
Molecular Characterization of Seasonal Influenza A and B from Hospitalized Patients in Thailand in 2018–2019

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Considerations
2.2. Specimen Collection and Influenza Virus Detection
2.3. Virus Isolation
2.4. Hemagglutination (HA) Assay
2.5. Viral RNA Extraction, Influenza Subtype Determination, and Genome Sequencing
2.6. Phylogenetic Analysis
2.7. Sensitivity to Anti-Neuraminidase Drugs
2.8. Homology Modelling
3. Results
3.1. Patient Demographic Data
3.2. Genome Characterization of Influenza Virus Isolates
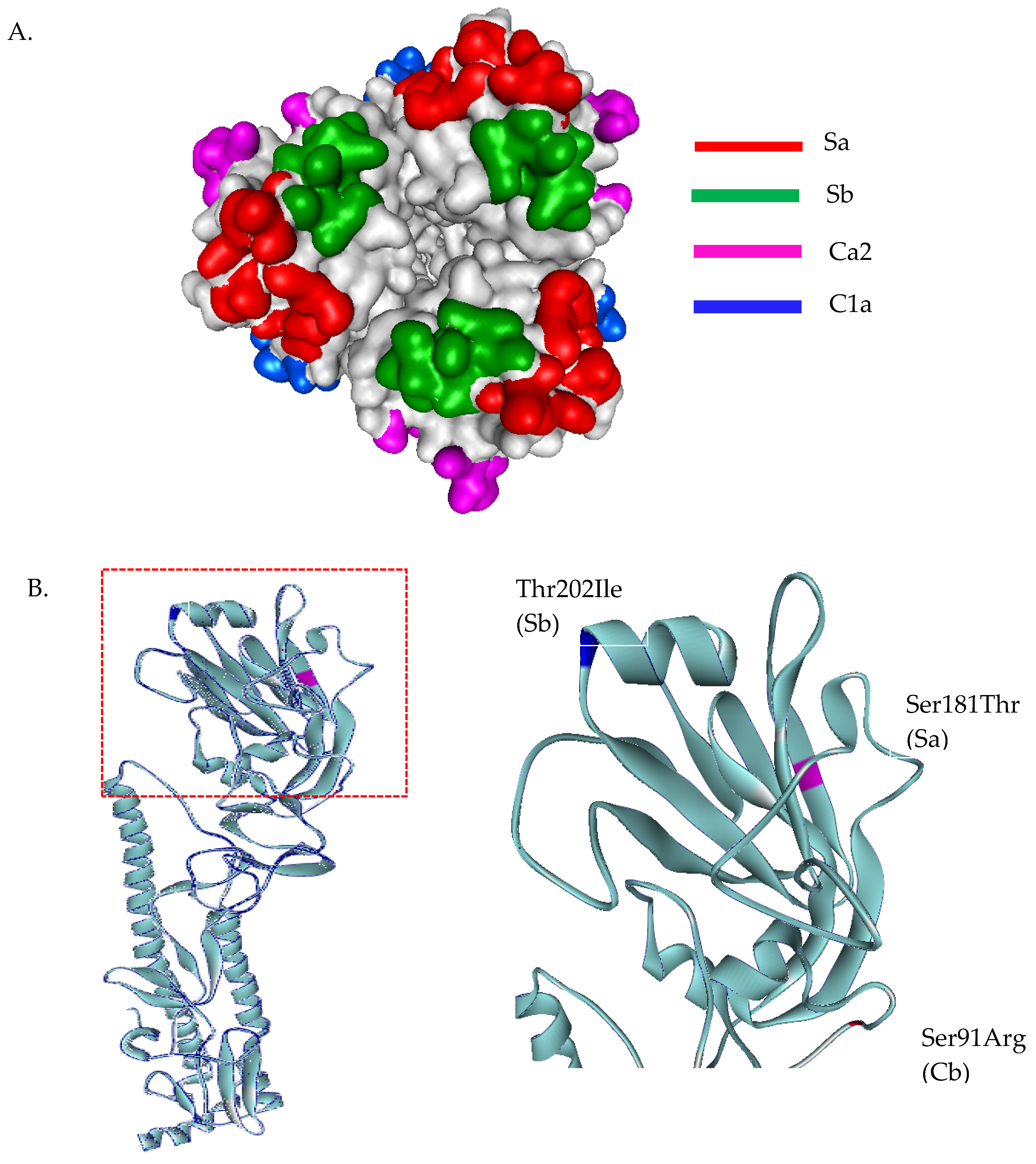
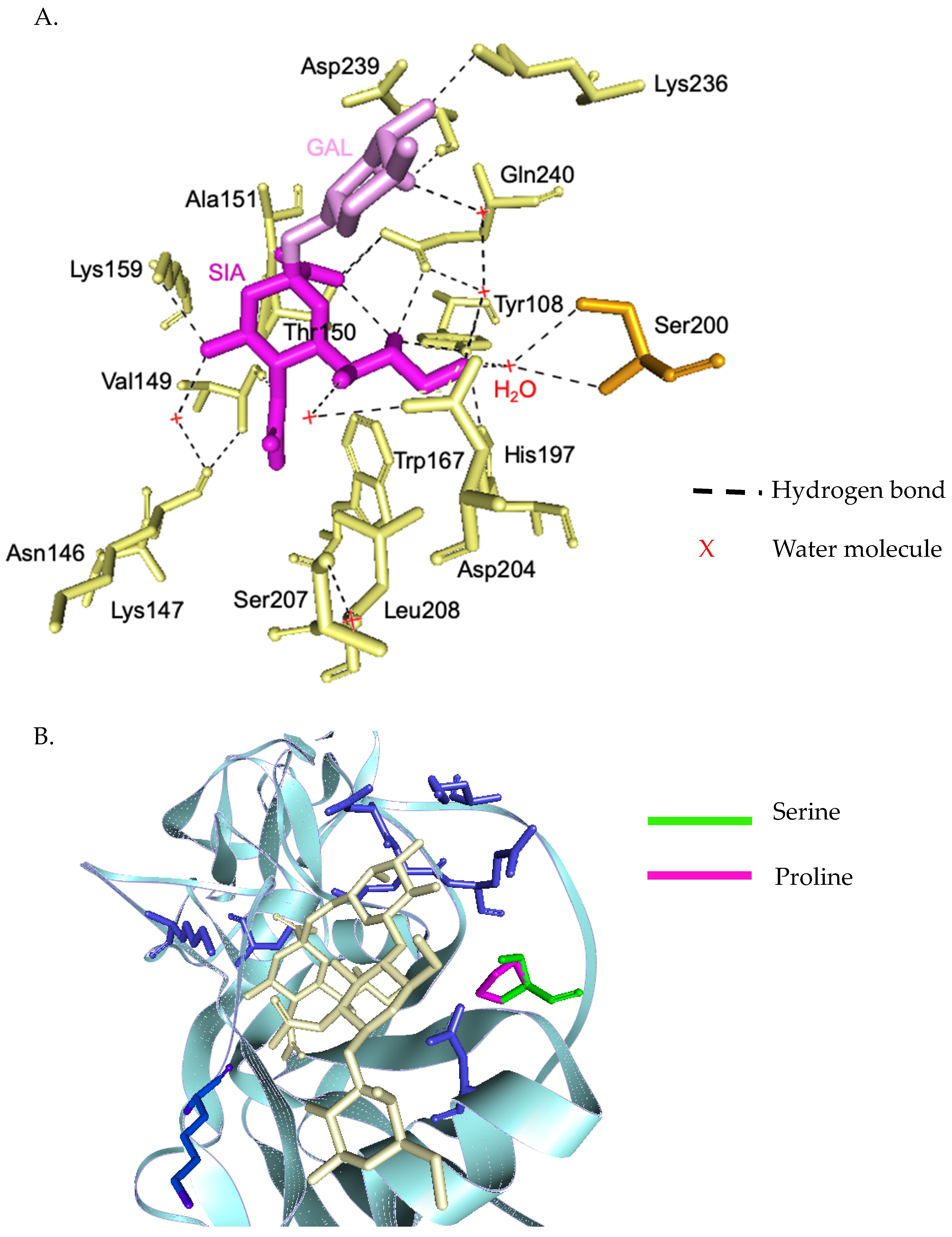
3.3. Evolutionary Characteristic and Structure Variations in the HA Domain of H1N1 Viruses
3.4. Evolutionary Characteristic and Structure Variations in the HA Domain of H3N2 Viruses
3.5. Evolutionary Characteristic and Structure Variations in the HA Domain of Influenza B Viruses
3.6. Analysis of Sensitivity to Anti-Neuraminidase Drugs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hannoun, C. The evolving history of influenza viruses and influenza vaccines. Expert Rev. Vaccines 2013, 12, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Brankston, G.; Gitterman, L.; Hirji, Z.; Lemieux, C.; Gardam, M. Transmission of influenza A in human beings. Lancet Infect. Dis. 2007, 7, 257–265. [Google Scholar] [CrossRef]
- Boni, M.F. Vaccination and antigenic drift in influenza. Vaccine 2008, 26 (Suppl. 3), C8–C14. [Google Scholar] [CrossRef]
- Doud, M.B.; Hensley, S.E.; Bloom, J.D. Complete mapping of viral escape from neutralizing antibodies. PLoS Pathog. 2017, 13, e1006271. [Google Scholar] [CrossRef]
- Forrest, H.L.; Webster, R.G. Perspectives on influenza evolution and the role of research. Anim Health Res. Rev. 2010, 11, 3–18. [Google Scholar] [CrossRef]
- Horimoto, T.; Kawaoka, Y. Influenza: Lessons from past pandemics, warnings from current incidents. Nat. Rev. Microbiol. 2005, 3, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Kilbourne, E.D. Influenza pandemics of the 20th century. Emerg Infect. Dis. 2006, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Oxford, J.S. Influenza A pandemics of the 20th century with special reference to 1918: Virology, pathology and epidemiology. Rev. Med. Virol. 2000, 10, 119–133. [Google Scholar] [CrossRef]
- Buckland, B.C. The development and manufacture of influenza vaccines. Hum. Vaccin. Immunother. 2015, 11, 1357–1360. [Google Scholar] [CrossRef]
- Lambert, L.C.; Fauci, A.S. Influenza vaccines for the future. N. Engl. J. Med. 2010, 363, 2036–2044. [Google Scholar] [CrossRef] [PubMed]
- Louisirirotchanakul, S.; Lerdsamran, H.; Wiriyarat, W.; Sangsiriwut, K.; Chaichoune, K.; Pooruk, P.; Songserm, T.; Kitphati, R.; Sawanpanyalert, P.; Komoltri, C.; et al. Erythrocyte binding preference of avian influenza H5N1 viruses. J. Clin. Microbiol. 2007, 45, 2284–2286. [Google Scholar] [CrossRef][Green Version]
- WHO. WHO Information for the Molecular Detection of Influenza Viruses. Available online: https://www.who.int/influenza/gisrs_laboratory/WHO_information_for_the_molecular_detection_of_influenza_viruses_20171023_Final.pdf?ua=1 (accessed on 24 August 2020).
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Lackenby, A.; Democratis, J.; Siqueira, M.M.; Zambon, M.C. Rapid quantitation of neuraminidase inhibitor drug resistance in influenza virus quasispecies. Antivir. Ther. 2008, 13, 809–820. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Moss, D.S.; Thornton, J.M. Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 1993, 231, 1049–1067. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Leang, S.K.; Hurt, A.C. Fluorescence-based Neuraminidase Inhibition Assay to Assess the Susceptibility of Influenza Viruses to The Neuraminidase Inhibitor Class of Antivirals. J. Vis. Exp. 2017, 2017, 55570. [Google Scholar] [CrossRef] [PubMed]
- Wetherall, N.T.; Trivedi, T.; Zeller, J.; Hodges-Savola, C.; McKimm-Breschkin, J.L.; Zambon, M.; Hayden, F.G. Evaluation of neuraminidase enzyme assays using different substrates to measure susceptibility of influenza virus clinical isolates to neuraminidase inhibitors: Report of the neuraminidase inhibitor susceptibility network. J. Clin. Microbiol. 2003, 41, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Price, O.H.; Spirason, N.; Rynehart, C.; Brown, S.K.; Todd, A.; Peck, H.; Patel, M.; Soppe, S.; Barr, I.G.; Chow, M.K. Report on influenza viruses received and tested by the Melbourne WHO Collaborating Centre for Reference and Research on Influenza in 2018. Commun. Dis. Intell. 2020, 44. [Google Scholar] [CrossRef] [PubMed]
- WHO. Recommended Composition of Influenza Virus Vaccines for Use in the 2020 Southern Hemisphere Influenza Season. Available online: https://www.who.int/influenza/vaccines/virus/recommendations/2020_south/en/ (accessed on 30 August 2020).
- Caton, A.J.; Brownlee, G.G.; Yewdell, J.W.; Gerhard, W. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 1982, 31, 417–427. [Google Scholar] [CrossRef]
- Das, S.R.; Hensley, S.E.; Ince, W.L.; Brooke, C.B.; Subba, A.; Delboy, M.G.; Russ, G.; Gibbs, J.S.; Bennink, J.R.; Yewdell, J.W. Defining influenza A virus hemagglutinin antigenic drift by sequential monoclonal antibody selection. Cell Host Microbe 2013, 13, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, Y.; Sugawara, K.; Nakauchi, M.; Takahashi, Y.; Onodera, T.; Tsunetsugu-Yokota, Y.; Matsumura, T.; Ato, M.; Kobayashi, K.; Shimotai, Y.; et al. Epitope mapping of the hemagglutinin molecule of A/(H1N1)pdm09 influenza virus by using monoclonal antibody escape mutants. J. Virol. 2014, 88, 12364–12373. [Google Scholar] [CrossRef]
- Jones, S.; Nelson-Sathi, S.; Wang, Y.; Prasad, R.; Rayen, S.; Nandel, V.; Hu, Y.; Zhang, W.; Nair, R.; Dharmaseelan, S.; et al. Evolutionary, genetic, structural characterization and its functional implications for the influenza A (H1N1) infection outbreak in India from 2009 to 2017. Sci. Rep. 2019, 9, 14690. [Google Scholar] [CrossRef] [PubMed]
- De Vries, R.P.; de Vries, E.; Martinez-Romero, C.; McBride, R.; van Kuppeveld, F.J.; Rottier, P.J.; Garcia-Sastre, A.; Paulson, J.C.; de Haan, C.A. Evolution of the hemagglutinin protein of the new pandemic H1N1 influenza virus: Maintaining optimal receptor binding by compensatory substitutions. J. Virol. 2013, 87, 13868–13877. [Google Scholar] [CrossRef]
- Virk, R.K.; Jayakumar, J.; Mendenhall, I.H.; Moorthy, M.; Lam, P.; Linster, M.; Lim, J.; Lin, C.; Oon, L.L.E.; Lee, H.K.; et al. Divergent evolutionary trajectories of influenza B viruses underlie their contemporaneous epidemic activity. Proc. Natl. Acad. Sci. USA 2020, 117, 619–628. [Google Scholar] [CrossRef]
- Hsu, K.C.; Hung, H.C.; HuangFu, W.C.; Sung, T.Y.; Eight Lin, T.; Fang, M.Y.; Chen, I.J.; Pathak, N.; Hsu, J.T.; Yang, J.M. Identification of neuraminidase inhibitors against dual H274Y/I222R mutant strains. Sci. Rep. 2017, 7, 12336. [Google Scholar] [CrossRef]
- Kim, H.M.; Lee, N.; Kim, M.S.; Kang, C.; Chung, Y.S. Characterization of neuraminidase inhibitor-resistant influenza virus isolates from immunocompromised patients in the Republic of Korea. Virol. J. 2020, 17, 94. [Google Scholar] [CrossRef]
- Eshaghi, A.; Shalhoub, S.; Rosenfeld, P.; Li, A.; Higgins, R.R.; Stogios, P.J.; Savchenko, A.; Bastien, N.; Li, Y.; Rotstein, C.; et al. Multiple influenza A (H3N2) mutations conferring resistance to neuraminidase inhibitors in a bone marrow transplant recipient. Antimicrob. Agents Chemother. 2014, 58, 7188–7197. [Google Scholar] [CrossRef]
- Takashita, E.; Daniels, R.S.; Fujisaki, S.; Gregory, V.; Gubareva, L.V.; Huang, W.; Hurt, A.C.; Lackenby, A.; Nguyen, H.T.; Pereyaslov, D.; et al. Global update on the susceptibilities of human influenza viruses to neuraminidase inhibitors and the cap-dependent endonuclease inhibitor baloxavir, 2017–2018. Antivir. Res. 2020, 175, 104718. [Google Scholar] [CrossRef] [PubMed]
- Bastien, N.; Gubbay, J.B.; Richardson, D.; Sleeman, K.; Gubareva, L.; Li, Y. Detection of an influenza B virus strain with reduced susceptibility to neuraminidase inhibitor drugs. J. Clin. Microbiol. 2011, 49, 4020–4021. [Google Scholar] [CrossRef]
- Burnham, A.J.; Baranovich, T.; Govorkova, E.A. Neuraminidase inhibitors for influenza B virus infection: Efficacy and resistance. Antiviral Res. 2013, 100, 520–534. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S.; Sugaya, N.; Ito, M.; Yamazaki, M.; Ichikawa, M.; Kimura, K.; Kiso, M.; Shimizu, H.; Kawakami, C.; Koike, K.; et al. Emergence of influenza B viruses with reduced sensitivity to neuraminidase inhibitors. JAMA 2007, 297, 1435–1442. [Google Scholar] [CrossRef]
- Lampejo, T. Influenza and antiviral resistance: An overview. Eur J. Clin. Microbiol. Infect. Dis. 2020, 39, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Moscona, A. Global transmission of oseltamivir-resistant influenza. N. Engl. J. Med. 2009, 360, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Paules, C.; Subbarao, K. Influenza. Lancet 2017, 390, 697–708. [Google Scholar] [CrossRef]
- Saunders-Hastings, P.R.; Krewski, D. Reviewing the History of Pandemic Influenza: Understanding Patterns of Emergence and Transmission. Pathogens 2016, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Awada, L.; Brown, I.; Chen, H.; Claes, F.; Dauphin, G.; Donis, R.; Culhane, M.; Hamilton, K.; Lewis, N.; et al. Review of influenza A virus in swine worldwide: A call for increased surveillance and research. Zoonoses Public Health 2014, 61, 4–17. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Number of Cases (n) |
|---|---|
| Gender | |
| Male | 7 |
| Female | 23 |
| Age groups | |
| 15–35 | 7 |
| 36–45 | 4 |
| 46–59 | 6 |
| ≥60 | 13 |
| Co- morbidities | |
| Cardiovascular diseases | 2 |
| Respiratory diseases | 3 |
| Chronic renal diseases | 2 |
| Diabetes mellitus | 4 |
| Hypertension | 8 |
| Cancer and hematological diseases | 2 |
| Stroke and neuromuscular diseases | 3 |
| Days in ICU | |
| 1–5 days | 0 |
| >5 days | 3 |
| Days in medical ventilation | |
| 1–5 days | 0 |
| >5 days | 3 |
| Outcome | |
| Death | 0 |
| Discharged | 30 |
| Viruses | Subtypes | Passage No. of Isolate | Mean IC50 ± SD (nM) |
|---|---|---|---|
| A/Thailand/TM-18334_61/2018 | H1N1 | 1,2,3 | 0.59 ± 0.07 |
| A/Thailand/TM-3627_55/2018 | H1N1 | 5,6,7 | 0.29 ± 0.08 |
| A/Thailand/TM-12108_51/2019 | H1N1 | 7,8,9 | 0.23 ± 0.05 |
| A/Thailand/TM-6375_40/2019 | H1N1 | 3,4,5 | 0.48 ± 0.07 |
| A/Thailand/TM-4042_61/2019 | H1N1 | 3,4,5 | 0.16 ± 0.01 |
| A/Thailand/TM-3418_56/2019 | H1N1 | 2,3,4 | 0.35 ± 0.04 |
| A/Thailand/TM-13108_62/2018 | H1N1 | 1,2,3 | 0.57 ± 0.01 |
| B/Thailand/TM-6610_57/2019 | B/Vic | 3,4,5 | 33.96 ± 4.12 |
| B/Thailand/TM-3038_62/2019 | B/Vic | 1,2,3 | 40.10 ± 0.91 |
| B/Thailand/TM-12875_57/2019 | B/Vic | 2,3,4 | 23.38 ± 2.76 |
| B/Thailand/TM-2580_62/2019 | B/Vic | 3,4,5 | 17.18 ± 3.38 |
| B/Thailand/TM-2953_55/2019 | B/Vic | 1,2,3 | 36.84 ± 5.28 |
| B/Thailand/TM-10944_62/2019 | B/Vic | 2,3,6 | 22.62 ± 1.15 |
| A/Thailand/TM-8453_54/2018 | H3N2 | 2,3,4 | 0.41 ± 0.04 |
| A/Thailand/TM-17617_61/2018 | H3N2 | 6,7,8 | 0.14 ± 0.02 |
| A/Thailand/TM-12054_56/2019 | H3N2 | 8,9,10 | 0.21 ± 0.04 |
| A/Thailand/TM-12909_43/2019 | H3N2 | 4,5,6 | 0.11 ± 0.02 |
| A/Thailand/TM-358_60/2018 | H3N2 | 7,8,9 | 0.05 ± 0.01 |
| A/Thailand/TM-54_54/2018 | H3N2 | 6,7,8 | 0.11 ± 0.01 |
| A/Thailand/TM-5434_51/2019 | H3N2 | 5,6,7 | 0.06 ± 0.01 |
| A/Mississippi/03/01 wt (274H) | H1N1 | 6 | 0.67 ± 0.03 |
| A/Mississippi/03/01mutant 274Y | H1N1 | 6 | 286.01 ± 14.02 |
| A/Fukui/45/04 wt (119E) | H3N2 | 5 | 0.23 ± 0.02 |
| A/Fukui/45/04 mutant (119V) | H3N2 | 5 | 30.66 ± 0.88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boonnak, K.; Mansanguan, C.; Schuerch, D.; Boonyuen, U.; Lerdsamran, H.; Jiamsomboon, K.; Sae Wang, F.; Huntrup, A.; Prasertsopon, J.; Kosoltanapiwat, N.; et al. Molecular Characterization of Seasonal Influenza A and B from Hospitalized Patients in Thailand in 2018–2019. Viruses 2021, 13, 977. https://doi.org/10.3390/v13060977
Boonnak K, Mansanguan C, Schuerch D, Boonyuen U, Lerdsamran H, Jiamsomboon K, Sae Wang F, Huntrup A, Prasertsopon J, Kosoltanapiwat N, et al. Molecular Characterization of Seasonal Influenza A and B from Hospitalized Patients in Thailand in 2018–2019. Viruses. 2021; 13(6):977. https://doi.org/10.3390/v13060977
Chicago/Turabian StyleBoonnak, Kobporn, Chayasin Mansanguan, Dennis Schuerch, Usa Boonyuen, Hatairat Lerdsamran, Kultida Jiamsomboon, Fanny Sae Wang, Arun Huntrup, Jarunee Prasertsopon, Nathamon Kosoltanapiwat, and et al. 2021. "Molecular Characterization of Seasonal Influenza A and B from Hospitalized Patients in Thailand in 2018–2019" Viruses 13, no. 6: 977. https://doi.org/10.3390/v13060977
APA StyleBoonnak, K., Mansanguan, C., Schuerch, D., Boonyuen, U., Lerdsamran, H., Jiamsomboon, K., Sae Wang, F., Huntrup, A., Prasertsopon, J., Kosoltanapiwat, N., & Puthavathana, P. (2021). Molecular Characterization of Seasonal Influenza A and B from Hospitalized Patients in Thailand in 2018–2019. Viruses, 13(6), 977. https://doi.org/10.3390/v13060977