Google Scholar] [CrossRef] [PubMed]
- Ishii, A.; Orba, Y.; Sasaki, M.; Kobayashi, S.; Moonga, L.; Hang’ombe, B.M.; Mweene, A.S.; Omori, R.; Ito, K.; Hall, W.W.; et al. ICTV Taxonomic Proposal 2016.019aM.A.v2.Mammarenavirus_sp. Create Species Solwezi Mamma-Renavirus in the Genus Mammarenavirus, Family Arenaviridae. 2016. Available online: http://www.ictv.global/proposals-16/2016.019aM.A.v2.Mammarenavirus_sp.pdf (accessed on 18 July 2016).
- Bryja, J.; Šumbera, R.; Peterhans, J.C.K.; Aghová, T.; Bryjová, A.; Mikula, O.; Nicolas, V.; Denys, C.; Verheyen, E. Evolutionary history of the thicket rats (genus Grammomys ) mirrors the evolution of African forests since late Miocene. J. Biogeogr. 2017, 44, 182–194. [Google Scholar] [CrossRef]
- Klempa, B.; Fichet-Calvet, E.; Lecompte, E.; Auste, B.; Aniskin, V.; Meisel, H.; Denys, C.; Koivogui, L.; Ter Meulen, J.; Krüger, D.H. Hantavirus in African Wood Mouse, Guinea. Emerg. Infect. Dis. 2006, 12, 838–840. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.P.; McCormick, J.B.; Baudon, D.; Gautun, J.P.; Meunier, D.Y.; Dournon, E.; Georges, A.J. Serological Evidence for Hantaan-related virus in Africa. Lancet 1984, 2, 1036–1037. [Google Scholar] [CrossRef]
- Sumibcay, L.; Kadjo, B.; Gu, S.H.; Kang, H.J.; Lim, B.K.; Cook, J.A.; Song, J.W.; Yanagihara, R. Divergent lineage of a novel hantavirus in the banana pipistrelle (Neoromicia nanus) in Côte d’Ivoire. Virol. J. 2012, 9, 34. [Google Scholar] [CrossRef]
- Weiss, S.; Witkowski, P.T.; Auste, B.; Nowak, K.; Weber, N.; Fahr, J.; Mombouli, J.-V.; Wolfe, N.D.; Drexler, J.F.; Drosten, C.; et al. Hantavirus in Bat, Sierra Leone. Emerg. Infect. Dis. 2012, 18, 159–161. [Google Scholar] [CrossRef]
- Witkowski, P.T.; Drexler, J.F.; Kallies, R.; Ličková, M.; Bokorová, S.; Mananga, G.D.; Szemes, T.; Leroy, E.M.; Krüger, D.H.; Drosten, C.; et al. Phylogenetic analysis of a newfound bat-borne hantavirus supports a laurasiatherian host association for ancestral mammalian hantaviruses. Infect. Genet. Evol. 2016, 41, 113–119. [Google Scholar] [CrossRef]
- Těšíková, J.; Bryjová, A.; Bryja, J.; Lavrenchenko, L.A.; Goüy de Bellocq, J. Hantavirus Strains in East Africa Related to Western African Hantaviruses. Vector-Borne Zoonotic Dis. 2017, 17, 278–280. [Google Scholar] [CrossRef]
- Klempa, B.; Fichet-Calvet, E.; Lecompte, E.; Auste, B.; Aniskin, V.; Meisel, H.; Barrière, P.; Koivogui, L.; Ter Meulen, J.; Krüger, D.H. Novel Hantavirus Sequences in Shrew, Guinea. Emerg. Infect. Dis. 2007, 13, 520–522. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Kadjo, B.; Dubey, S.; Jacquet, F.; Yanagihara, R. Molecular evolution of Azagny virus, a newfound hantavirus harbored by the West African pygmy shrew (Crocidura obscurior) in Côte d’Ivoire. Virol. J. 2011, 8, 373. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.H.; Nicolas, V.; Lalis, A.; Sathirapongsasuti, N.; Yanagihara, R. Complete genome sequence and molecular phylogeny of a newfound hantavirus harbored by the Doucet’s musk shrew (Crocidura douceti) in Guinea. Infect. Genet. Evol. 2013, 20, 118–123. [Google Scholar] [CrossRef]
- Kang, H.J.; Stanley, W.T.; Esselstyn, J.A.; Gu, S.H.; Yanagihara, R. Expanded Host Diversity and Geographic Distribution of Hantaviruses in Sub-Saharan Africa. J. Virol. 2014, 88, 7663–7667. [Google Scholar] [CrossRef] [PubMed]
- Meheretu, Y.; Stanley, W.T.; Craig, E.W.; Goüy de Bellocq, J.; Bryja, J.; Leirs, H.; Pahlmann, M.; Günther, S. Tigray Orthohantavirus Infects Two Related Rodent Species Adapted to Different Elevations in Ethiopia. Vector-Borne Zoonotic Dis. 2019, 19, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Diagne, M.M.; Dieng, I.; Granjon, L.; Lucaccioni, H.; Sow, A.; Ndiaye, O.; Faye, M.; Bâ, K.; Bâ, Y.; Diallo, M.; et al. Seoul Orthohantavirus in Wild Black Rats, Senegal, 2012–2013. Emerg. Infect. Dis. 2020, 26, 2460–2464. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.M.; Kocher, T.D.; Wilson, A.C. Evolution of the cytochromeb gene of mammals. J. Mol. Evol. 1991, 32, 128–144. [Google Scholar] [CrossRef]
- Krásová, J.; Mikula, O.; Mazoch, V.; Bryja, J.; Říčan, O.; Šumbera, R. Evolution of the Grey-bellied pygmy mouse group: Highly structured molecular diversity with predictable geographic ranges but morphological crypsis. Mol. Phylogenetics Evol. 2019, 130, 143–155. [Google Scholar] [CrossRef]
- Vieth, S.; Drosten, C.; Lenz, O.; Vincent, M.; Omilabu, S.; Hass, M.; Becker-Ziaja, B.; Ter Meulen, J.; Nichol, S.T.; Schmitz, H.; et al. RT-PCR assay for detection of Lassa virus and related Old World arenaviruses targeting the L gene. Trans. R. Soc. Trop. Med. Hyg. 2007, 101, 1253–1264. [Google Scholar] [CrossRef]
- Ehichioya, D.U.; Hass, M.; Becker-Ziaja, B.; Ehimuan, J.; Asogun, D.A.; Fichet-Calvet, E.; Kleinsteuber, K.; Lelke, M.; Ter Meulen, J.; Akpede, G.O.; et al. Current Molecular Epidemiology of Lassa Virus in Nigeria. J. Clin. Microbiol. 2010, 49, 1157–1161. [Google Scholar] [CrossRef]
- Rózsa, L.; Reiczigel, J.; Majoros, G. Quantifying parasites in samples of hosts. J. Parasitol. 2000, 86, 228–232. [Google Scholar] [CrossRef]
- Reiczigel, J. Confidence intervals for the binomial parameter: Some new considerations. Stat. Med. 2003, 22, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Goüy de Bellocq, J.; Bryjová, A.; Martynov, A.A.; Lavrenchenko, L.A. Dhati Welel virus, the missing mammarenavirus of the widespread Mastomys natalensis. J. Vertebr. Biol. 2020, 69, 1–11. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. In Babraham Bioinformatics; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Jiang, H.; Lei, R.; Ding, S.-W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, D.R.; Blaxter, M.L. BlobTools: Interrogation of genome assemblies [version 1; peer review: 2 approved with reservations]. F1000Research 2017, 6, 1287. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and high-performance computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Bao, Y.; Chetvernin, V.; Tatusova, T. Improvements to pairwise sequence comparison (PASC): A genome-based web tool for virus classification. Arch. Virol. 2014, 159, 3293–3304. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Morin, B.; Coutard, B.; Lelke, M.; Ferron, F.; Kerber, R.; Jamal, S.; Frangeul, A.; Baronti, C.; Charrel, R.; De Lamballerie, X.; et al. The N-Terminal Domain of the Arenavirus L Protein Is an RNA Endonuclease Essential in mRNA Transcription. PLoS Pathog. 2010, 6, e1001038. [Google Scholar] [CrossRef]
- Whitton, J.L.; Tishon, A.; Lewicki, H.; Gebhard, J.; Cook, T.; Salvato, M.; Joly, E.; Oldstone, M.B. Molecular analyses of a five-amino-acid cytotoxic T-lymphocyte (CTL) epitope: An immunodominant region which induces nonreciprocal CTL cross-reactivity. J. Virol. 1989, 63, 4303–4310. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.P.; Sanchez, A.; Rico-Hesse, R. Molecular phylogeny of Guanarito virus, an emerging arenavirus affecting hu-mans. Am. J. Trop. Med. Hyg. 1995, 53, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, C.J.; Capul, A.A.; Lauron, E.J.; Bederka, L.H.; Knopp, K.A.; Buchmeier, M.J. Glycosylation modulates arenavirus glycoprotein expression and function. Virology 2011, 409, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Gryseels, S.; Rieger, T.; Oestereich, L.; Cuypers, B.; Borremans, B.; Makundi, R.; Leirs, H.; Günther, S.; Goüy de Bellocq, J. Gairo virus, a novel arenavirus of the widespread Mastomys natalensis: Genetically divergent, but ecologically similar to Lassa and Morogoro viruses. Virology 2015, 476, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, V.; Fabre, P.-H.; Bryja, J.; Denys, C.; Verheyen, E.; Missoup, A.D.; Olayemi, A.; Katuala, P.; Dudu, A.; Colyn, M.; et al. The phylogeny of the African wood mice (Muridae, Hylomyscus) based on complete mitochondrial genomes and five nuclear genes reveals their evolutionary history and undescribed diversity. Mol. Phylogenetics Evol. 2020, 144, 106703. [Google Scholar] [CrossRef]
- Russo, I.-R.M.; Chimimba, C.T.; Bloomer, P. Bioregion heterogeneity correlates with extensive mitochondrial DNA diversity in the Namaqua rock mouse, Micaelamys namaquensis (Rodentia: Muridae) from southern Africa—Evidence for a species complex. BMC Evol. Biol. 2010, 10, 307. [Google Scholar] [CrossRef]
- Ishii, A.; Thomas, Y.; Moonga, L.; Nakamura, I.; Ohnuma, A.; Hang’Ombe, B.M.; Takada, A.; Mweene, A.S.; Sawa, H. Molecular surveillance and phylogenetic analysis of Old World arenaviruses in Zambia. J. Gen. Virol. 2012, 93, 2247–2251. [Google Scholar] [CrossRef]
- Lecompte, E.; Ter Meulen, J.; Emonet, S.; Daffis, S.; Charrel, R.N. Genetic identification of Kodoko virus, a novel arenavirus of the African pigmy mouse (Mus Nannomys minutoides) in West Africa. Virology 2007, 364, 178–183. [Google Scholar] [CrossRef]
- Coulibaly-N’Golo, D.; Allali, B.; Kouassi, S.K.; Fichet-Calvet, E.; Becker-Ziaja, B.; Rieger, T.; Ölschläger, S.; Dosso, H.; Denys, C.; Ter Meulen, J.; et al. Novel Arenavirus Sequences in Hylomyscus sp. and Mus (Nannomys) setulosus from Côte d’Ivoire: Implications for Evolution of Arenaviruses in Africa. PLoS ONE 2011, 6, e20893. [Google Scholar] [CrossRef]
- Moreno, H.; Rastrojo, A.; Pryce, R.; Fedeli, C.; Zimmer, G.; Bowden, T.A.; Gerold, G.; Kunz, S. A novel circulating tamiami mammarenavirus shows potential for zoonotic spillover. PLoS Negl. Trop. Dis. 2020, 14, e0009004. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Virus | Bitu (BITV) | Bitu (BITV) | Luna (LUAV) | Kwanza (KWAV) |
|---|---|---|---|---|
| Sample | ANG0052 | ANG0070 | ANG0117 | ANG0206 |
| Locality | Tundavala | Tundavala | Bicuar NP | 20 km SW of Cassongue |
| Host species | Micaelamys namaquensis | Micaelamys namaquensis | Mastomys natalensis | Mus triton |
| GenBank # Host Cyt b | MZ065491 | MW544581 | MZ065496 | MK011523 |
| GenBank # L segment | MZ065536 | MZ065538 | MZ065542 | MZ065540 |
| GenBank # S segment | MZ065537 | MZ065539 | MZ065543 | MZ065541 |
| Coverage L segment | 46 ± 20 | 98 ± 41 | 319 ± 113 | 1674 ± 1339 |
| Coverage S segment | 51 ± 24 | 63 ± 30 | 761 ± 263 | 1660 ± 1240 |
| Length L segment (nt) | 7187 | 7223 | 7296 | 7222 |
| Length L ORF (aa) | 2225 | 2225 | 2219 | 2220 |
| Length Z ORF (aa) | 93 | 93 | 98 | 99 |
| Length S segment (nt) | 3374 | 3373 | 3476 | 3421 |
| Length GPC ORF (aa) | 501 | 501 | 491 | 489 |
| Length NP ORF (aa) | 562 | 562 | 569 | 569 |
| Late domains in Z | PTCP-none | PTCP-none | PTAP-PPPY | PTAP-PPAY |
| CTL epitope in NP * | GVYMGNL | GVYMGNL | GVYMGNL | GIYMGNL |
| Antigenic site in NP ** | RKDKRDD | RKDKRDD | RKEKRDD | RKDKRDD |
| GP1/GP2 cleavage site | RRLR | RRLR | RRLM | RRLL |
| N-glycosylation GP1 | 6 | 6 | 4 | 5 |
| N-glycosylation GP2 | 3 | 4 | 3 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Těšíková, J.; Krásová, J.; Goüy de Bellocq, J. Multiple Mammarenaviruses Circulating in Angolan Rodents. Viruses 2021, 13, 982. https://doi.org/10.3390/v13060982
Těšíková J, Krásová J, Goüy de Bellocq J. Multiple Mammarenaviruses Circulating in Angolan Rodents. Viruses. 2021; 13(6):982. https://doi.org/10.3390/v13060982
Chicago/Turabian StyleTěšíková, Jana, Jarmila Krásová, and Joëlle Goüy de Bellocq. 2021. "Multiple Mammarenaviruses Circulating in Angolan Rodents" Viruses 13, no. 6: 982. https://doi.org/10.3390/v13060982
APA StyleTěšíková, J., Krásová, J., & Goüy de Bellocq, J. (2021). Multiple Mammarenaviruses Circulating in Angolan Rodents. Viruses, 13(6), 982. https://doi.org/10.3390/v13060982