
Clinical Use of Improved Diagnostic Testing for Detection of Prion Disease


Abstract
1. Introduction
2. Background
3. Clinical Phenomena
4. Markers for Rapid Neurodegeneration
5. Non-Laboratory Based Diagnostic Tests for Prion Disease
6. Prion Specific Assays
7. Genetics
8. Atypical Prion Disease
9. The Potential for Emerging Zoonotic Prion Disease
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of Sporadic Creutzfeldt-Jakob Disease Based on Molecular and Phenotypic Analysis of 300 Subjects. Ann. Neurol. 1999, 46, 224–233. [Google Scholar] [CrossRef]
- Cali, I.; Castellani, R.; Alshekhlee, A.; Cohen, Y.; Blevins, J.; Yuan, J.; Langeveld, J.P.M.; Parchi, P.; Safar, J.G.; Zou, W.-Q.; et al. Co-Existence of Scrapie Prion Protein Types 1 and 2 in Sporadic Creutzfeldt-Jakob Disease: Its Effect on the Phenotype and Prion-Type Characteristics. Brain J. Neurol. 2009, 132, 2643–2658. [Google Scholar] [CrossRef] [PubMed]
- Schoch, G.; Seeger, H.; Bogousslavsky, J.; Tolnay, M.; Janzer, R.C.; Aguzzi, A.; Glatzel, M. Analysis of Prion Strains by PrPSc Profiling in Sporadic Creutzfeldt-Jakob Disease. PLoS Med. 2006, 3, e14. [Google Scholar] [CrossRef] [PubMed]
- Appleby, B.S.; Rhoads, D.D.; Mente, K.; Cohen, M.L. A Practical Primer on Prion Pathology. J. Neuropathol. Exp. Neurol. 2018, 77, 346–352. [Google Scholar] [CrossRef]
- Notari, S.; Appleby, B.S.; Gambetti, P. Variably Protease-Sensitive Prionopathy. Handb. Clin. Neurol. 2018, 153, 175–190. [Google Scholar] [CrossRef]
- Lloyd, S.E.; Mead, S.; Collinge, J. Genetics of Prion Diseases. Curr. Opin. Genet. Dev. 2013, 23, 345–351. [Google Scholar] [CrossRef]
- Mallucci, G.R.; Campbell, T.A.; Dickinson, A.; Beck, J.; Holt, M.; Plant, G.; de Pauw, K.W.; Hakin, R.N.; Clarke, C.E.; Howell, S.; et al. Inherited Prion Disease with an Alanine to Valine Mutation at Codon 117 in the Prion Protein Gene. Brain J. Neurol. 1999, 122, 1823–1837. [Google Scholar] [CrossRef]
- Saitoh, Y.; Ogawa, M.; Naito, Y.; Komatsuzaki, Y.; Tagaya, H.; Arima, K.; Tamaoka, A.; Kitamoto, T.; Murata, M. Discordant Clinicopathologic Phenotypes in a Japanese Kindred of Fatal Familial Insomnia. Neurology 2010, 74, 86–89. [Google Scholar] [CrossRef]
- Sparkes, R.S.; Simon, M.; Cohn, V.H.; Fournier, R.E.; Lem, J.; Klisak, I.; Heinzmann, C.; Blatt, C.; Lucero, M.; Mohandas, T. Assignment of the Human and Mouse Prion Protein Genes to Homologous Chromosomes. Proc. Natl. Acad. Sci. USA 1986, 83, 7358–7362. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Bauer, P.; Schöls, L. Prion Mutation D178N with Highly Variable Disease Onset and Phenotype. J. Neurol. Neurosurg. Psychiatry 2009, 80, 345–346. [Google Scholar] [CrossRef]
- Goldfarb, L.; Petersen, R.; Tabaton, M.; Brown, P.; LeBlanc, A.; Montagna, P.; Cortelli, P.; Julien, J.; Vital, C.; Pendelbury, W.; et al. Fatal Familial Insomnia and Familial Creutzfeldt-Jakob Disease: Disease Phenotype Determined by a DNA Polymorphism. Science 1992, 258, 806–808. [Google Scholar] [CrossRef]
- Brown, P.; Brandel, J.-P.; Sato, T.; Nakamura, Y.; MacKenzie, J.; Will, R.G.; Ladogana, A.; Pocchiari, M.; Leschek, E.W.; Schonberger, L.B. Iatrogenic Creutzfeldt-Jakob Disease, Final Assessment. Emerg. Infect. Dis. 2012, 18, 901–907. [Google Scholar] [CrossRef]
- Klug, G.M.J.A.; Wand, H.; Simpson, M.; Boyd, A.; Law, M.; Masters, C.L.; Matěj, R.; Howley, R.; Farrell, M.; Breithaupt, M.; et al. Intensity of Human Prion Disease Surveillance Predicts Observed Disease Incidence. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1372–1377. [Google Scholar] [CrossRef]
- Maddox, R.A.; Person, M.K.; Blevins, J.E.; Abrams, J.Y.; Appleby, B.S.; Schonberger, L.B.; Belay, E.D. Prion Disease Incidence in the United States: 2003–2015. Neurology 2020, 94, e153–e157. [Google Scholar] [CrossRef] [PubMed]
- Rabinovici, G.D.; Wang, P.N.; Levin, J.; Cook, L.; Pravdin, M.; Davis, J.; DeArmond, S.J.; Barbaro, N.M.; Martindale, J.; Miller, B.L.; et al. First Symptom in Sporadic Creutzfeldt-Jakob Disease. Neurology 2006, 66, 286–287. [Google Scholar] [CrossRef] [PubMed]
- CDC’s Diagnostic Criteria for Creutzfeldt-Jakob Disease (CJD). Available online: https://www.cdc.gov/prions/cjd/diagnostic-criteria.html (accessed on 24 February 2021).
- Zeidler, M.; Green, A. Advances in Diagnosing Creutzfeldt-Jakob Disease with MRI and CSF 14-3-3 Protein Analysis. Neurology 2004, 63, 410–411. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Kurohara, K.; Yukitake, M.; Kuroda, Y. The 14-3-3 Protein Detectable in the Cerebrospinal Fluid of Patients with Prion-Unrelated Neurological Diseases Is Expressed Constitutively in Neurons and Glial Cells in Culture. Eur. Neurol. 1999, 41, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Chohan, G.; Pennington, C.; Mackenzie, J.M.; Andrews, M.; Everington, D.; Will, R.G.; Knight, R.S.G.; Green, A.J.E. The Role of Cerebrospinal Fluid 14-3-3 and Other Proteins in the Diagnosis of Sporadic Creutzfeldt-Jakob Disease in the UK: A 10-Year Review. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1243–1248. [Google Scholar] [CrossRef]
- Bahl, J.M.C.; Heegaard, N.H.H.; Falkenhorst, G.; Laursen, H.; Høgenhaven, H.; Mølbak, K.; Jespersgaard, C.; Hougs, L.; Waldemar, G.; Johannsen, P.; et al. The Diagnostic Efficiency of Biomarkers in Sporadic Creutzfeldt-Jakob Disease Compared to Alzheimer’s Disease. Neurobiol. Aging 2009, 30, 1834–1841. [Google Scholar] [CrossRef]
- Sanchez-Juan, P.; Green, A.; Ladogana, A.; Cuadrado-Corrales, N.; Sáanchez-Valle, R.; Mitrováa, E.; Stoeck, K.; Sklaviadis, T.; Kulczycki, J.; Hess, K.; et al. CSF Tests in the Differential Diagnosis of Creutzfeldt-Jakob Disease. Neurology 2006, 67, 637–643. [Google Scholar] [CrossRef]
- Coulthart, M.B.; Jansen, G.H.; Olsen, E.; Godal, D.L.; Connolly, T.; Choi, B.C.; Wang, Z.; Cashman, N.R. Diagnostic Accuracy of Cerebrospinal Fluid Protein Markers for Sporadic Creutzfeldt-Jakob Disease in Canada: A 6-Year Prospective Study. BMC Neurol. 2011, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.J.; Sanchez-Juan, P.; Masters, C.L.; Klug, G.M.; van Duijn, C.; Poleggi, A.; Pocchiari, M.; Almonti, S.; Cuadrado-Corrales, N.; de Pedro-Cuesta, J.; et al. Determinants of Diagnostic Investigation Sensitivities across the Clinical Spectrum of Sporadic Creutzfeldt-Jakob Disease. Brain J. Neurol. 2006, 129, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Stoeck, K.; Sanchez-Juan, P.; Gawinecka, J.; Green, A.; Ladogana, A.; Pocchiari, M.; Sanchez-Valle, R.; Mitrova, E.; Sklaviadis, T.; Kulczycki, J.; et al. Cerebrospinal Fluid Biomarker Supported Diagnosis of Creutzfeldt-Jakob Disease and Rapid Dementias: A Longitudinal Multicentre Study over 10 Years. Brain J. Neurol. 2012, 135, 3051–3061. [Google Scholar] [CrossRef] [PubMed]
- Muayqil, T.; Gronseth, G.; Camicioli, R. Evidence-Based Guideline: Diagnostic Accuracy of CSF 14-3-3 Protein in Sporadic Creutzfeldt-Jakob Disease: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2012, 79, 1499–1506. [Google Scholar] [CrossRef]
- Pennington, C.; Chohan, G.; Mackenzie, J.; Andrews, M.; Will, R.; Knight, R.; Green, A. The Role of Cerebrospinal Fluid Proteins as Early Diagnostic Markers for Sporadic Creutzfeldt–Jakob Disease. Neurosci. Lett. 2009, 455, 56–59. [Google Scholar] [CrossRef]
- Beaudry, P.; Cohen, P.; Brandel, J.P.; Delasnerie-Lauprêtre, N.; Richard, S.; Launay, J.M.; Laplanche, J.L. 14-3-3 Protein, Neuron-Specific Enolase, and S-100 Protein in Cerebrospinal Fluid of Patients with Creutzfeldt-Jakob Disease. Dement. Geriatr. Cogn. Disord. 1999, 10, 40–46. [Google Scholar] [CrossRef]
- Hamlin, C.; Puoti, G.; Berri, S.; Sting, E.; Harris, C.; Cohen, M.; Spear, C.; Bizzi, A.; Debanne, S.M.; Rowland, D.Y. A Comparison of Tau and 14-3-3 Protein in the Diagnosis of Creutzfeldt-Jakob Disease. Neurology 2012, 79, 547–552. [Google Scholar] [CrossRef]
- Forner, S.A.; Takada, L.T.; Bettcher, B.M.; Lobach, I.V.; Tartaglia, M.C.; Torres-Chae, C.; Haman, A.; Thai, J.; Vitali, P.; Neuhaus, J.; et al. Comparing CSF Biomarkers and Brain MRI in the Diagnosis of Sporadic Creutzfeldt-Jakob Disease. Neurol. Clin. Pract. 2015, 5, 116–125. [Google Scholar] [CrossRef]
- Castellani, R.J.; Colucci, M.; Xie, Z.; Zou, W.; Li, C.; Parchi, P.; Capellari, S.; Pastore, M.; Rahbar, M.H.; Chen, S.G.; et al. Sensitivity of 14-3-3 Protein Test Varies in Subtypes of Sporadic Creutzfeldt-Jakob Disease. Neurology 2004, 63, 436–442. [Google Scholar] [CrossRef]
- Krasnianski, A.; Schulz-Schaeffer, W.J.; Kallenberg, K.; Meissner, B.; Collie, D.A.; Roeber, S.; Bartl, M.; Heinemann, U.; Varges, D.; Kretzschmar, H.A.; et al. Clinical Findings and Diagnostic Tests in the MV2 Subtype of Sporadic CJD. Brain J. Neurol. 2006, 129, 2288–2296. [Google Scholar] [CrossRef]
- Zerr, I.; Bodemer, M.; Gefeller, O.; Otto, M.; Poser, S.; Wiltfang, J.; Windl, O.; Kretzschmar, H.A.; Weber, T. Detection of 14-3-3 Protein in the Cerebrospinal Fluid Supports the Diagnosis of Creutzfeldt-Jakob Disease. Ann. Neurol. 1998, 43, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Chapman, T.; McKeel, D.W.; Morris, J.C. Misleading Results with the 14-3-3 Assay for the Diagnosis of Creutzfeldt-Jakob Disease. Neurology 2000, 55, 1396–1397. [Google Scholar] [CrossRef] [PubMed]
- Abu-Rumeileh, S.; Baiardi, S.; Polischi, B.; Mammana, A.; Franceschini, A.; Green, A.; Capellari, S.; Parchi, P. Diagnostic Value of Surrogate CSF Biomarkers for Creutzfeldt-Jakob Disease in the Era of RT-QuIC. J. Neurol. 2019, 266, 3136–3143. [Google Scholar] [CrossRef]
- Lattanzio, F.; Abu-Rumeileh, S.; Franceschini, A.; Kai, H.; Amore, G.; Poggiolini, I.; Rossi, M.; Baiardi, S.; McGuire, L.; Ladogana, A.; et al. Prion-Specific and Surrogate CSF Biomarkers in Creutzfeldt-Jakob Disease: Diagnostic Accuracy in Relation to Molecular Subtypes and Analysis of Neuropathological Correlates of p-Tau and Aβ42 Levels. Acta Neuropathol. 2017, 133, 559–578. [Google Scholar] [CrossRef] [PubMed]
- Koscova, S.; Zakova Slivarichova, D.; Tomeckova, I.; Melicherova, K.; Stelzer, M.; Janakova, A.; Kosorinova, D.; Belay, G.; Mitrova, E. Cerebrospinal Fluid Biomarkers in the Diagnosis of Creutzfeldt-Jakob Disease in Slovak Patients: Over 10-Year Period Review. Mol. Neurobiol. 2017, 54, 5919–5927. [Google Scholar] [CrossRef] [PubMed]
- Skillbäck, T.; Rosén, C.; Asztely, F.; Mattsson, N.; Blennow, K.; Zetterberg, H. Diagnostic Performance of Cerebrospinal Fluid Total Tau and Phosphorylated Tau in Creutzfeldt-Jakob Disease: Results from the Swedish Mortality Registry. JAMA Neurol. 2014, 71, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Baldeiras, I.E.; Ribeiro, M.H.; Pacheco, P.; Machado, Á.; Santana, I.; Cunha, L.; Oliveira, C.R. Diagnostic Value of CSF Protein Profile in a Portuguese Population of SCJD Patients. J. Neurol. 2009, 256, 1540–1550. [Google Scholar] [CrossRef]
- Skinningsrud, A.; Stenset, V.; Gundersen, A.S.; Fladby, T. Cerebrospinal Fluid Markers in Creutzfeldt-Jakob Disease. Cereb. Fluid Res. 2008, 5, 14. [Google Scholar] [CrossRef]
- Abu-Rumeileh, S.; Baiardi, S.; Ladogana, A.; Zenesini, C.; Bartoletti-Stella, A.; Poleggi, A.; Mammana, A.; Polischi, B.; Pocchiari, M.; Capellari, S.; et al. Comparison between Plasma and Cerebrospinal Fluid Biomarkers for the Early Diagnosis and Association with Survival in Prion Disease. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1181–1188. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Andreasson, U.; Liman, V.; Regelsberger, G.; Lutz, M.I.; Danics, K.; Keller, E.; Zetterberg, H.; Blennow, K. Plasma and Cerebrospinal Fluid Tau and Neurofilament Concentrations in Rapidly Progressive Neurological Syndromes: A Neuropathology-Based Cohort. Eur. J. Neurol. 2017, 24, 1326-e77. [Google Scholar] [CrossRef]
- Staffaroni, A.M.; Kramer, A.O.; Casey, M.; Kang, H.; Rojas, J.C.; Orrú, C.D.; Caughey, B.; Allen, I.E.; Kramer, J.H.; Rosen, H.J.; et al. Association of Blood and Cerebrospinal Fluid Tau Level and Other Biomarkers With Survival Time in Sporadic Creutzfeldt-Jakob Disease. JAMA Neurol. 2019, 76, 969. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Diaz-Lucena, D.; Zetterberg, H.; Villar-Pique, A.; Karch, A.; Vidal, E.; Hermann, P.; Schmitz, M.; Ferrer Abizanda, I.; Zerr, I.; et al. CSF Neurogranin as a Neuronal Damage Marker in CJD: A Comparative Study with AD. J. Neurol. Neurosurg. Psychiatry 2019, 90, 846–853. [Google Scholar] [CrossRef]
- Ermann, N.; Lewczuk, P.; Schmitz, M.; Lange, P.; Knipper, T.; Goebel, S.; Kornhuber, J.; Zerr, I.; Llorens, F. CSF Nonphosphorylated Tau as a Biomarker for the Discrimination of AD from CJD. Ann. Clin. Transl. Neurol. 2018, 5, 883–887. [Google Scholar] [CrossRef]
- Johnson, E.B.; Byrne, L.M.; Gregory, S.; Rodrigues, F.B.; Blennow, K.; Durr, A.; Leavitt, B.R.; Roos, R.A.; Zetterberg, H.; Tabrizi, S.J.; et al. TRACK-HD Study Group. Neurofilament Light Protein in Blood Predicts Regional Atrophy in Huntington Disease. Neurology 2018, 90, e717–e723. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, M.; Blomberg, M.; Jonsson, M.; Wahlund, L.O.; Edman, A.; Lind, K.; Rosengren, L.; Blennow, K.; Wallin, A. Neurofilament Protein in Cerebrospinal Fluid: A Marker of White Matter Changes. J. Neurosci. Res. 2001, 66, 510–516. [Google Scholar] [CrossRef]
- Bergman, J.; Dring, A.; Zetterberg, H.; Blennow, K.; Norgren, N.; Gilthorpe, J.; Bergenheim, T.; Svenningsson, A. Neurofilament Light in CSF and Serum Is a Sensitive Marker for Axonal White Matter Injury in MS. Neurol. Neuroimmunol. Neuroinflammation 2016, 3, e271. [Google Scholar] [CrossRef]
- Olsson, B.; Portelius, E.; Cullen, N.C.; Sandelius, Å.; Zetterberg, H.; Andreasson, U.; Höglund, K.; Irwin, D.J.; Grossman, M.; Weintraub, D.; et al. Association of Cerebrospinal Fluid Neurofilament Light Protein Levels with Cognition in Patients With Dementia, Motor Neuron Disease, and Movement Disorders. JAMA Neurol. 2019, 76, 318–325. [Google Scholar] [CrossRef]
- Hampel, H.; Toschi, N.; Baldacci, F.; Zetterberg, H.; Blennow, K.; Kilimann, I.; Teipel, S.J.; Cavedo, E.; Melo Dos Santos, A.; Epelbaum, S.; et al. Alzheimer Precision Medicine Initiative (APMI). Alzheimer’s Disease Biomarker-Guided Diagnostic Workflow Using the Added Value of Six Combined Cerebrospinal Fluid Candidates: Aβ1-42, Total-Tau, Phosphorylated-Tau, NFL, Neurogranin, and YKL-40. Alzheimers Dement. J. Alzheimers Assoc. 2018, 14, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Skillbäck, T.; Mattsson, N.; Trojanowski, J.Q.; Portelius, E.; Shaw, L.M.; Weiner, M.W.; Blennow, K. For the Alzheimer’s Disease Neuroimaging Initiative. Association of Cerebrospinal Fluid Neurofilament Light Concentration with Alzheimer Disease Progression. JAMA Neurol. 2016, 73, 60. [Google Scholar] [CrossRef]
- Mattsson, N.; Insel, P.S.; Palmqvist, S.; Portelius, E.; Zetterberg, H.; Weiner, M.; Blennow, K.; Hansson, O. The Alzheimer’s Disease Neuroimaging Initiative. Cerebrospinal Fluid Tau, Neurogranin, and Neurofilament Light in Alzheimer’s Disease. EMBO Mol. Med. 2016, 8, 1184–1196. [Google Scholar] [CrossRef]
- Skillback, T.; Farahmand, B.; Bartlett, J.W.; Rosen, C.; Mattsson, N.; Nagga, K.; Kilander, L.; Religa, D.; Wimo, A.; Winblad, B.; et al. CSF Neurofilament Light Differs in Neurodegenerative Diseases and Predicts Severity and Survival. Neurology 2014, 83, 1945–1953. [Google Scholar] [CrossRef]
- Oeckl, P.; Jardel, C.; Salachas, F.; Lamari, F.; Andersen, P.M.; Bowser, R.; de Carvalho, M.; Costa, J.; van Damme, P.; Gray, E.; et al. Multicenter Validation of CSF Neurofilaments as Diagnostic Biomarkers for ALS. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 404–413. [Google Scholar] [CrossRef]
- Feneberg, E.; Oeckl, P.; Steinacker, P.; Verde, F.; Barro, C.; Van Damme, P.; Gray, E.; Grosskreutz, J.; Jardel, C.; Kuhle, J.; et al. Multicenter Evaluation of Neurofilaments in Early Symptom Onset Amyotrophic Lateral Sclerosis. Neurology 2018, 90, e22–e30. [Google Scholar] [CrossRef]
- Rossi, D.; Volanti, P.; Brambilla, L.; Colletti, T.; Spataro, R.; La Bella, V. CSF Neurofilament Proteins as Diagnostic and Prognostic Biomarkers for Amyotrophic Lateral Sclerosis. J. Neurol. 2018, 265, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Meeter, L.H.H.; Gendron, T.F.; Sias, A.C.; Jiskoot, L.C.; Russo, S.P.; Donker Kaat, L.; Papma, J.M.; Panman, J.L.; van der Ende, E.L.; Dopper, E.G.; et al. Poly(GP), Neurofilament and Grey Matter Deficits in C9orf72 Expansion Carriers. Ann. Clin. Transl. Neurol. 2018, 5, 583–597. [Google Scholar] [CrossRef]
- Goossens, J.; Bjerke, M.; Van Mossevelde, S.; Van den Bossche, T.; Goeman, J.; De Vil, B.; Sieben, A.; Martin, J.-J.; Cras, P.; De Deyn, P.P.; et al. Diagnostic Value of Cerebrospinal Fluid Tau, Neurofilament, and Progranulin in Definite Frontotemporal Lobar Degeneration. Alzheimers Res. Ther. 2018, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Meeter, L.H.; Dopper, E.G.; Jiskoot, L.C.; Sanchez-Valle, R.; Graff, C.; Benussi, L.; Ghidoni, R.; Pijnenburg, Y.A.; Borroni, B.; Galimberti, D.; et al. Neurofilament Light Chain: A Biomarker for Genetic Frontotemporal Dementia. Ann. Clin. Transl. Neurol. 2016, 3, 623–636. [Google Scholar] [CrossRef]
- Alcolea, D.; Vilaplana, E.; Suárez-Calvet, M.; Illán-Gala, I.; Blesa, R.; Clarimón, J.; Lladó, A.; Sánchez-Valle, R.; Molinuevo, J.L.; García-Ribas, G.; et al. CSF SAPPβ, YKL-40, and Neurofilament Light in Frontotemporal Lobar Degeneration. Neurology 2017, 89, 178–188. [Google Scholar] [CrossRef]
- Van Eijk, J.J.J.; van Everbroeck, B.; Abdo, W.F.; Kremer, B.P.H.; Verbeek, M.M. CSF Neurofilament Proteins Levels Are Elevated in Sporadic Creutzfeldt-Jakob Disease. J. Alzheimers Dis. JAD 2010, 21, 569–576. [Google Scholar] [CrossRef]
- Steinacker, P.; Blennow, K.; Halbgebauer, S.; Shi, S.; Ruf, V.; Oeckl, P.; Giese, A.; Kuhle, J.; Slivarichova, D.; Zetterberg, H.; et al. Neurofilaments in Blood and CSF for Diagnosis and Prediction of Onset in Creutzfeldt-Jakob Disease. Sci. Rep. 2016, 6, 38737. [Google Scholar] [CrossRef] [PubMed]
- Zerr, I.; Schmitz, M.; Karch, A.; Villar-Piqué, A.; Kanata, E.; Golanska, E.; Díaz-Lucena, D.; Karsanidou, A.; Hermann, P.; Knipper, T.; et al. Cerebrospinal Fluid Neurofilament Light Levels in Neurodegenerative Dementia: Evaluation of Diagnostic Accuracy in the Differential Diagnosis of Prion Diseases. Alzheimers Dement. J. Alzheimers Assoc. 2018, 14, 751–763. [Google Scholar] [CrossRef]
- Antonell, A.; Tort-Merino, A.; Ríos, J.; Balasa, M.; Borrego-Écija, S.; Auge, J.M.; Muñoz-García, C.; Bosch, B.; Falgàs, N.; Rami, L.; et al. Synaptic, Axonal Damage and Inflammatory Cerebrospinal Fluid Biomarkers in Neurodegenerative Dementias. Alzheimers Dement. J. Alzheimers Assoc. 2020, 16, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Abu-Rumeileh, S.; Capellari, S.; Stanzani-Maserati, M.; Polischi, B.; Martinelli, P.; Caroppo, P.; Ladogana, A.; Parchi, P. The CSF Neurofilament Light Signature in Rapidly Progressive Neurodegenerative Dementias. Alzheimers Res. Ther. 2018, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Kanata, E.; Golanska, E.; Villar-Piqué, A.; Karsanidou, A.; Dafou, D.; Xanthopoulos, K.; Schmitz, M.; Ferrer, I.; Karch, A.; Sikorska, B.; et al. Cerebrospinal Fluid Neurofilament Light in Suspected Sporadic Creutzfeldt-Jakob Disease. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2019, 60, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.G.B.; Luk, C.; Heslegrave, A.J.; Zetterberg, H.; Mead, S.H.; Collinge, J.; Jackson, G.S. Neurofilament Light Chain and Tau Concentrations Are Markedly Increased in the Serum of Patients with Sporadic Creutzfeldt-Jakob Disease, and Tau Correlates with Rate of Disease Progression. J. Neurol. Neurosurg. Psychiatry 2018, 89, 955–961. [Google Scholar] [CrossRef]
- Thompson, A.G.B.; Anastasiadis, P.; Druyeh, R.; Whitworth, I.; Nayak, A.; Nihat, A.; Mok, T.H.; Rudge, P.; Wadsworth, J.D.F.; Rohrer, J.; et al. Evaluation of Plasma Tau and Neurofilament Light Chain Biomarkers in a 12-Year Clinical Cohort of Human Prion Diseases. Mol. Psychiatry 2021. [Google Scholar] [CrossRef]
- Vallabh, S.M.; Minikel, E.V.; Williams, V.J.; Carlyle, B.C.; McManus, A.J.; Wennick, C.D.; Bolling, A.; Trombetta, B.A.; Urick, D.; Nobuhara, C.K.; et al. Cerebrospinal Fluid and Plasma Biomarkers in Individuals at Risk for Genetic Prion Disease. BMC Med. 2020, 18, 140. [Google Scholar] [CrossRef]
- Llorens, F.; Kruse, N.; Karch, A.; Schmitz, M.; Zafar, S.; Gotzmann, N.; Sun, T.; Köchy, S.; Knipper, T.; Cramm, M.; et al. Validation of α-Synuclein as a CSF Biomarker for Sporadic Creutzfeldt-Jakob Disease. Mol. Neurobiol. 2018, 55, 2249–2257. [Google Scholar] [CrossRef]
- Llorens, F.; Kruse, N.; Schmitz, M.; Gotzmann, N.; Golanska, E.; Thüne, K.; Zejneli, O.; Kanata, E.; Knipper, T.; Cramm, M.; et al. Evaluation of α-Synuclein as a Novel Cerebrospinal Fluid Biomarker in Different Forms of Prion Diseases. Alzheimers Dement. 2017, 13, 710–719. [Google Scholar] [CrossRef]
- Schmitz, M.; Villar-Piqué, A.; Llorens, F.; Gmitterová, K.; Hermann, P.; Varges, D.; Zafar, S.; Lingor, P.; Vanderstichele, H.; Demeyer, L.; et al. Cerebrospinal Fluid Total and Phosphorylated α-Synuclein in Patients with Creutzfeldt–Jakob Disease and Synucleinopathy. Mol. Neurobiol. 2019, 56, 3476–3483. [Google Scholar] [CrossRef]
- Ascari, L.M.; Rocha, S.C.; Gonçalves, P.B.; Vieira, T.C.R.G.; Cordeiro, Y. Challenges and Advances in Antemortem Diagnosis of Human Transmissible Spongiform Encephalopathies. Front. Bioeng. Biotechnol. 2020, 8, 585896. [Google Scholar] [CrossRef]
- Aksamit, A.J.; Preissner, C.M.; Homburger, H.A. Quantitation of 14-3-3 and Neuron-Specific Enolase Proteins in CSF in Creutzfeldt-Jakob Disease. Neurology 2001, 57, 728–730. [Google Scholar] [CrossRef]
- Zerr, I.; Bodemer, M.; Räcker, S.; Grosche, S.; Poser, S.; Weber, T.; Kretzschmar, H.A. Cerebrospinal Fluid Concentration of Neuron-Specific Enolase in Diagnosis of Creutzfeldt-Jakob Disease. Lancet 1995, 345, 1609–1610. [Google Scholar] [CrossRef]
- Otto, M.; Wiltfang, J.; Schutz, E.; Zerr, I.; Otto, A.; Pfahlberg, A.; Gefeller, O.; Uhr, M.; Giese, A.; Weber, T.; et al. Diagnosis of Creutzfeldt-Jakob Disease by Measurement of S100 Protein in Serum: Prospective Case-Control Study. BMJ 1998, 316, 577–582. [Google Scholar] [CrossRef]
- Ladogana, A.; Sanchez-Juan, P.; Mitrová, E.; Green, A.; Cuadrado-Corrales, N.; Sánchez-Valle, R.; Koscova, S.; Aguzzi, A.; Sklaviadis, T.; Kulczycki, J.; et al. Cerebrospinal Fluid Biomarkers in Human Genetic Transmissible Spongiform Encephalopathies. J. Neurol. 2009, 256, 1620–1628. [Google Scholar] [CrossRef]
- Krasnianski, A.; Meissner, B.; Schulz-Schaeffer, W.; Kallenberg, K.; Bartl, M.; Heinemann, U.; Varges, D.; Kretzschmar, H.A.; Zerr, I. Clinical Features and Diagnosis of the MM2 Cortical Subtype of Sporadic Creutzfeldt-Jakob Disease. Arch. Neurol. 2006, 63, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Le Pera, M.; Urso, E.; Sprovieri, T.; Bossio, S.; Aguglia, U.; Manna, I.; Cupidi, C.; Ferraro, T.; Gambardella, A.; Qualtieri, A.; et al. Contribution of Cerebrospinal Fluid Thymosin Β4 Levels to the Clinical Differentiation of Creutzfeldt-Jakob Disease. Arch. Neurol. 2012, 69, 868–872. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Steinhoff, B.J.; Räcker, S.; Herrendorf, G.; Poser, S.; Grosche, S.; Zerr, I.; Kretzschmar, H.; Weber, T. Accuracy and Reliability of Periodic Sharp Wave Complexes in Creutzfeldt-Jakob Disease. Arch. Neurol. 1996, 53, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Zerr, I.; Kallenberg, K.; Summers, D.M.; Romero, C.; Taratuto, A.; Heinemann, U.; Breithaupt, M.; Varges, D.; Meissner, B.; Ladogana, A.; et al. Updated Clinical Diagnostic Criteria for Sporadic Creutzfeldt-Jakob Disease. Brain 2009, 132, 2659–2668. [Google Scholar] [CrossRef] [PubMed]
- Bizzi, A.; Pascuzzo, R.; Blevins, J.; Grisoli, M.; Lodi, R.; Moscatelli, M.E.M.; Castelli, G.; Cohen, M.L.; Schonberger, L.B.; Foutz, A.; et al. Evaluation of a New Criterion for Detecting Prion Disease with Diffusion Magnetic Resonance Imaging. JAMA Neurol. 2020, 77, 1141. [Google Scholar] [CrossRef] [PubMed]
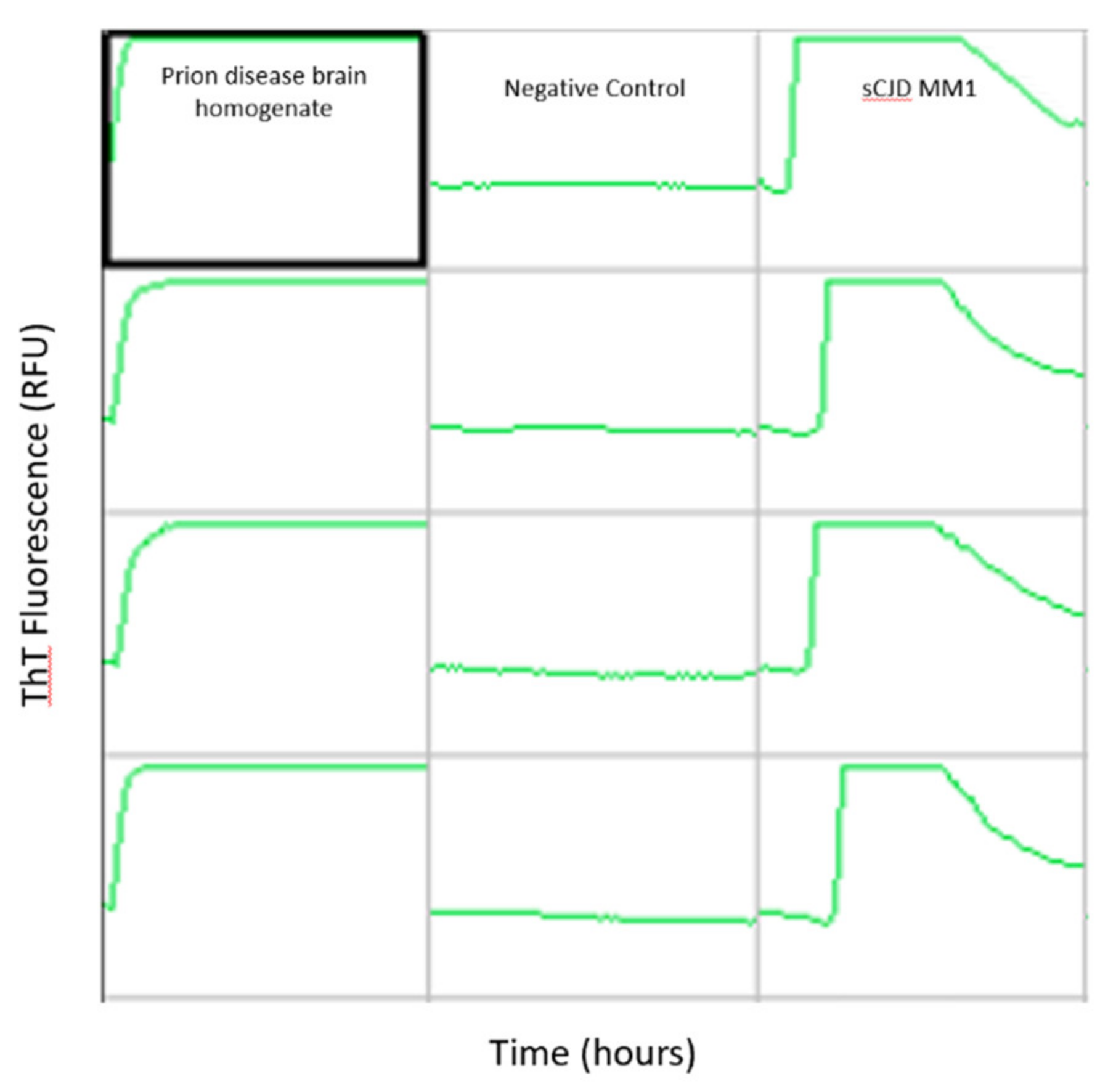
- Foutz, A.; Appleby, B.S.; Hamlin, C.; Liu, X.; Yang, S.; Cohen, Y.; Chen, W.; Blevins, J.; Fausett, C.; Wang, H.; et al. Diagnostic and Prognostic Value of Human Prion Detection in Cerebrospinal Fluid: CSF RT-QuIC Test. Ann. Neurol. 2017, 81, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Baiardi, S.; Hughson, A.G.; McKenzie, N.; Moda, F.; Rossi, M.; Capellari, S.; Green, A.; Giaccone, G.; Caughey, B.; et al. High Diagnostic Value of Second Generation CSF RT-QuIC across the Wide Spectrum of CJD Prions. Sci. Rep. 2017, 7, 10655. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, D.D.; Wrona, A.; Foutz, A.; Blevins, J.; Glisic, K.; Person, M.; Maddox, R.A.; Belay, E.D.; Schonberger, L.B.; Tatsuoka, C.; et al. Diagnosis of Prion Diseases by RT-QuIC Results in Improved Surveillance. Neurology 2020, 95, e1017–e1026. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Hua, Y.X.; Quan, Z.W.; Ping, D.X.; Qi, S. Assessment of the Sensitivity and Specificity of the Established Real-Time Quaking-Induced Conversion (RT-QuIC) Technique in Chinese CJD Surveillance. Biomed. Environ. Sci. 2020, 33, 620–622. [Google Scholar]
- Appleby, B.S.; Yobs, D.R. Symptomatic Treatment, Care, and Support of CJD Patients. Handb. Clin. Neurol. 2018, 153, 399–408. [Google Scholar] [CrossRef]
- Gmitterová, K.; Heinemann, U.; Bodemer, M.; Krasnianski, A.; Meissner, B.; Kretzschmar, H.A.; Zerr, I. 14-3-3 CSF Levels in Sporadic Creutzfeldt–Jakob Disease Differ across Molecular Subtypes. Neurobiol. Aging 2009, 30, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Shirabe, S.; Tsujino, A.; Eguchi, H.; Motomura, M.; Honda, H.; Tomita, I.; Satoh, A.; Tsujihata, M.; Matsuo, H.; et al. Total Tau Protein in Cerebrospinal Fluid and Diffusion-Weighted MRI as an Early Diagnostic Marker for Creutzfeldt-Jakob Disease. Dement. Geriatr. Cogn. Disord. 2007, 24, 207–212. [Google Scholar] [CrossRef]
- Sanchez-Juan, P.; Sánchez-Valle, R.; Green, A.; Ladogana, A.; Cuadrado-Corrales, N.; Mitrová, E.; Stoeck, K.; Sklaviadis, T.; Kulczycki, J.; Hess, K.; et al. Influence of Timing on CSF Tests Value for Creutzfeldt-Jakob Disease Diagnosis. J. Neurol. 2007, 254, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Savard, M.; Irani, S.R.; Guillemette, A.; Gosselin-Lefebvre, S.; Geschwind, M.; Jansen, G.H.; Gould, P.V.; Laforce, R. Creutzfeldt-Jakob Disease-Like Periodic Sharp Wave Complexes in Voltage-Gated Potassium Channel-Complex Antibodies Encephalitis: A Case Report. J. Clin. Neurophysiol. Off. Publ. Am. Electroencephalogr. Soc. 2016, 33, e1–e4. [Google Scholar] [CrossRef]
- Wieser, H.; Schindler, K.; Zumsteg, D. EEG in Creutzfeldt–Jakob Disease. Clin. Neurophysiol. 2006, 117, 935–951. [Google Scholar] [CrossRef]
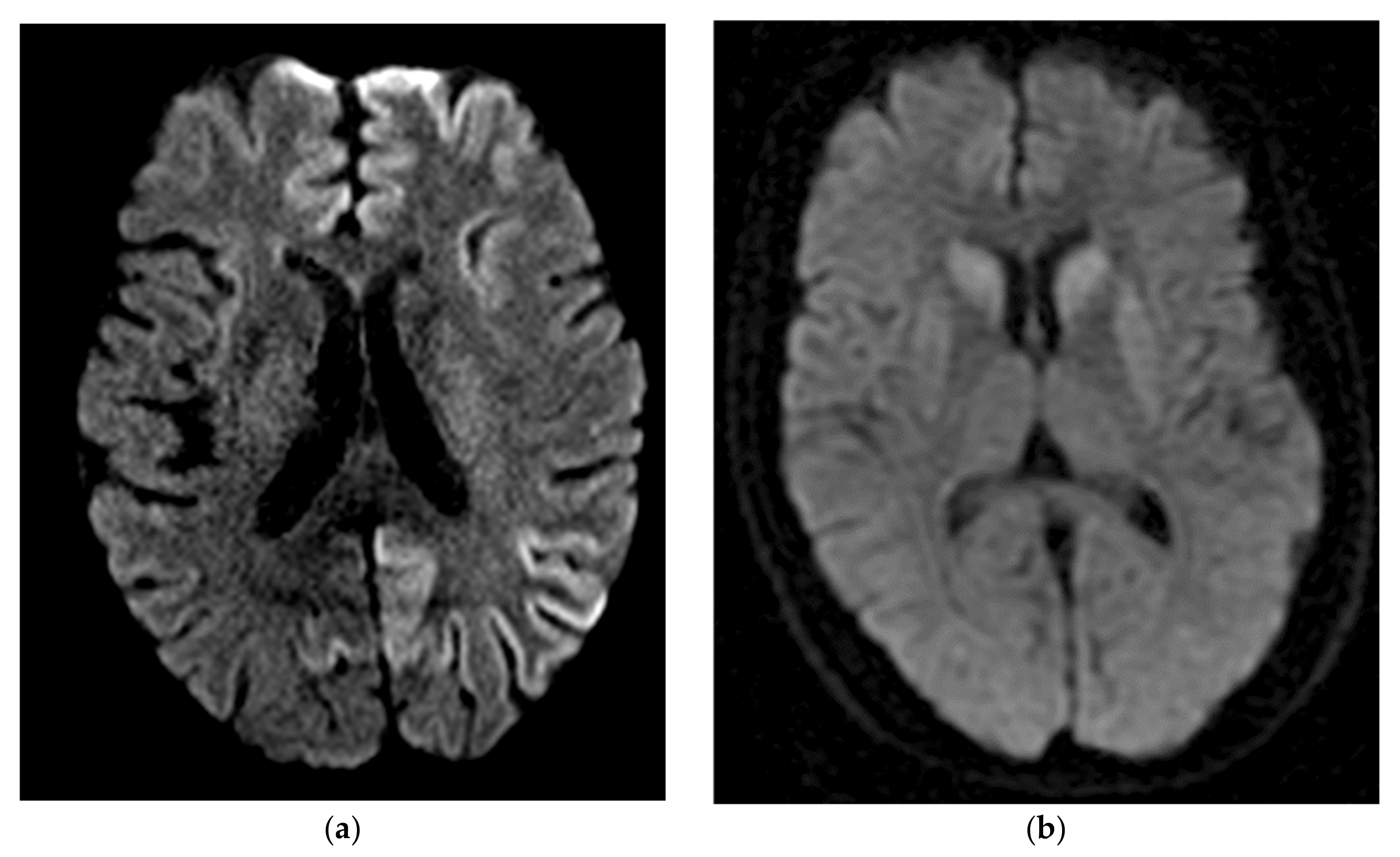
- Vitali, P.; Maccagnano, E.; Caverzasi, E.; Henry, R.G.; Haman, A.; Torres-Chae, C.; Johnson, D.Y.; Miller, B.L.; Geschwind, M.D. Diffusion-Weighted MRI Hyperintensity Patterns Differentiate CJD from Other Rapid Dementias. Neurology 2011, 76, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Zeidler, M.; Sellar, R.J.; Collie, D.A.; Knight, R.; Stewart, G.; Macleod, M.A.; Ironside, J.W.; Cousens, S.; Colchester, A.C.; Hadley, D.M.; et al. The Pulvinar Sign on Magnetic Resonance Imaging in Variant Creutzfeldt-Jakob Disease. Lancet 2000, 355, 1412–1418. [Google Scholar] [CrossRef]
- Collie, D.A.; Summers, D.M.; Sellar, R.J.; Ironside, J.W.; Cooper, S.; Zeidler, M.; Knight, R.; Will, R.G. Diagnosing Variant Creutzfeldt-Jakob Disease with the Pulvinar Sign: MR Imaging Findings in 86 Neuropathologically Confirmed Cases. AJNR Am. J. Neuroradiol. 2003, 24, 1560–1569. [Google Scholar]
- Rudge, P.; Hyare, H.; Green, A.; Collinge, J.; Mead, S. Imaging and CSF Analyses Effectively Distinguish CJD from Its Mimics. J. Neurol. Neurosurg. Psychiatry 2018, 89, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Chitravas, N.; Jung, R.S.; Kofskey, D.M.; Blevins, J.E.; Gambetti, P.; Leigh, R.J.; Cohen, M.L. Treatable Neurological Disorders Misdiagnosed as Creutzfeldt-Jakob Disease. Ann. Neurol. 2011, 70, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Bizzi, A.; Pascuzzo, R.; Blevins, J.; Moscatelli, M.E.M.; Grisoli, M.; Lodi, R.; Doniselli, F.M.; Castelli, G.; Cohen, M.L.; Stamm, A.; et al. Subtype Diagnosis of Sporadic CREUTZFELDT–JAKOB Disease with Diffusion MAGNETIC RESONANCE IMAGING. Ann. Neurol. 2021, 89, 560–572. [Google Scholar] [CrossRef]
- Meissner, B.; Kallenberg, K.; Sanchez-Juan, P.; Collie, D.; Summers, D.M.; Almonti, S.; Collins, S.J.; Smith, P.; Cras, P.; Jansen, G.H.; et al. MRI Lesion Profiles in Sporadic Creutzfeldt-Jakob Disease. Neurology 2009, 72, 1994–2001. [Google Scholar] [CrossRef]
- Green, A.J.E. RT-QuIC: A New Test for Sporadic CJD. Pract. Neurol. 2019, 19, 49–55. [Google Scholar] [CrossRef]
- Zerr, I.; Cramm, M.; da Silva Correia, S.M.; Zafar, S.; Villar-Piqué, A.; Llorens, F.; Schmitz, M. Optimization of the Real-Time Quaking-Induced Conversion Assay for Prion Disease Diagnosis. Front. Bioeng. Biotechnol. 2020, 8, 586890. [Google Scholar] [CrossRef]
- Hermann, P.; Appleby, B.; Brandel, J.-P.; Caughey, B.; Collins, S.; Geschwind, M.D.; Green, A.; Haïk, S.; Kovacs, G.G.; Ladogana, A.; et al. Biomarkers and Diagnostic Guidelines for Sporadic Creutzfeldt-Jakob Disease. Lancet Neurol. 2021, 20, 235–246. [Google Scholar] [CrossRef]
- Orrú, C.D.; Yuan, J.; Appleby, B.S.; Li, B.; Li, Y.; Winner, D.; Wang, Z.; Zhan, Y.-A.; Rodgers, M.; Rarick, J.; et al. Prion Seeding Activity and Infectivity in Skin Samples from Patients with Sporadic Creutzfeldt-Jakob Disease. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Orrù, C.D.; Soldau, K.; Cordano, C.; Llibre-Guerra, J.; Green, A.J.; Sanchez, H.; Groveman, B.R.; Edland, S.D.; Safar, J.G.; Lin, J.H.; et al. Prion Seeds Distribute throughout the Eyes of Sporadic Creutzfeldt-Jakob Disease Patients. mBio 2018, 9. [Google Scholar] [CrossRef]
- Quadrio, I.; Ugnon-Café, S.; Dupin, M.; Esposito, G.; Streichenberger, N.; Krolak-Salmon, P.; Vital, A.; Pellissier, J.-F.; Perret-Liaudet, A.; Perron, H. Rapid Diagnosis of Human Prion Disease Using Streptomycin with Tonsil and Brain Tissues. Lab. Investig. J. Tech. Methods Pathol. 2009, 89, 406–413. [Google Scholar] [CrossRef]
- Glatzel, M.; Abela, E.; Maissen, M.; Aguzzi, A. Extraneural Pathologic Prion Protein in Sporadic Creutzfeldt-Jakob Disease. N. Engl. J. Med. 2003, 349, 1812–1820. [Google Scholar] [CrossRef] [PubMed]
- Favereaux, A.; Quadrio, I.; Vital, C.; Perret-Liaudet, A.; Anne, O.; Laplanche, J.-L.; Petry, K.G.; Vital, A. Pathologic Prion Protein Spreading in the Peripheral Nervous System of a Patient with Sporadic Creutzfeldt-Jakob Disease. Arch. Neurol. 2004, 61, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Head, M.W.; Kouverianou, E.; Taylor, L.; Green, A.; Knight, R. Evaluation of Urinary PrPSc as a Diagnostic Test for Sporadic, Variant, and Familial CJD. Neurology 2005, 64, 1794–1796. [Google Scholar] [CrossRef]
- Zanusso, G.; Ferrari, S.; Cardone, F.; Zampieri, P.; Gelati, M.; Fiorini, M.; Farinazzo, A.; Gardiman, M.; Cavallaro, T.; Bentivoglio, M.; et al. Detection of Pathologic Prion Protein in the Olfactory Epithelium in Sporadic Creutzfeldt-Jakob Disease. N. Engl. J. Med. 2003, 348, 711–719. [Google Scholar] [CrossRef]
- Shaked, G.M.; Shaked, Y.; Kariv-Inbal, Z.; Halimi, M.; Avraham, I.; Gabizon, R. A Protease-Resistant Prion Protein Isoform Is Present in Urine of Animals and Humans Affected with Prion Diseases. J. Biol. Chem. 2001, 276, 31479–31482. [Google Scholar] [CrossRef] [PubMed]
- Orrú, C.D.; Bongianni, M.; Tonoli, G.; Ferrari, S.; Hughson, A.G.; Groveman, B.R.; Fiorini, M.; Pocchiari, M.; Monaco, S.; Caughey, B.; et al. A Test for Creutzfeldt-Jakob Disease Using Nasal Brushings. N. Engl. J. Med. 2014, 371, 519–529. [Google Scholar] [CrossRef]
- Bongianni, M.; Orrù, C.; Groveman, B.R.; Sacchetto, L.; Fiorini, M.; Tonoli, G.; Triva, G.; Capaldi, S.; Testi, S.; Ferrari, S.; et al. Diagnosis of Human Prion Disease Using Real-Time Quaking-Induced Conversion Testing of Olfactory Mucosa and Cerebrospinal Fluid Samples. JAMA Neurol. 2017, 74, 155–162. [Google Scholar] [CrossRef]
- Mammana, A.; Baiardi, S.; Rossi, M.; Franceschini, A.; Donadio, V.; Capellari, S.; Caughey, B.; Parchi, P. Detection of Prions in Skin Punch Biopsies of Creutzfeldt-Jakob Disease Patients. Ann. Clin. Transl. Neurol. 2020, 7, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Medori, R.; Tritschler, H.J.; LeBlanc, A.; Villare, F.; Manetto, V.; Chen, H.Y.; Xue, R.; Leal, S.; Montagna, P.; Cortelli, P. Fatal Familial Insomnia, a Prion Disease with a Mutation at Codon 178 of the Prion Protein Gene. N. Engl. J. Med. 1992, 326, 444–449. [Google Scholar] [CrossRef]
- Minikel, E.V.; Vallabh, S.M.; Lek, M.; Estrada, K.; Samocha, K.E.; Sathirapongsasuti, J.F.; McLean, C.Y.; Tung, J.Y.; Yu, L.P.C.; Gambetti, P.; et al. Quantifying Prion Disease Penetrance Using Large Population Control Cohorts. Sci. Transl. Med. 2016, 8, 322ra9. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.S.; Dryden, A.J.; Hughes, J.T.; Collinge, J. Homozygous Prion Protein Genotype Predisposes to Sporadic Creutzfeldt-Jakob Disease. Nature 1991, 352, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Mead, S.; Burnell, M.; Lowe, J.; Thompson, A.; Lukic, A.; Porter, M.-C.; Carswell, C.; Kaski, D.; Kenny, J.; Mok, T.H.; et al. Clinical Trial Simulations Based on Genetic Stratification and the Natural History of a Functional Outcome Measure in Creutzfeldt-Jakob Disease. JAMA Neurol. 2016, 73, 447. [Google Scholar] [CrossRef] [PubMed]
- Carswell, C.; Thompson, A.; Lukic, A.; Stevens, J.; Rudge, P.; Mead, S.; Collinge, J.; Hyare, H. MRI Findings Are Often Missed in the Diagnosis of Creutzfeldt-Jakob Disease. BMC Neurol. 2012, 12, 153. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Yu, M.M.; Strutt, A.M. Variably Protease-Sensitive Prionopathy: A Differential Diagnostic Consideration for Dementia. Neurol. Clin. Pract. 2019, 9, 145–151. [Google Scholar] [CrossRef]
- Zou, W.-Q.; Puoti, G.; Xiao, X.; Yuan, J.; Qing, L.; Cali, I.; Shimoji, M.; Langeveld, J.P.M.; Castellani, R.; Notari, S.; et al. Variably Protease-Sensitive Prionopathy: A New Sporadic Disease of the Prion Protein. Ann. Neurol. 2010, 68, 162–172. [Google Scholar] [CrossRef]
- Head, M.W.; Yull, H.M.; Ritchie, D.L.; Langeveld, J.P.; Fletcher, N.A.; Knight, R.S.; Ironside, J.W. Variably Protease-Sensitive Prionopathy in the UK: A Retrospective Review 1991–2008. Brain J. Neurol. 2013, 136, 1102–1115. [Google Scholar] [CrossRef]
- Aizpurua, M.; Selvackadunco, S.; Yull, H.; Kipps, C.M.; Ironside, J.W.; Bodi, I. Variably Protease-sensitive Prionopathy Mimicking Frontotemporal Dementia. Neuropathology 2019, 39, 135–140. [Google Scholar] [CrossRef]
- Gambetti, P.; Dong, Z.; Yuan, J.; Xiao, X.; Zheng, M.; Alshekhlee, A.; Castellani, R.; Cohen, M.; Barria, M.A.; Gonzalez-Romero, D.; et al. A Novel Human Disease with Abnormal Prion Protein Sensitive to Protease. Ann. Neurol. 2008, 63, 697–708. [Google Scholar] [CrossRef]
- Jansen, C.; Head, M.W.; van Gool, W.A.; Baas, F.; Yull, H.; Ironside, J.W.; Rozemuller, A.J.M. The First Case of Protease-Sensitive Prionopathy (PSPr) in The Netherlands: A Patient with an Unusual GSS-like Clinical Phenotype. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1052–1055. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.-Q.; Gambetti, P.; Xiao, X.; Yuan, J.; Langeveld, J.; Pirisinu, L. Prions in Variably Protease-Sensitive Prionopathy: An Update. Pathogens 2013, 2, 457–471. [Google Scholar] [CrossRef]
- Plazzi, G.; Schutz, Y.; Cortelli, P.; Provini, F.; Avoni, P.; Heikkila, E.; Tinuper, P.; Solieri, L.; Lugaresi, E.; Montagna, P. Motor Overactivity and Loss of Motor Circadian Rhythm in Fatal Familial Insomnia: An Actigraphic Study. Sleep 1997, 20, 739–742. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gallassi, R.; Morreale, A.; Montagna, P.; Gambetti, P.; Lugaresi, E. “Fatal Familial Insomnia”: Neuropsychological Study of a Disease with Thalamic Degeneration. Cortex J. Devoted Study Nerv. Syst. Behav. 1992, 28, 175–187. [Google Scholar] [CrossRef]
- Pedroso, J.L.; Pinto, W.B.V.D.R.; De Souza, P.V.S.; Ricarte, I.F.; Landemberger, M.C.; Martins, V.R.; Prado, L.B.; Prado, G.F.; Barsottini, O.G. Complex movement disorders in fatal familial insomnia: A clinical and genetic discussion. Neurology 2013, 81, 1098–1099. [Google Scholar] [CrossRef] [PubMed]
- Cortelli, P.; Fabbri, M.; Calandra-Buonaura, G.; Capellari, S.; Tinuper, P.; Parchi, P.; Lugaresi, E. Gait Disorders in Fatal Familial Insomnia. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 420–424. [Google Scholar] [CrossRef]
- Manetto, V.; Medori, R.; Cortelli, P.; Montagna, P.; Tinuper, P.; Baruzzi, A.; Rancurel, G.; Hauw, J.J.; Vanderhaeghen, J.J.; Mailleux, P. Fatal Familial Insomnia: Clinical and Pathologic Study of Five New Cases. Neurology 1992, 42, 312–319. [Google Scholar] [CrossRef]
- Lugaresi, E.; Medori, R.; Montagna, P.; Baruzzi, A.; Cortelli, P.; Lugaresi, A.; Tinuper, P.; Zucconi, M.; Gambetti, P. Fatal Familial Insomnia and Dysautonomia with Selective Degeneration of Thalamic Nuclei. N. Engl. J. Med. 1986, 315, 997–1003. [Google Scholar] [CrossRef]
- Zerr, I.; Giese, A.; Windl, O.; Kropp, S.; Schulz-Schaeffer, W.; Riedemann, C.; Skworc, K.; Bodemer, M.; Kretzschmar, H.A.; Poser, S. Phenotypic Variability in Fatal Familial Insomnia (D178N-129M) Genotype. Neurology 1998, 51, 1398–1405. [Google Scholar] [CrossRef]
- Cortelli, P.; Perani, D.; Montagna, P.; Gallassi, R.; Tinuper, P.; Provini, F.; Federica, P.; Avoni, P.; Ferrillo, F.; Anchisi, D.; et al. Pre-Symptomatic Diagnosis in Fatal Familial Insomnia: Serial Neurophysiological and 18FDG-PET Studies. Brain J. Neurol. 2006, 129, 668–675. [Google Scholar] [CrossRef]
- Perani, D.; Cortelli, P.; Lucignani, G.; Montagna, P.; Tinuper, P.; Gallassi, R.; Gambetti, P.; Lenzi, G.L.; Lugaresi, E.; Fazio, F. [18F]FDG PET in Fatal Familial Insomnia: The Functional Effects of Thalamic Lesions. Neurology 1993, 43, 2565–2569. [Google Scholar] [CrossRef] [PubMed]
- Cortelli, P.; Perani, D.; Parchi, P.; Grassi, F.; Montagna, P.; De Martin, M.; Castellani, R.; Tinuper, P.; Gambetti, P.; Lugaresi, E.; et al. Cerebral Metabolism in Fatal Familial Insomnia: Relation to Duration, Neuropathology, and Distribution of Protease-Resistant Prion Protein. Neurology 1997, 49, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Krasnianski, A.; Bartl, M.; Sanchez Juan, P.J.; Heinemann, U.; Meissner, B.; Varges, D.; Schulze-Sturm, U.; Kretzschmar, H.A.; Schulz-Schaeffer, W.J.; Zerr, I. Fatal Familial Insomnia: Clinical Features and Early Identification. Ann. Neurol. 2008, 63, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Abu-Rumeileh, S.; Redaelli, V.; Baiardi, S.; Mackenzie, G.; Windl, O.; Ritchie, D.L.; Didato, G.; Hernandez-Vara, J.; Rossi, M.; Capellari, S.; et al. Sporadic Fatal Insomnia in Europe: Phenotypic Features and Diagnostic Challenges: SFI in Europe. Ann. Neurol. 2018, 84, 347–360. [Google Scholar] [CrossRef]
- Krasnianski, A.; Kallenberg, K.; Collie, D.A.; Meissner, B.; Schulz-Schaeffer, W.J.; Heinemann, U.; Varges, D.; Summers, D.M.; Kretzschmar, H.A.; Talbot, T.; et al. MRI in the Classical MM1 and the Atypical MV2 Subtypes of Sporadic CJD: An Inter-Observer Agreement Study. Eur. J. Neurol. 2008, 15, 762–771. [Google Scholar] [CrossRef]
- Meissner, B.; Westner, I.M.; Kallenberg, K.; Krasnianski, A.; Bartl, M.; Varges, D.; Bosenberg, C.; Kretzschmar, H.A.; Knauth, M.; Schulz-Schaeffer, W.J.; et al. Sporadic Creutzfeldt-Jakob Disease: Clinical and Diagnostic Characteristics of the Rare VV1 Type. Neurology 2005, 65, 1544–1550. [Google Scholar] [CrossRef] [PubMed]
- Goodall, C.A.; Head, M.W.; Everington, D.; Ironside, J.W.; Knight, R.S.G.; Green, A.J.E. Raised CSF Phospho-Tau Concentrations in Variant Creutzfeldt-Jakob Disease: Diagnostic and Pathological Implications. J. Neurol. Neurosurg. Psychiatry 2006, 77, 89–91. [Google Scholar] [CrossRef][Green Version]
- Green, A.J.; Thompson, E.J.; Stewart, G.E.; Zeidler, M.; McKenzie, J.M.; MacLeod, M.A.; Ironside, J.W.; Will, R.G.; Knight, R.S. Use of 14-3-3 and Other Brain-Specific Proteins in CSF in the Diagnosis of Variant Creutzfeldt-Jakob Disease. J. Neurol. Neurosurg. Psychiatry 2001, 70, 744–748. [Google Scholar] [CrossRef]
- Binelli, S.; Agazzi, P.; Giaccone, G.; Will, R.G.; Bugiani, O.; Franceschetti, S.; Tagliavini, F. Periodic Electroencephalogram Complexes in a Patient with Variant Creutzfeldt-Jakob Disease. Ann. Neurol. 2006, 59, 423–427. [Google Scholar] [CrossRef]
- Will, R.G.; Zeidler, M.; Stewart, G.E.; Macleod, M.A.; Ironside, J.W.; Cousens, S.N.; Mackenzie, J.; Estibeiro, K.; Green, A.J.; Knight, R.S. Diagnosis of New Variant Creutzfeldt-Jakob Disease. Ann. Neurol. 2000, 47, 575–582. [Google Scholar] [CrossRef]
- Hill, A.F.; Zeidler, M.; Ironside, J.; Collinge, J. Diagnosis of New Variant Creutzfeldt-Jakob Disease by Tonsil Biopsy. Lancet 1997, 349, 99–100. [Google Scholar] [CrossRef]
- Wadsworth, J.D.; Joiner, S.; Hill, A.F.; Campbell, T.A.; Desbruslais, M.; Luthert, P.J.; Collinge, J. Tissue Distribution of Protease Resistant Prion Protein in Variant Creutzfeldt-Jakob Disease Using a Highly Sensitive Immunoblotting Assay. Lancet 2001, 358, 171–180. [Google Scholar] [CrossRef]
- Williams, E.S.; Young, S. Chronic Wasting Disease of Captive Mule Deer: A Spongiform Encephalopathy. J. Wildl. Dis. 1980, 16, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.S.; Miller, M.W.; Kreeger, T.J.; Kahn, R.H.; Thorne, E.T. Chronic Wasting Disease of Deer and Elk: A Review with Recommendations for Management. J. Wildl. Manag. 2002, 66, 551. [Google Scholar] [CrossRef]
- Williams, E.S.; Young, S. Spongiform Encephalopathy of Rocky Mountain Elk. J. Wildl. Dis. 1982, 18, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.S.; Young, S. Spongiform Encephalopathies in Cervidae. Rev. Sci. Tech. Int. Off. Epizoot. 1992, 11, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Chronic Wasting Disease (CWD). Available online: https://www.cdc.gov/prions/cwd/index.html (accessed on 19 February 2021).
- Pritzkow, S.; Morales, R.; Moda, F.; Khan, U.; Telling, G.C.; Hoover, E.; Soto, C. Grass Plants Bind, Retain, Uptake, and Transport Infectious Prions. Cell Rep. 2015, 11, 1168–1175. [Google Scholar] [CrossRef]
- Barria, M.A.; Libori, A.; Mitchell, G.; Head, M.W. Susceptibility of Human Prion Protein to Conversion by Chronic Wasting Disease Prions. Emerg. Infect. Dis. 2018, 24, 1482–1489. [Google Scholar] [CrossRef]
- Morales, R. Prion Strains in Mammals: Different Conformations Leading to Disease. PLoS Pathog. 2017, 13, e1006323. [Google Scholar] [CrossRef]
- Zabel, M.D.; Reid, C. A Brief History of Prions. Pathog. Dis. 2015, 73, ftv087. [Google Scholar] [CrossRef] [PubMed]
- Duque Velásquez, C.; Kim, C.; Herbst, A.; Daude, N.; Garza, M.C.; Wille, H.; Aiken, J.; McKenzie, D. Deer Prion Proteins Modulate the Emergence and Adaptation of Chronic Wasting Disease Strains. J. Virol. 2015, 89, 12362–12373. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.; Velásquez, C.D.; Triscott, E.; Aiken, J.M.; McKenzie, D. Chronic Wasting Disease Prion Strain Emergence and Host Range Expansion. Emerg. Infect. Dis. 2017, 23, 1598–1600. [Google Scholar] [CrossRef] [PubMed]
- Waddell, L.; Greig, J.; Mascarenhas, M.; Otten, A.; Corrin, T.; Hierlihy, K. Current Evidence on the Transmissibility of Chronic Wasting Disease Prions to Humans-A Systematic Review. Transbound. Emerg. Dis. 2018, 65, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Raymond, G.J.; Raymond, L.D.; Meade-White, K.D.; Hughson, A.G.; Favara, C.; Gardner, D.; Williams, E.S.; Miller, M.W.; Race, R.E.; Caughey, B. Transmission and Adaptation of Chronic Wasting Disease to Hamsters and Transgenic Mice: Evidence for Strains. J. Virol. 2007, 81, 4305–4314. [Google Scholar] [CrossRef]
- Sigurdson, C.J.; Manco, G.; Schwarz, P.; Liberski, P.; Hoover, E.A.; Hornemann, S.; Polymenidou, M.; Miller, M.W.; Glatzel, M.; Aguzzi, A. Strain Fidelity of Chronic Wasting Disease upon Murine Adaptation. J. Virol. 2006, 80, 12303–12311. [Google Scholar] [CrossRef]
- Hamir, A.N.; Kunkle, R.A.; Cutlip, R.C.; Miller, J.M.; O’Rourke, K.I.; Williams, E.S.; Miller, M.W.; Stack, M.J.; Chaplin, M.J.; Richt, J.A. Experimental Transmission of Chronic Wasting Disease Agent from Mule Deer to Cattle by the Intracerebral Route. J. Veter. Diagn. Investig. 2005, 17, 276–281. [Google Scholar] [CrossRef]
- Hamir, A.N.; Kunkle, R.A.; Cutlip, R.C.; Miller, J.M.; Williams, E.S.; Richt, J.A. Transmission of Chronic Wasting Disease of Mule Deer to Suffolk Sheep following Intracerebral Inoculation. J. Veter. Diagn. Investig. 2006, 18, 558–565. [Google Scholar] [CrossRef]
- Race, B.; Meade-White, K.D.; Miller, M.W.; Barbian, K.D.; Rubenstein, R.; LaFauci, G.; Cervenakova, L.; Favara, C.; Gardner, D.; Long, D.; et al. Susceptibilities of Nonhuman Primates to Chronic Wasting Disease. Emerg. Infect. Dis. 2009, 15, 1366–1376. [Google Scholar] [CrossRef]
- Kong, Q.; Huang, S.; Zou, W.; Vanegas, D.; Wang, M.; Wu, D.; Yuan, J.; Zheng, M.; Bai, H.; Deng, H.; et al. Chronic Wasting Disease of Elk: Transmissibility to Humans Examined by Transgenic Mouse Models. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 7944–7949. [Google Scholar] [CrossRef]
- Tamgüney, G.; Giles, K.; Bouzamondo-Bernstein, E.; Bosque, P.J.; Miller, M.W.; Safar, J.; DeArmond, S.J.; Prusiner, S.B. Transmission of Elk and Deer Prions to Transgenic Mice. J. Virol. 2006, 80, 9104–9114. [Google Scholar] [CrossRef] [PubMed]
- Race, B.; Williams, K.; Orru, C.D.; Hughson, A.G.; Lubke, L.; Chesebro, B. Lack of Transmission of Chronic Wasting Disease to Cynomolgus Macaques. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Czub, S.; Schulz-Schaeffer, W.; Stahl-Hennig, C.; Beekes, M.; Schaetz, H.; Motzkus, D. First Evidence of Intracranial and Peroral Transmission of Chronic Wasting Disease (CWD) into Cynomolgus Macaques: A Work in Progress. Available online: https://cjdfoundation.org/files/pdf/CWD%20study%20oral%20transmission%20of%20CWD%20to%20primates.pdf (accessed on 24 February 2021).
- Haley, N.J.; Hoover, E.A. Chronic Wasting Disease of Cervids: Current Knowledge and Future Perspectives. Annu. Rev. Anim. Biosci. 2015, 3, 305–325. [Google Scholar] [CrossRef]
- Haley, N.J.; Siepker, C.; Walter, W.D.; Thomsen, B.V.; Greenlee, J.J.; Lehmkuhl, A.D.; Richt, J.A. Antemortem Detection of Chronic Wasting Disease Prions in Nasal Brush Collections and Rectal Biopsy Specimens from White-Tailed Deer by Real-Time Quaking-Induced Conversion. J. Clin. Microbiol. 2016, 54, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.C.; Charco, J.M.; Plagenz, J.; Orru, C.D.; Denkers, N.D.; Metrick, M.A.; Hughson, A.G.; Griffin, K.A.; Race, B.; Hoover, E.A.; et al. Detection of Chronic Wasting Disease in Mule and White-Tailed Deer by RT-QuIC Analysis of Outer Ear. Sci. Rep. 2021, 11, 7702. [Google Scholar] [CrossRef] [PubMed]
- Bodin, M. Chronic Wasting Disease: Real Risk or Irrational Hype? Available online: https://undark.org/2018/04/18/chronic-wasting-disease-cwd/ (accessed on 20 February 2021).
- Osterholm, M.T.; Anderson, C.J.; Zabel, M.D.; Scheftel, J.M.; Moore, K.A.; Appleby, B.S. Chronic Wasting Disease in Cervids: Implications for Prion Transmission to Humans and Other Animal Species. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Definite CJD Requires Neuropathological Diagnosis, Immunohistochemical Confirmation, Tissue Western Blotting for Proteinase-Resistant PrP, or the Presence of Scrapie-Associated Fibrils. |
|---|
Probable CJD criteria are fulfilled by a neuropsychiatric disorder plus a positive real-time quaking-induced conversion (RT-QuIC) test OR a rapidly progressive dementia with at least 2 of the 4 following clinical symptom criteria:
|
Possible CJD is defined as a progressive dementia with at least 2 of 4 of the following criteria:
AND duration of illness less than two years AND without routine investigations indicating an alternative diagnosis |
| Iatrogenic CJD is defined as a progressive cerebellar syndrome in a patient who received human cadaver-derived pituitary hormone or sCJD with a known high-risk exposure |
| Genetic CJD is classified as definite/probable CJD plus a first-degree relative with definite/probable CJD or a neuropsychiatric disorder with a disease-specific PRNP mutation. |
| Test | Sensitivity | Specificity | References |
|---|---|---|---|
| Non-specific biomarkers | |||
| CSF 14-3-3 | 85–95% | 40–100% | [19,20,21,22,23,24,25,26,27,28,29] |
| CSF Tau | 67–91% | 87–95% | [20,21,22,28,29,34,35,36] |
| CSF T-tau/P-tau ratio | 75–94% | 94–97% | [20,37,38,39] |
| Serum/plasma Tau | 57–91% | 83–97% | [61,66,67] |
| CSF NfL | 86–97% | 43–95% | [34,61,64] |
| Serum/plasma NfL | 93–100% | 57–100% | [61,66,67] |
| Alpha-synuclein | 94–98% | 96–97% | [69,70] |
| S100B | 78–94% | 81–87% | [27,75] |
| NSE | 53–80% | 83–98% | [29,73,74] |
| Thymosin β4 | 100% | 98.5% | [78] |
| 14-3-3 + T-tau | 84–86% | 57–96% | [20,21] |
| +NSE or S100B | 93% | ||
| Neurodiagnostic Tests | |||
| EEG | ~65% | ~90% | [79] |
| MRI | 94.7–98% | 90–100% | [80,81] |
| Prion-specific Tests | |||
| RT-QuIC (2nd generation) | 90.3–97.2% | 98.5–100% | [82,83,84,85] |
| Brain tissue PrPSc Western blotting | 20–60% | [86] |
| Disease | Tests with High Disease-Specific Diagnostic Utility | Tests with Lower Disease-Specific Diagnostic Utility |
|---|---|---|
| VPSPr | None | 14-3-3, RT-QuIC, EEG, MRI |
| sCJD MV2 | RT-QuIC, MRI | 14-3-3, tau, EEG |
| sCJD VV1 | MRI, 14-3-3 | RT-QuIC, EEG |
| sFI | Brain FDG-PET, polysomnography, PRNP gene sequencing | 14-3-3, tau, RT-QuIC, EEG, MRI |
| FFI | Brain FDG-PET, polysomnography, PRNP gene sequencing | 14-3-3, tau, RT-QuIC, EEG, MRI |
| GSS | PRNP gene sequencing | 14-3-3, tau, RT-QuIC, EEG, MRI |
| vCJD | Tonsil biopsy, MRI (pulvinar or hockey-stick sign) | 14-3-3, RT-QuIC, EEG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figgie, M.P., Jr.; Appleby, B.S. Clinical Use of Improved Diagnostic Testing for Detection of Prion Disease. Viruses 2021, 13, 789. https://doi.org/10.3390/v13050789
Figgie MP Jr., Appleby BS. Clinical Use of Improved Diagnostic Testing for Detection of Prion Disease. Viruses. 2021; 13(5):789. https://doi.org/10.3390/v13050789
Chicago/Turabian StyleFiggie, Mark P., Jr., and Brian S. Appleby. 2021. "Clinical Use of Improved Diagnostic Testing for Detection of Prion Disease" Viruses 13, no. 5: 789. https://doi.org/10.3390/v13050789
APA StyleFiggie, M. P., Jr., & Appleby, B. S. (2021). Clinical Use of Improved Diagnostic Testing for Detection of Prion Disease. Viruses, 13(5), 789. https://doi.org/10.3390/v13050789