
Human Retrotransposons and the Global Shutdown of Homeostatic Innate Immunity by Oncolytic Parvovirus H-1PV in Pancreatic Cancer

 , , , ,
, , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. H-1PV Production and Infection
2.3. Transfection-Based Experiments
2.4. Western Blot Analysis
2.5. ELISA
2.6. Real-Time qRT-PCR
2.7. RNA Sequencing
2.8. Statistical Analyses
3. Results
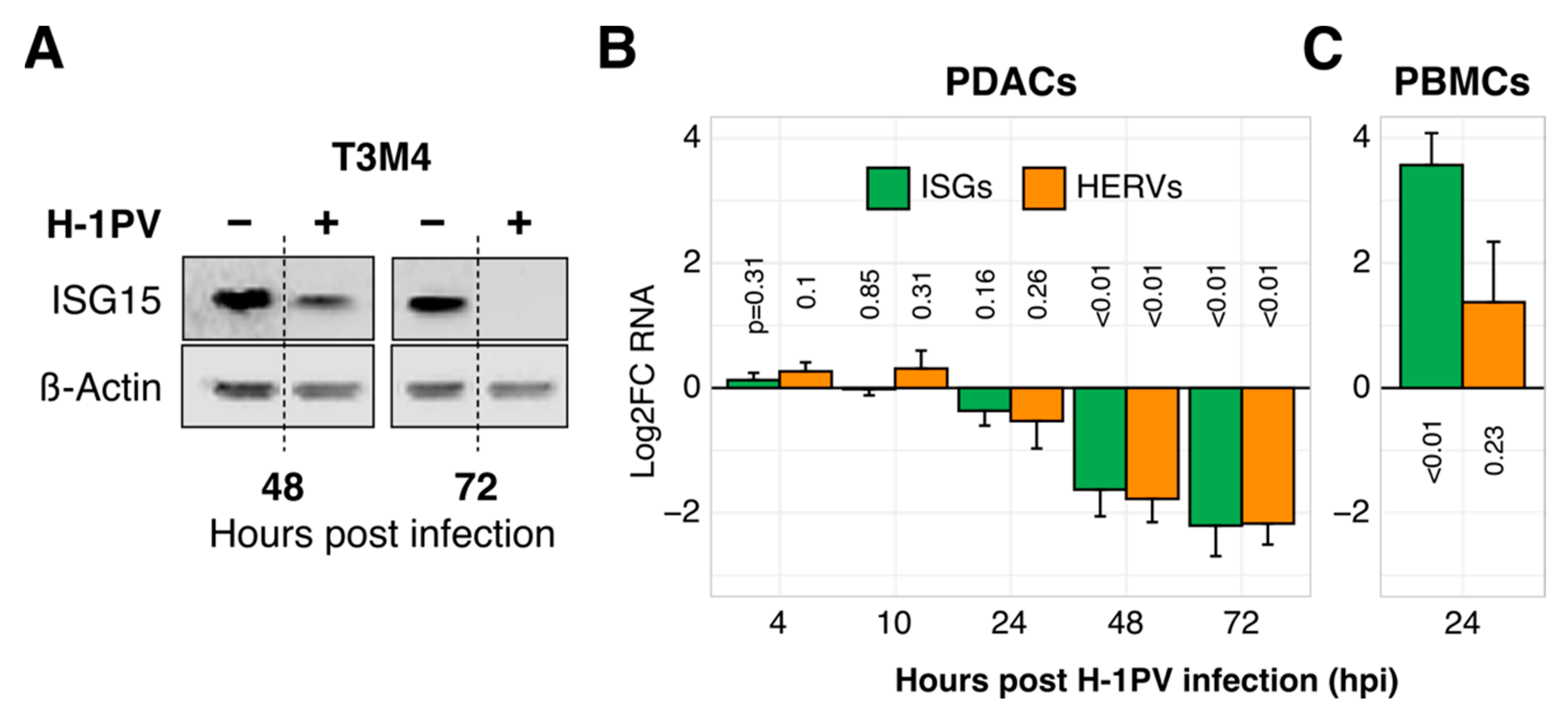
3.1. Concomitant Suppression of ISGs and HERVs in H-1PV-Infected PDAC Cells
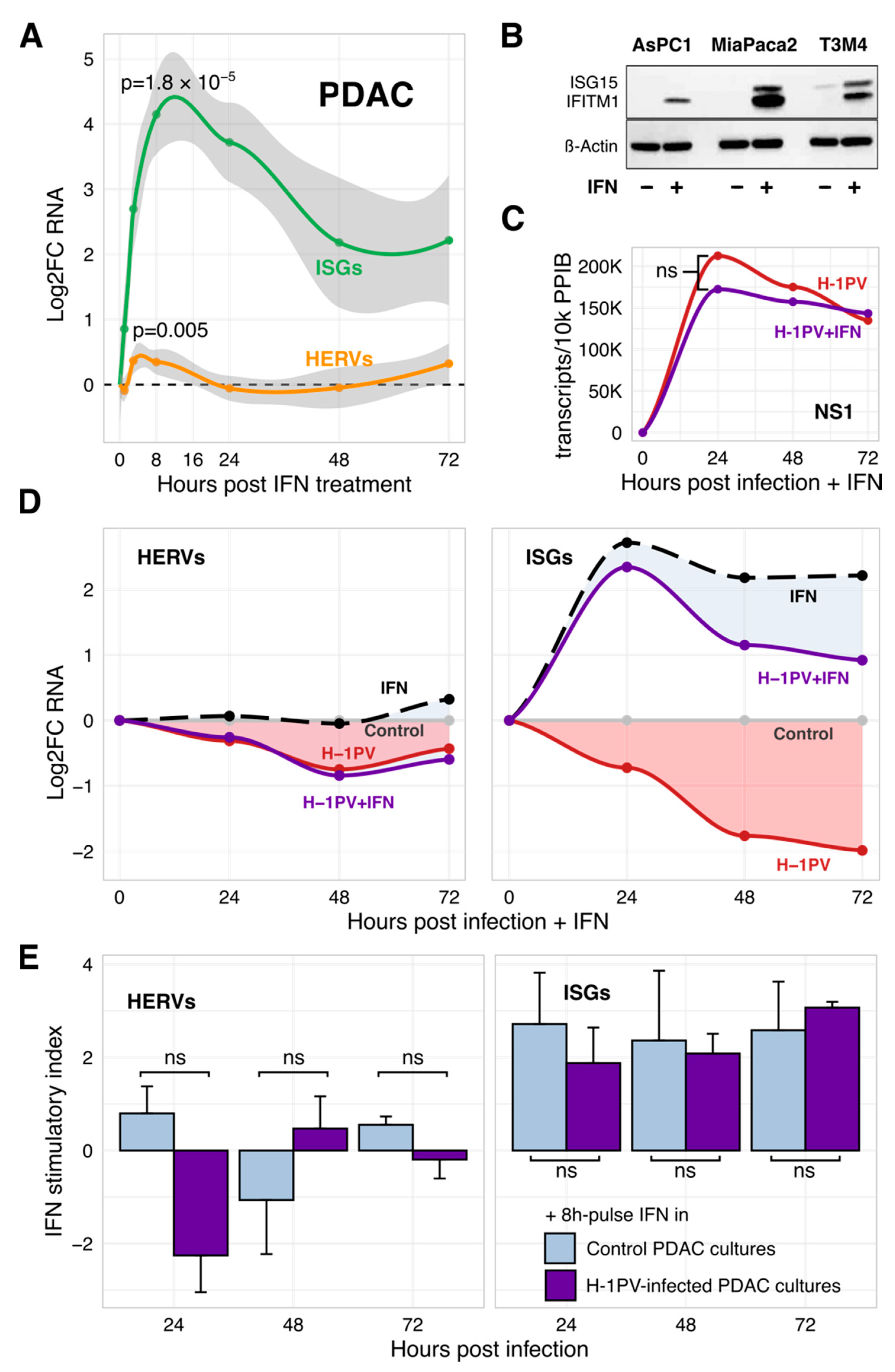
3.2. Suppressed Type I and III IFNs Are Co-Targets Rather Than Regulators of ISGs
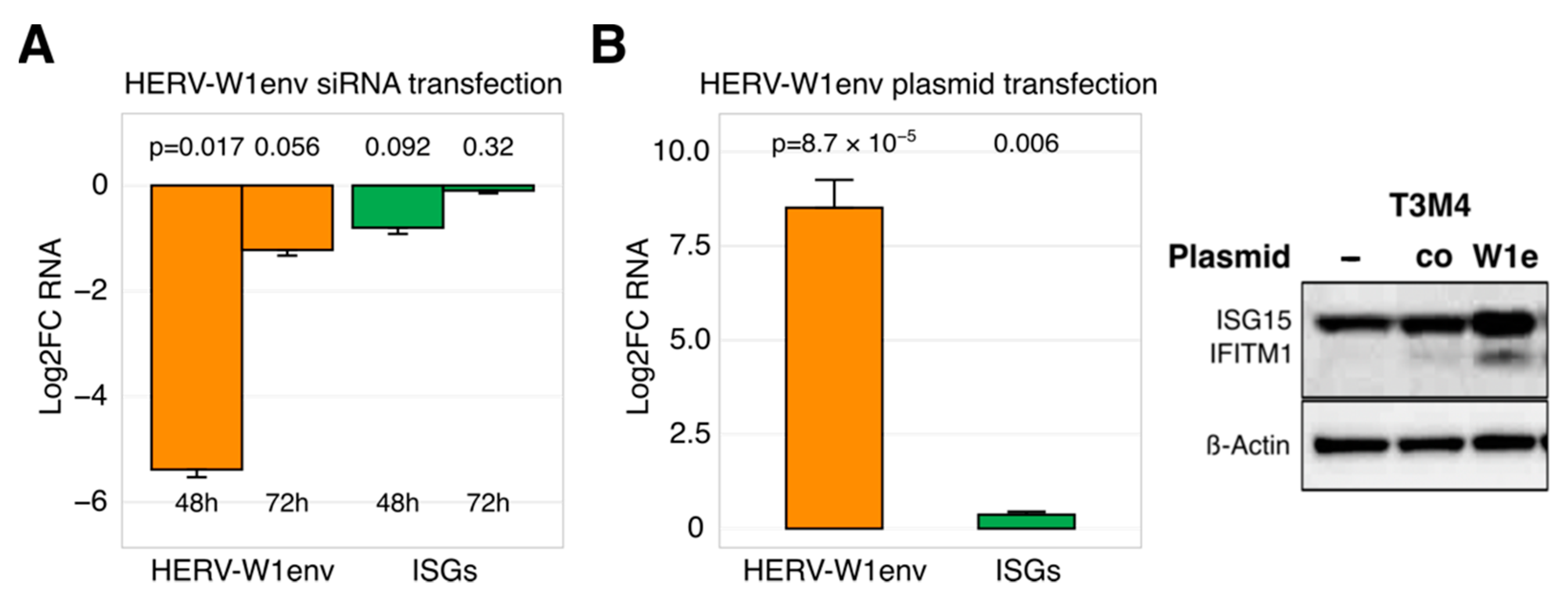
3.3. Restricted Ability of HERVs to Regulate ISG Expression
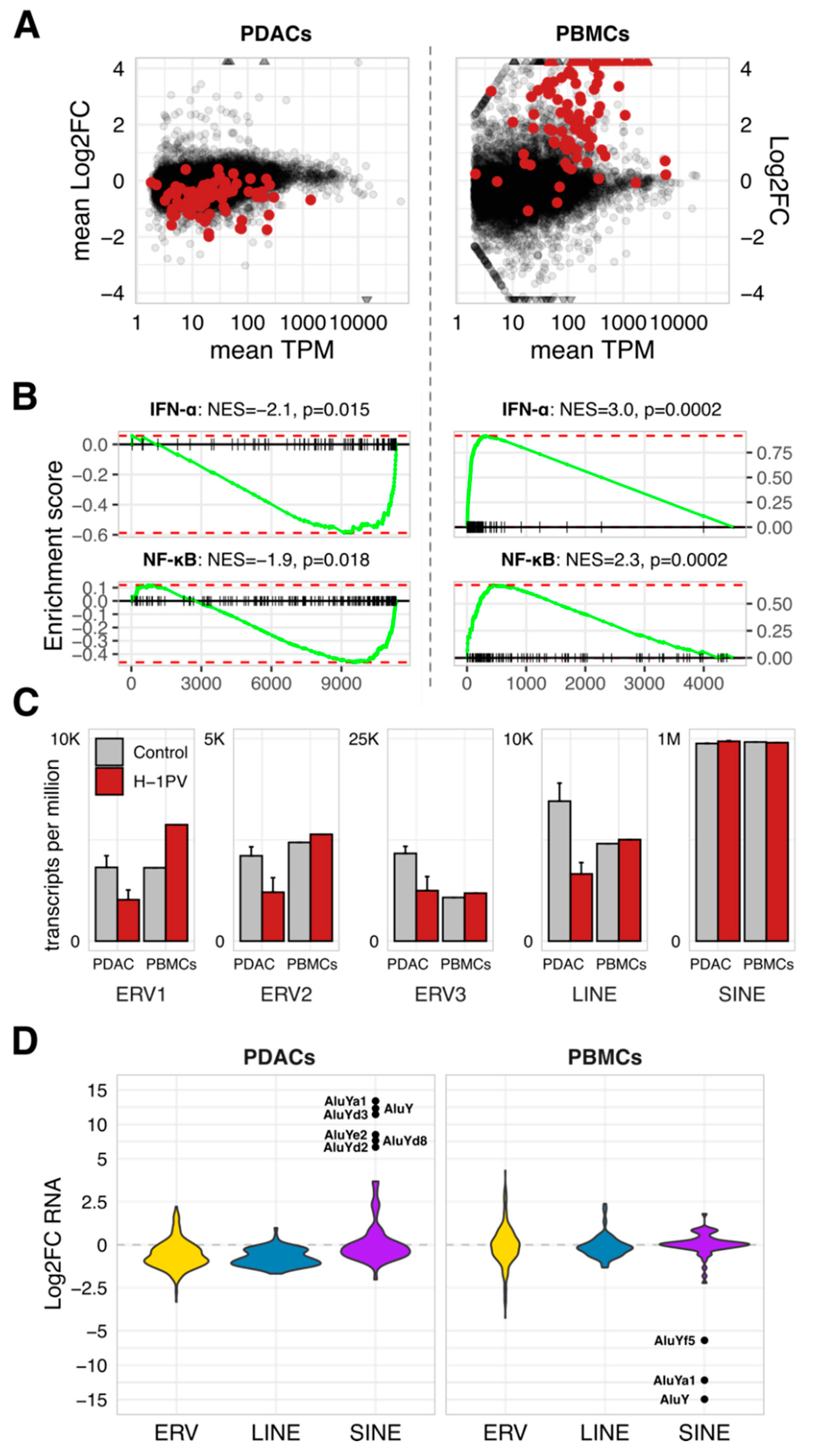
3.4. Global Disturbance of Innate Immunity by H-1PV Associates with a Clade-Specific Distortion of Retrotransposons
3.5. H-1PV Does Not Succumb to IFN but Blunts Its Triggering by Gemcitabine
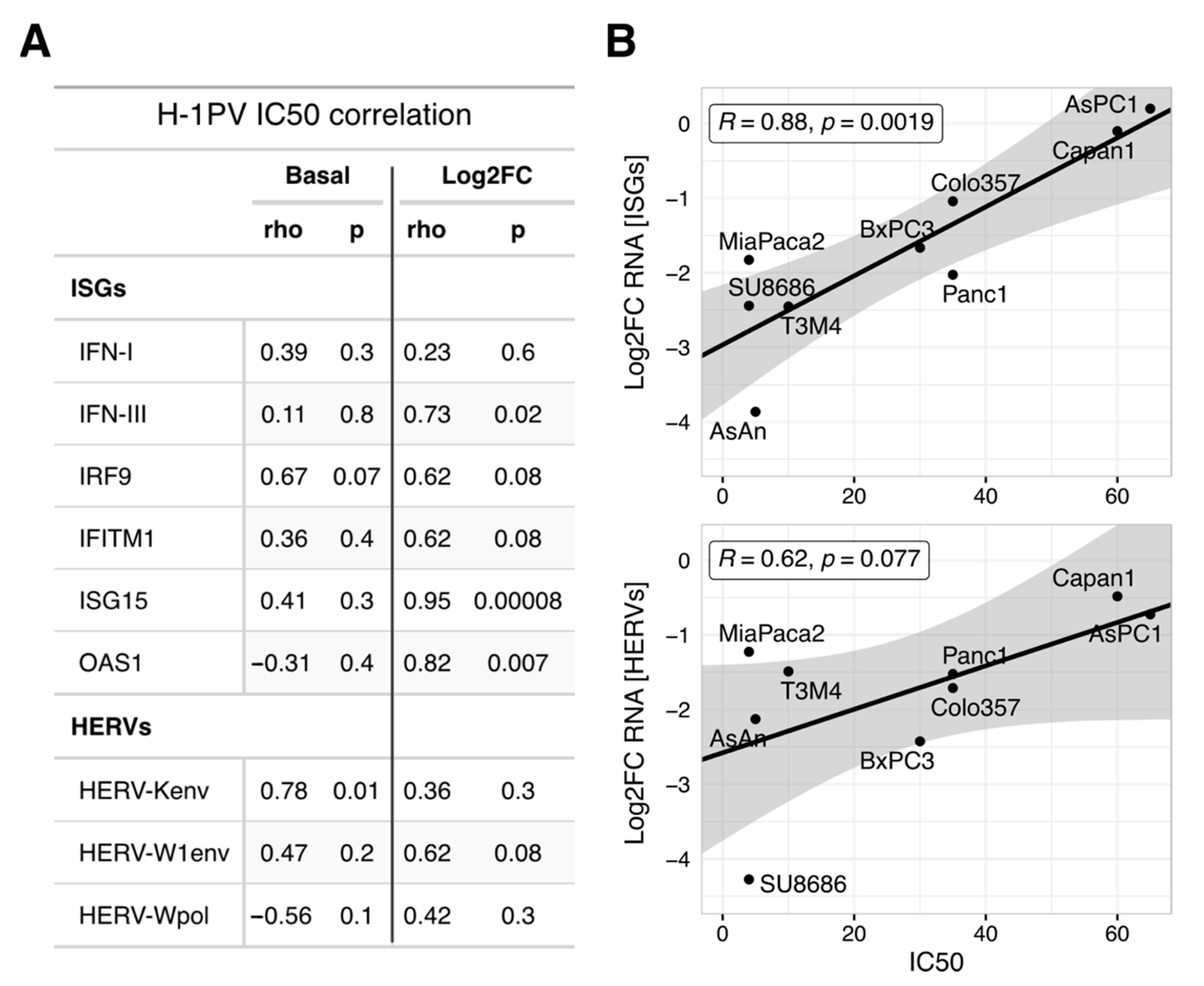
3.6. Oncolytic Efficacy of H-1PV in PDAC Cells Correlates with the Degree of ISGs Suppression but Not with Their Absolute Levels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.O.; Schoning, T.; Husing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef] [PubMed]
- Hajda, J.; Lehmann, M.; Krebs, O.; Kieser, M.; Geletneky, K.; Jager, D.; Dahm, M.; Huber, B.; Schoning, T.; Sedlaczek, O.; et al. A non-controlled, single arm, open label, phase II study of intravenous and intratumoral administration of ParvOryx in patients with metastatic, inoperable pancreatic cancer: ParvOryx02 protocol. BMC Cancer 2017, 17, 576. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [PubMed]
- Angelova, A.L.; Aprahamian, M.; Grekova, S.P.; Hajri, A.; Leuchs, B.; Giese, N.A.; Dinsart, C.; Herrmann, A.; Balboni, G.; Rommelaere, J.; et al. Improvement of Gemcitabine-Based Therapy of Pancreatic Carcinoma by Means of Oncolytic Parvovirus H-1PV. Clin. Cancer Res. 2009, 15, 511–519. [Google Scholar] [CrossRef]
- Moehler, M.H.; Zeidler, M.; Wilsberg, V.; Cornelis, J.J.; Woelfel, T.; Rommelaere, J.; Galle, P.R.; Heike, M. Parvovirus H-1-induced tumor cell death enhances human immune response in vitro via increased phagocytosis, maturation, and cross-presentation by dendritic cells. Hum. Gene Ther. 2005, 16, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Grekova, S.; Aprahamian, M.; Giese, N.; Schmitt, S.; Giese, T.; Falk, C.S.; Daeffler, L.; Cziepluch, C.; Rommelaere, J.; Raykov, Z. Immune cells participate in the oncosuppressive activity of parvovirus H-1PV and are activated as a result of their abortive infection with this agent. Cancer Biol. Ther. 2010, 10, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, T.; Iwama, M.; Ueno, Y.; Sugiyama, F.; Nakajima, T.; Fukamizu, A.; Yagami, K. Induction of apoptosis in vitro and in vivo by H-1 parvovirus infection. J. Gen. Virol. 1998, 79 Pt 12, 3067–3071. [Google Scholar] [CrossRef]
- Ueno, Y.; Harada, T.; Iseki, H.; Ohshima, T.; Sugiyama, F.; Yagami, K. Propagation of rat parvovirus in thymic lymphoma cell line C58(NT)d and subsequent appearance of a resistant cell clone after lytic infection. J. Virol. 2001, 75, 3965–3970. [Google Scholar] [CrossRef]
- Angelova, A.L.; Grekova, S.P.; Heller, A.; Kuhlmann, O.; Soyka, E.; Giese, T.; Aprahamian, M.; Bour, G.; Ruffer, S.; Cziepluch, C.; et al. Complementary Induction of Immunogenic Cell Death by Oncolytic Parvovirus H-1PV and Gemcitabine in Pancreatic Cancer. J. Virol. 2014, 88, 5263–5276. [Google Scholar] [CrossRef]
- Chen, X.; Fosco, D.; Kline, D.E.; Kline, J. Calreticulin promotes immunity and type I interferon-dependent survival in mice with acute myeloid leukemia. Oncoimmunology 2017, 6, e1278332. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kroemer, G. Calreticulin and type I interferon: An unsuspected connection. Oncoimmunology 2017, 6, e1288334. [Google Scholar] [CrossRef]
- Nuesch, J.P.; Dettwiler, S.; Corbau, R.; Rommelaere, J. Replicative functions of minute virus of mice NS1 protein are regulated in vitro by phosphorylation through protein kinase C. J. Virol. 1998, 72, 9966–9977. [Google Scholar] [CrossRef]
- Dettwiler, S.; Rommelaere, J.; Nuesch, J.P. DNA unwinding functions of minute virus of mice NS1 protein are modulated specifically by the lambda isoform of protein kinase C. J. Virol. 1999, 73, 7410–7420. [Google Scholar] [CrossRef]
- Lachmann, S.; Rommeleare, J.; Nuesch, J.P. Novel PKCeta is required to activate replicative functions of the major nonstructural protein NS1 of minute virus of mice. J. Virol. 2003, 77, 8048–8060. [Google Scholar] [CrossRef] [PubMed]
- Nuesch, J.P.; Lacroix, J.; Marchini, A.; Rommelaere, J. Molecular pathways: Rodent parvoviruses—Mechanisms of oncolysis and prospects for clinical cancer treatment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 3516–3523. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. Parvoviruses: Small Does Not Mean Simple. Annu. Rev. Virol. 2014, 1, 517–537. [Google Scholar] [CrossRef]
- Lang, S.I.; Giese, N.A.; Rommelaere, J.; Dinsart, C.; Cornelis, J.J. Humoral immune responses against minute virus of mice vectors. J. Gene Med. 2006, 8, 1141–1150. [Google Scholar] [CrossRef]
- Raykov, Z.; Grekova, S.P.; Horlein, R.; Leuchs, B.; Giese, T.; Giese, N.A.; Rommelaere, J.; Zawatzky, R.; Daeffler, L. TLR-9 contributes to the antiviral innate immune sensing of rodent parvoviruses MVMp and H-1PV by normal human immune cells. PLoS ONE 2013, 8, e55086. [Google Scholar] [CrossRef] [PubMed]
- Schlehofer, J.R.; Rentrop, M.; Mannel, D.N. Parvoviruses are inefficient in inducing interferon-beta, tumor necrosis factor-alpha, or interleukin-6 in mammalian cells. Med. Microbiol. Immunol. 1992, 181, 153–164. [Google Scholar] [CrossRef]
- Paglino, J.C.; Andres, W.; van den Pol, A.N. Autonomous parvoviruses neither stimulate nor are inhibited by the type I interferon response in human normal or cancer cells. J. Virol. 2014, 88, 4932–4942. [Google Scholar] [CrossRef]
- Grekova, S.; Zawatzky, R.; Horlein, R.; Cziepluch, C.; Mincberg, M.; Davis, C.; Rommelaere, J.; Daeffler, L. Activation of an antiviral response in normal but not transformed mouse cells: A new determinant of minute virus of mice oncotropism. J. Virol. 2010, 84, 516–531. [Google Scholar] [CrossRef]
- Heller, A.; Gaida, M.M.; Mannle, D.; Giese, T.; Scarpa, A.; Neoptolemos, J.P.; Hackert, T.; Strobel, O.; Hoheisel, J.D.; Giese, N.A.; et al. Stratification of pancreatic tissue samples for molecular studies: RNA-based cellular annotation procedure. Pancreatol. Off. J. Int. Assoc. Pancreatol. 2015, 15, 423–431. [Google Scholar] [CrossRef]
- Mahajan, U.M.; Langhoff, E.; Goni, E.; Costello, E.; Greenhalf, W.; Halloran, C.; Ormanns, S.; Kruger, S.; Boeck, S.; Ribback, S.; et al. Immune Cell and Stromal Signature Associated With Progression-Free Survival of Patients With Resected Pancreatic Ductal Adenocarcinoma. Gastroenterology 2018, 155, 1625–1639. [Google Scholar] [CrossRef]
- Hartmann, N.; Giese, N.A.; Giese, T.; Poschke, I.; Offringa, R.; Werner, J.; Ryschich, E. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 3422–3433. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Vail, P.; Balaji, U.; Ngo, H.; Botros, I.W.; Makarov, V.; Riaz, N.; Balachandran, V.; Leach, S.; Thompson, D.M.; et al. Stratification of Pancreatic Ductal Adenocarcinoma: Combinatorial Genetic, Stromal, and Immunologic Markers. Clin. Cancer Res. Off. J.Am. Assoc. Cancer Res. 2017, 23, 4429–4440. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, X.; Ye, H.; Yao, M.; Li, S.; Chen, L. Nonstructural protein (NS1) of human parvovirus B19 stimulates host innate immunity and blunts the exogenous type I interferon signaling in vitro. Virus Res. 2016, 222, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Mattei, L.M.; Cotmore, S.F.; Tattersall, P.; Iwasaki, A. Parvovirus evades interferon-dependent viral control in primary mouse embryonic fibroblasts. Virology 2013, 442, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Ventoso, I.; Berlanga, J.J.; Almendral, J.M. Translation control by protein kinase R restricts minute virus of mice infection: Role in parvovirus oncolysis. J. Virol. 2010, 84, 5043–5051. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Liu, D.; Tian, J.; Hu, X.; Zhang, X.; Yin, H.; Wu, H.; Liu, C.; Guo, D.; Li, Z.; et al. Feline Panleucopenia Virus NS2 Suppresses the Host IFN-beta Induction by Disrupting the Interaction between TBK1 and STING. Viruses 2017, 9. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Mitzel, D.N.; Lowry, V.; Shirali, A.C.; Liu, Y.; Stout-Delgado, H.W. Age-enhanced endoplasmic reticulum stress contributes to increased Atg9A inhibition of STING-mediated IFN-beta production during Streptococcus pneumoniae infection. J. Immunol. 2014, 192, 4273–4283. [Google Scholar] [CrossRef]
- Lee, K.; Kim, D.E.; Jang, K.S.; Kim, S.J.; Cho, S.; Kim, C. Gemcitabine, a broad-spectrum antiviral drug, suppresses enterovirus infections through innate immunity induced by the inhibition of pyrimidine biosynthesis and nucleotide depletion. Oncotarget 2017, 8, 115315–115325. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Shin, H.J.; Kim, C.; Cho, S. Gemcitabine and Nucleos(t)ide Synthesis Inhibitors Are Broad-Spectrum Antiviral Drugs that Activate Innate Immunity. Viruses 2018, 10, 211. [Google Scholar] [CrossRef]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Moerdyk-Schauwecker, M.; Shah, N.R.; Murphy, A.M.; Hastie, E.; Mukherjee, P.; Grdzelishvili, V.Z. Resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus: Role of type I interferon signaling. Virology 2013, 436, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; van Eijck, C.H.; van Koetsveld Ing, P.M.; Erdmann, J.I.; Speel, E.J.; van der Wansem Ing, K.; Mooij, D.M.; Colao, A.; Lombardi, G.; Croze, E.; et al. Type I interferons in the treatment of pancreatic cancer: Mechanisms of action and role of related receptors. Ann. Surg. 2007, 246, 259–268. [Google Scholar] [CrossRef]
- Faisst, S.R.; Faisst, S.; Grangette, C.; Schlehofer, J.R.; Rommelaere, J. NF kappa B upstream regulatory sequences of the HIV-1 LTR are involved in the inhibition of HIV-1 promoter activity by the NS proteins of autonomous parvoviruses H-1 and MVMp. Virology 1993, 197, 770–773. [Google Scholar] [CrossRef] [PubMed]
- Kassiotis, G.; Stoye, J.P. Immune responses to endogenous retroelements: Taking the bad with the good. Nat. Rev. Immunol. 2016, 16, 207–219. [Google Scholar] [CrossRef]
- Solovyov, A.; Vabret, N.; Arora, K.S.; Snyder, A.; Funt, S.A.; Bajorin, D.F.; Rosenberg, J.E.; Bhardwaj, N.; Ting, D.T.; Greenbaum, B.D. Global Cancer Transcriptome Quantifies Repeat Element Polarization between Immunotherapy Responsive and T Cell Suppressive Classes. Cell Rep. 2018, 23, 512–521. [Google Scholar] [CrossRef]
- Attig, J.; Young, G.R.; Stoye, J.P.; Kassiotis, G. Physiological and Pathological Transcriptional Activation of Endogenous Retroelements Assessed by RNA-Sequencing of B Lymphocytes. Front. Microbiol. 2017, 8, 2489. [Google Scholar] [CrossRef]
- Canadas, I.; Thummalapalli, R.; Kim, J.W.; Kitajima, S.; Jenkins, R.W.; Christensen, C.L.; Campisi, M.; Kuang, Y.; Zhang, Y.; Gjini, E.; et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat. Med. 2018, 24, 1143–1150. [Google Scholar] [CrossRef]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Alcazer, V.; Bonaventura, P.; Depil, S. Human Endogenous Retroviruses (HERVs): Shaping the Innate Immune Response in Cancers. Cancers 2020, 12, 610. [Google Scholar] [CrossRef]
- Rejiba, S.; Bigand, C.; Parmentier, C.; Masmoudi, A.; Hajri, A. Oncosuppressive suicide gene virotherapy "PVH1-yCD/5-FC" for pancreatic peritoneal carcinomatosis treatment: NFkappaB and Akt/PI3K involvement. PLoS ONE 2013, 8, e70594. [Google Scholar] [CrossRef] [PubMed]
- Heller, A.; Angelova, A.L.; Bauer, S.; Grekova, S.P.; Aprahamian, M.; Rommelaere, J.; Volkmar, M.; Janssen, J.W.; Bauer, N.; Herr, I.; et al. Establishment and Characterization of a Novel Cell Line, ASAN-PaCa, Derived From Human Adenocarcinoma Arising in Intraductal Papillary Mucinous Neoplasm of the Pancreas. Pancreas 2016, 45, 1452–1460. [Google Scholar] [CrossRef]
- Wetzel, K.; Menten, P.; Opdenakker, G.; Van Damme, J.; Grone, H.J.; Giese, N.; Vecchi, A.; Sozzani, S.; Cornelis, J.J.; Rommelaere, J.; et al. Transduction of human MCP-3 by a parvoviral vector induces leukocyte infiltration and reduces growth of human cervical carcinoma cell xenografts. J. Gene Med. 2001, 3, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Leuchs, B.; Roscher, M.; Muller, M.; Kurschner, K.; Rommelaere, J. Standardized large-scale H-1PV production process with efficient quality and quantity monitoring. J. Virol. Methods 2016, 229, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Leuchs, B.; Frehtman, V.; Riese, M.; Muller, M.; Rommelaere, J. A novel scalable, robust downstream process for oncolytic rat parvovirus: Isoelectric point-based elimination of empty particles. Appl. Microbiol. Biotechnol. 2017, 101, 3143–3152. [Google Scholar] [CrossRef]
- Heller, A.; Zornig, I.; Muller, T.; Giorgadze, K.; Frei, C.; Giese, T.; Bergmann, F.; Schmidt, J.; Werner, J.; Buchler, M.W.; et al. Immunogenicity of SEREX-identified antigens and disease outcome in pancreatic cancer. Cancer Immunol. Immunother. 2010, 59, 1389–1400. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 23 April 2020).
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Jeong, H.H.; Yalamanchili, H.K.; Guo, C.; Shulman, J.M.; Liu, Z. An Ultra-Fast and Scalable Quantification Pipeline for Transposable Elements from Next Generation Sequencing Data. In Proceedings of the Pacific Symposium on Biocomputing 2018, Kohala, HI, USA, 3–7 January 2018; Volume 23, pp. 168–179. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize Implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Williams, W.P.; Tamburic, L.; Astell, C.R. Increased levels of B1 and B2 SINE transcripts in mouse fibroblast cells due to minute virus of mice infection. Virology 2004, 327, 233–241. [Google Scholar] [CrossRef]
- Deininger, P. Alu elements: Know the SINEs. Genome Biol. 2011, 12, 236. [Google Scholar] [CrossRef] [PubMed]
- Ina, S.; Hirono, S.; Noda, T.; Yamaue, H. Identifying molecular markers for chemosensitivity to gemcitabine in pancreatic cancer: Increased expression of interferon-stimulated gene 15 kd is associated with intrinsic chemoresistance. Pancreas 2010, 39, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Pertusati, F.; Pileggi, E.; Richards, J.; Wootton, M.; Van Leemputte, T.; Persoons, L.; De Coster, D.; Villanueva, X.; Daelemans, D.; Steenackers, H.; et al. Drug repurposing: Phosphate prodrugs of anticancer and antiviral FDA-approved nucleosides as novel antimicrobials. J. Antimicrob. Chemother. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ling, W.H.; Soe, P.P.; Pang, A.S.; Lee, S.C. Hepatitis B virus reactivation risk varies with different chemotherapy regimens commonly used in solid tumours. Br. J. Cancer 2013, 108, 1931–1935. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remedios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nature Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Hunger, A.; Medrano, R.F.; Zanatta, D.B.; Del Valle, P.R.; Merkel, C.A.; Salles, T.A.; Ferrari, D.G.; Furuya, T.K.; Bustos, S.O.; de Freitas Saito, R.; et al. Reestablishment of p53/Arf and interferon-beta pathways mediated by a novel adenoviral vector potentiates antiviral response and immunogenic cell death. Cell Death Discov. 2017, 3, 17017. [Google Scholar] [CrossRef]
- Zhou, H.; Forveille, S.; Sauvat, A.; Yamazaki, T.; Senovilla, L.; Ma, Y.; Liu, P.; Yang, H.; Bezu, L.; Muller, K.; et al. The oncolytic peptide LTX-315 triggers immunogenic cell death. Cell Death Dis. 2016, 7, e2134. [Google Scholar] [CrossRef] [PubMed]
- Zevini, A.; Olagnier, D.; Hiscott, J. Crosstalk between Cytoplasmic RIG-I and STING Sensing Pathways. Trends Immunol. 2017, 38, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Herzner, A.M.; Hagmann, C.A.; Goldeck, M.; Wolter, S.; Kubler, K.; Wittmann, S.; Gramberg, T.; Andreeva, L.; Hopfner, K.P.; Mertens, C.; et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat. Immunol. 2015, 16, 1025–1033. [Google Scholar] [CrossRef]
- Andreeva, L.; Hiller, B.; Kostrewa, D.; Lassig, C.; de Oliveira Mann, C.C.; Jan Drexler, D.; Maiser, A.; Gaidt, M.; Leonhardt, H.; Hornung, V.; et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 2017, 549, 394–398. [Google Scholar] [CrossRef]
- Mankan, A.K.; Schmidt, T.; Chauhan, D.; Goldeck, M.; Honing, K.; Gaidt, M.; Kubarenko, A.V.; Andreeva, L.; Hopfner, K.P.; Hornung, V. Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J. 2014, 33, 2937–2946. [Google Scholar] [CrossRef]
- Lachmann, S.; Bar, S.; Rommelaere, J.; Nuesch, J.P. Parvovirus interference with intracellular signalling: Mechanism of PKCeta activation in MVM-infected A9 fibroblasts. Cell Microbiol. 2008, 10, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Ban, T.; Taniguchi, T. High-mobility group box family of proteins: Ligand and sensor for innate immunity. Trends Immunol. 2012, 33, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.L.; Chen, F.X.; Rhode, S.L. Parvovirus H-1 P38 promoter requires the trans-activation region (tar), an SP1 site, and a TATA box for full activity. Virology 1992, 187, 10–17. [Google Scholar] [CrossRef]
- Lorson, C.; Pearson, J.; Burger, L.; Pintel, D.J. An Sp1-binding site and TATA element are sufficient to support full transactivation by proximally bound NS1 protein of minute virus of mice. Virology 1998, 240, 326–337. [Google Scholar] [CrossRef][Green Version]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Wu, X.; Dao Thi, V.L.; Huang, Y.; Billerbeck, E.; Saha, D.; Hoffmann, H.H.; Wang, Y.; Silva, L.A.V.; Sarbanes, S.; Sun, T.; et al. Intrinsic Immunity Shapes Viral Resistance of Stem Cells. Cell 2018, 172, 423–438 e425. [Google Scholar] [CrossRef]
- Haas, S.; Trumpp, A. An Intrinsic Interferon Program Protects Stem Cells from Viral Infection. Dev. Cell 2018, 44, 279–280. [Google Scholar] [CrossRef]
- Li, M.; Radvanyi, L.; Yin, B.; Li, J.; Chivukula, R.; Lin, K.; Lu, Y.; Shen, J.; Chang, D.Z.; Li, D.; et al. Downregulation of Human Endogenous Retrovirus Type K (HERV-K) Viral env RNA in Pancreatic Cancer Cells Decreases Cell Proliferation and Tumor Growth. Clinical Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 5892–5911. [Google Scholar] [CrossRef] [PubMed]
- Monsurro, V.; Beghelli, S.; Wang, R.; Barbi, S.; Coin, S.; Di Pasquale, G.; Bersani, S.; Castellucci, M.; Sorio, C.; Eleuteri, S.; et al. Anti-viral state segregates two molecular phenotypes of pancreatic adenocarcinoma: Potential relevance for adenoviral gene therapy. J. Transl. Med. 2010, 8, 10. [Google Scholar] [CrossRef]
- Harris, R.E.; Coleman, P.H.; Morahan, P.S. Erythrocyte association and interferon production by minute virus of mice. Soc. Exp. Biol. Med. 1974, 145, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Bretscher, C.; Marchini, A. H-1 Parvovirus as a Cancer-Killing Agent: Past, Present, and Future. Viruses 2019, 11. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neulinger-Muñoz, M.; Schaack, D.; Grekova, S.P.; Bauer, A.S.; Giese, T.; Salg, G.A.; Espinet, E.; Leuchs, B.; Heller, A.; Nüesch, J.P.F.; et al. Human Retrotransposons and the Global Shutdown of Homeostatic Innate Immunity by Oncolytic Parvovirus H-1PV in Pancreatic Cancer. Viruses 2021, 13, 1019. https://doi.org/10.3390/v13061019
Neulinger-Muñoz M, Schaack D, Grekova SP, Bauer AS, Giese T, Salg GA, Espinet E, Leuchs B, Heller A, Nüesch JPF, et al. Human Retrotransposons and the Global Shutdown of Homeostatic Innate Immunity by Oncolytic Parvovirus H-1PV in Pancreatic Cancer. Viruses. 2021; 13(6):1019. https://doi.org/10.3390/v13061019
Chicago/Turabian StyleNeulinger-Muñoz, Matthias, Dominik Schaack, Svetlana P. Grekova, Andrea S. Bauer, Thomas Giese, Gabriel A. Salg, Elisa Espinet, Barbara Leuchs, Anette Heller, Jürg P. F. Nüesch, and et al. 2021. "Human Retrotransposons and the Global Shutdown of Homeostatic Innate Immunity by Oncolytic Parvovirus H-1PV in Pancreatic Cancer" Viruses 13, no. 6: 1019. https://doi.org/10.3390/v13061019
APA StyleNeulinger-Muñoz, M., Schaack, D., Grekova, S. P., Bauer, A. S., Giese, T., Salg, G. A., Espinet, E., Leuchs, B., Heller, A., Nüesch, J. P. F., Schenk, M., Volkmar, M., & Giese, N. A. (2021). Human Retrotransposons and the Global Shutdown of Homeostatic Innate Immunity by Oncolytic Parvovirus H-1PV in Pancreatic Cancer. Viruses, 13(6), 1019. https://doi.org/10.3390/v13061019