
Glycan Recognition in Human Norovirus Infections

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Norovirus Disease Burden, Classification, and Epidemiology
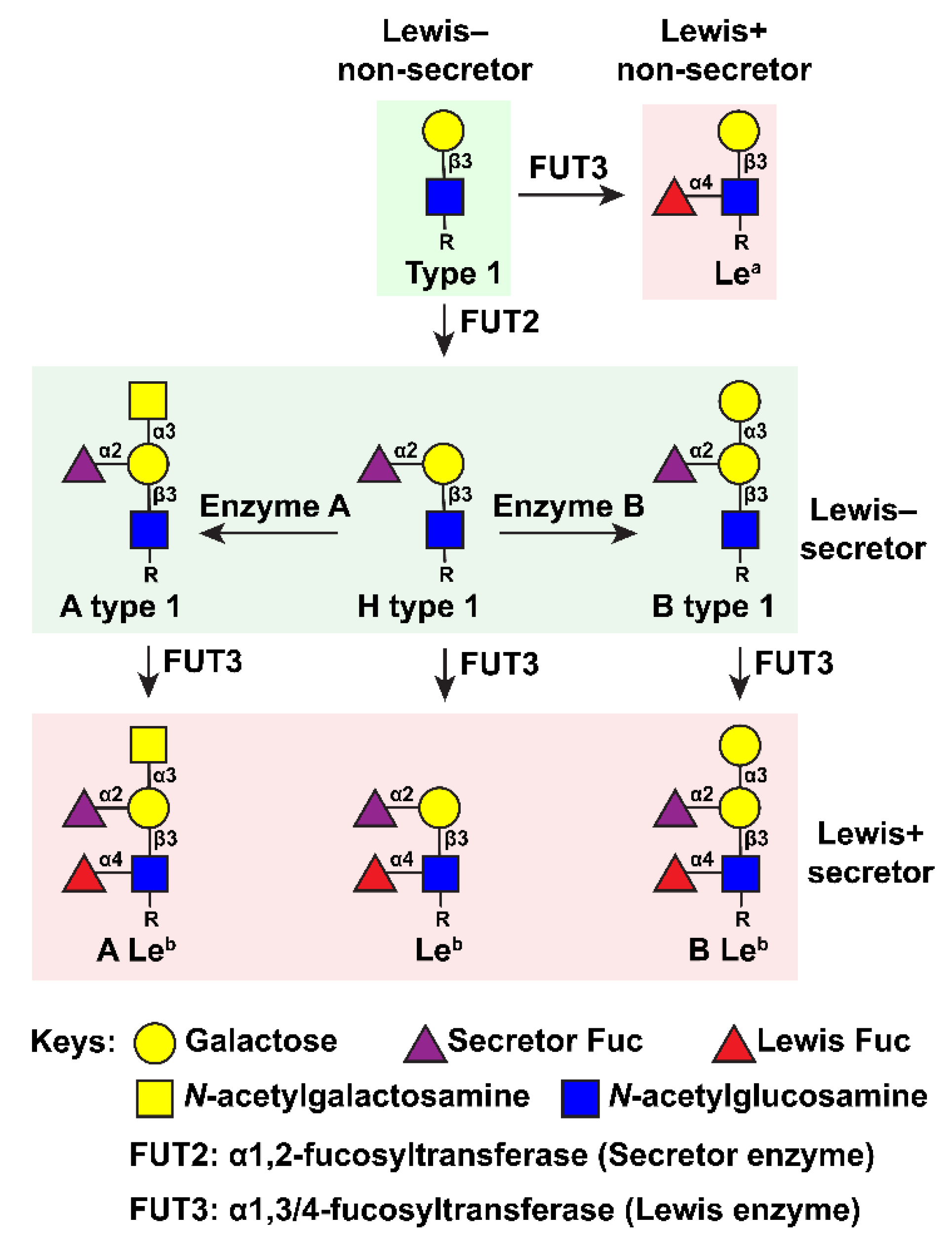
3. Early Discoveries on the Genetic Basis of Glycan Involvement in HuNoV Infection
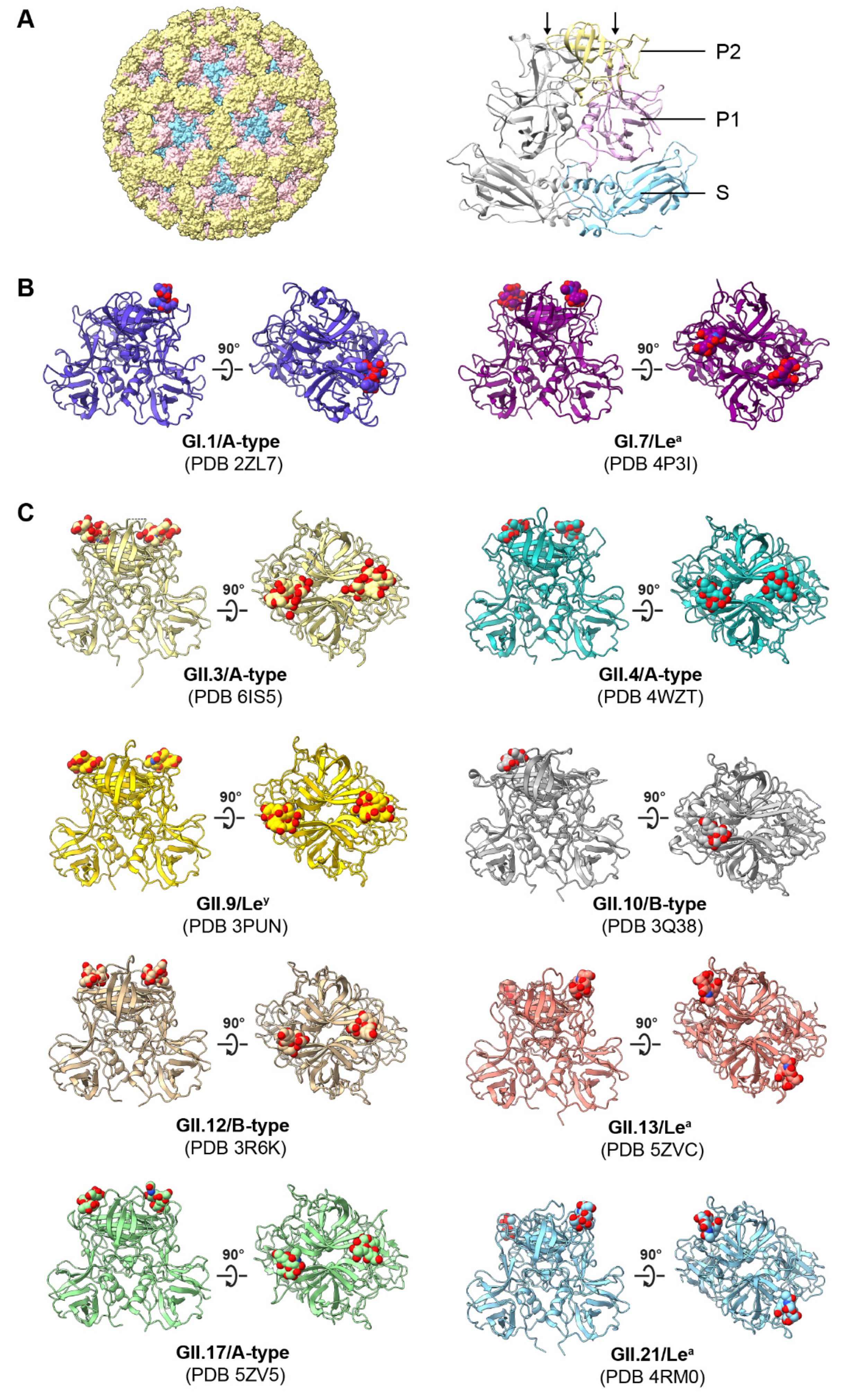
4. Norovirus–Glycan Binding Specificity and Structural Interactions
4.1. Binding Patterns to HBGAs and Related Glycans
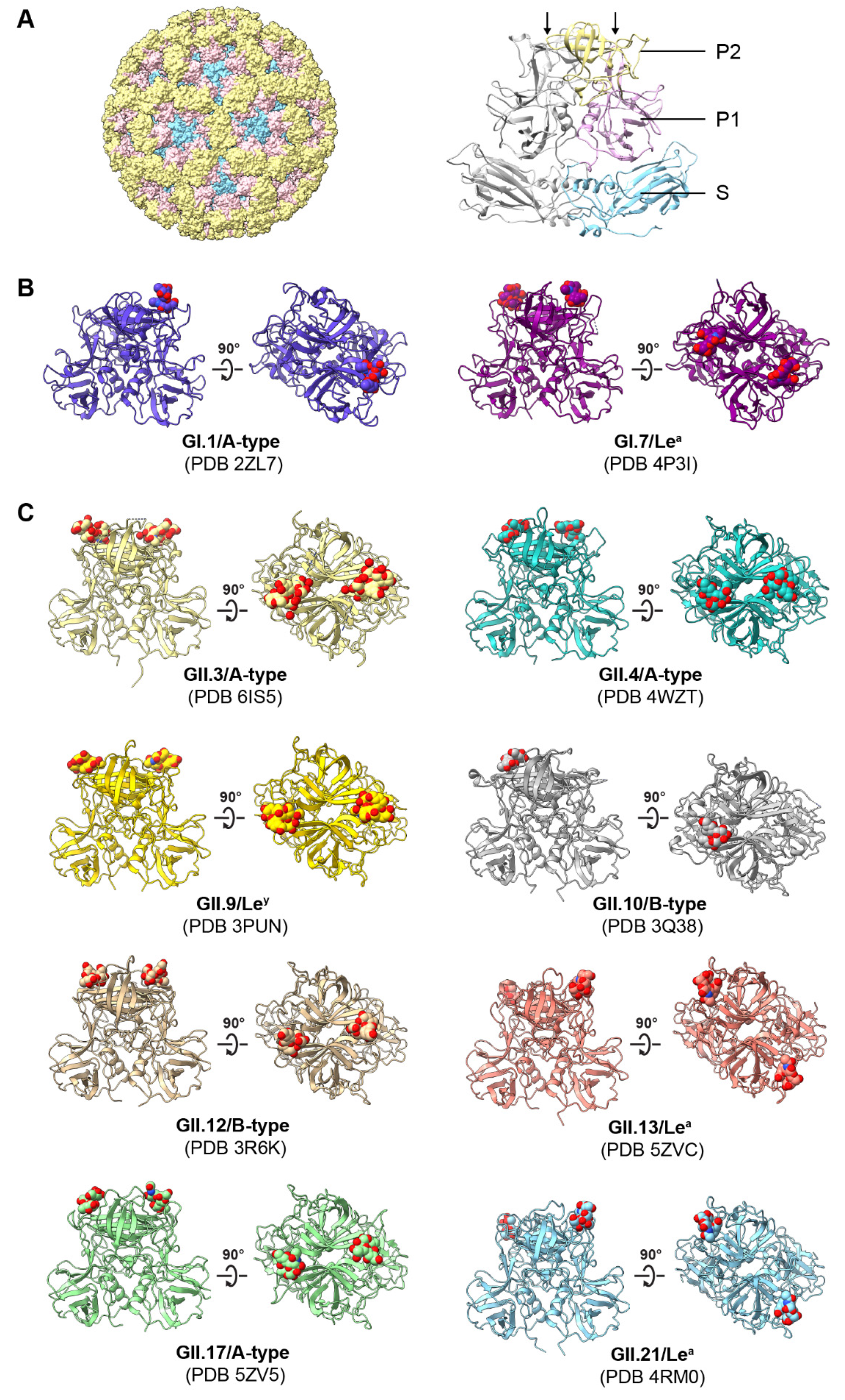
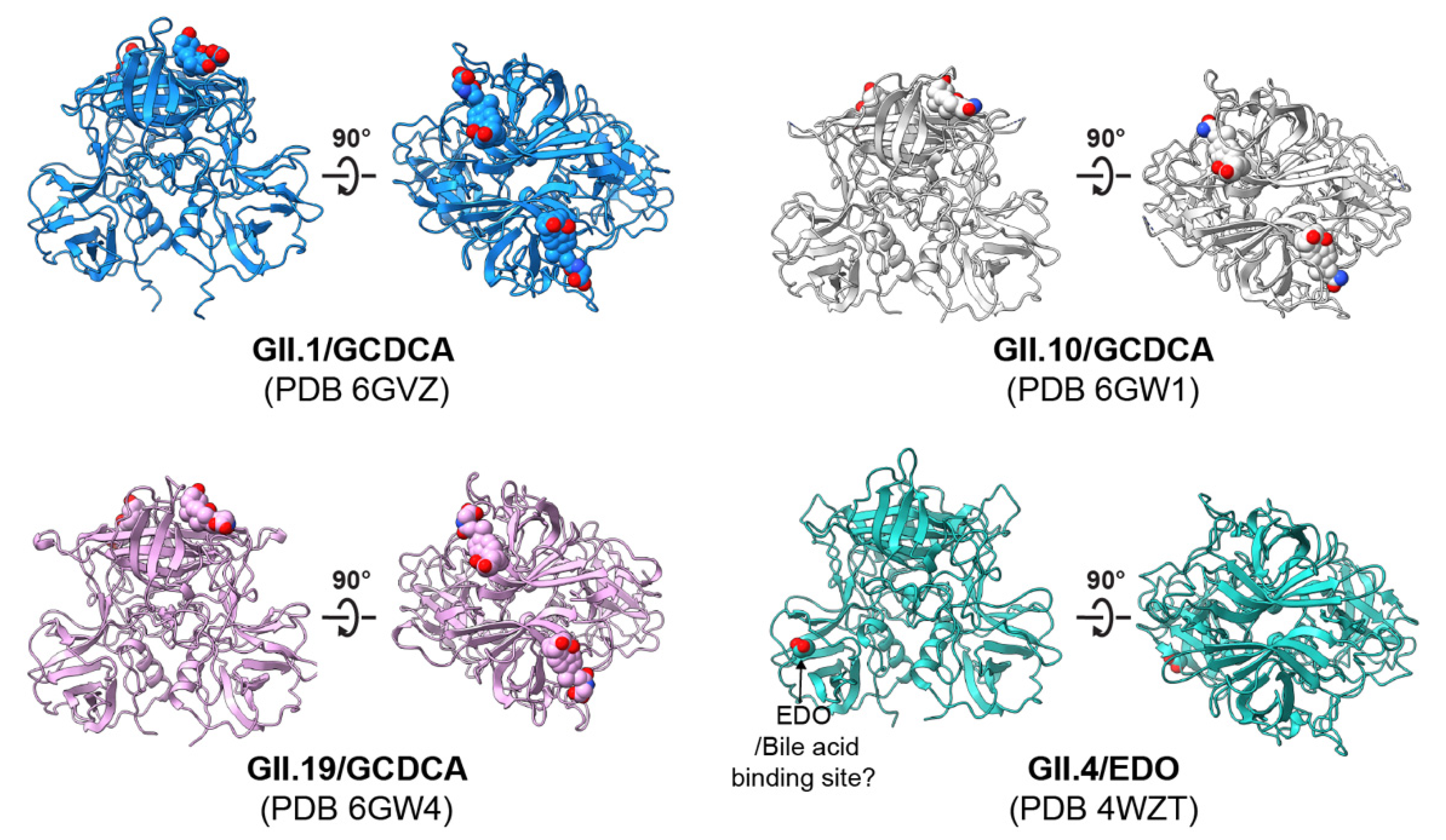
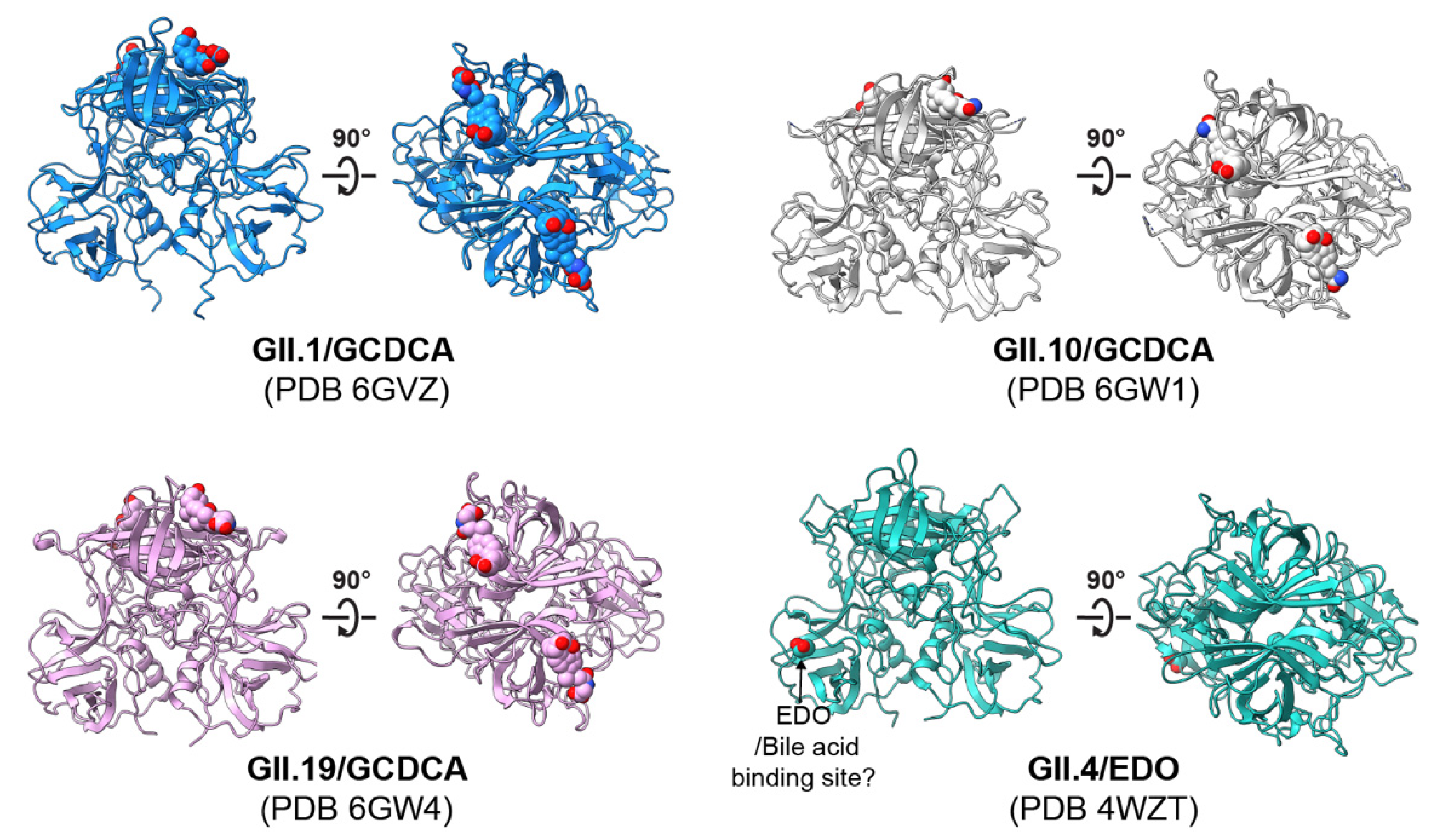
4.2. Structural Basis of HuNoV–HBGA Interactions
5. HIEs Are a Useful Platform to Study Intestinal Glycan Expression and the Role of Glycans in Viral Infection
5.1. Human Intestinal Enteroids Reflect Host Genetic Susceptibility to HuNoVs
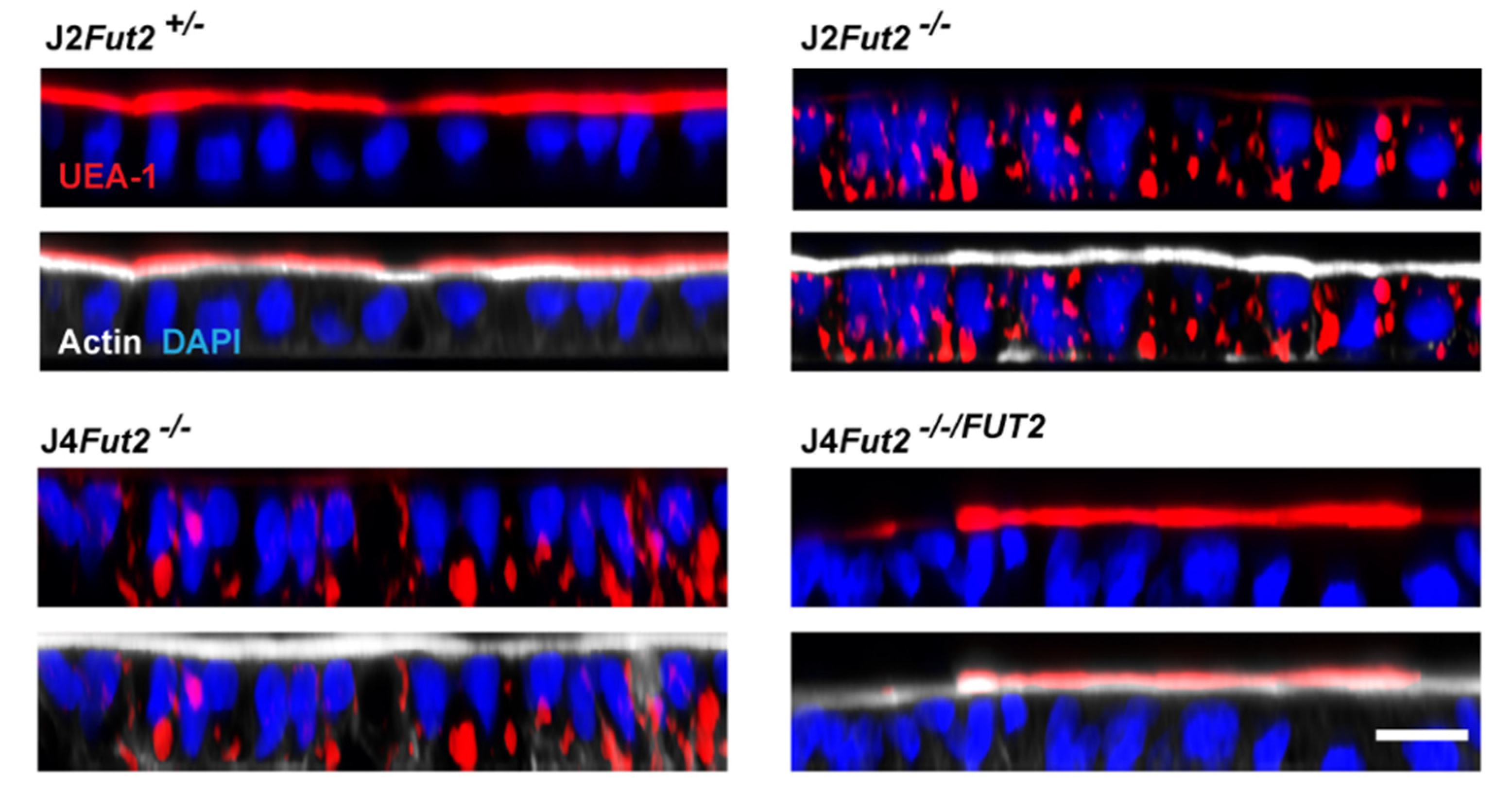
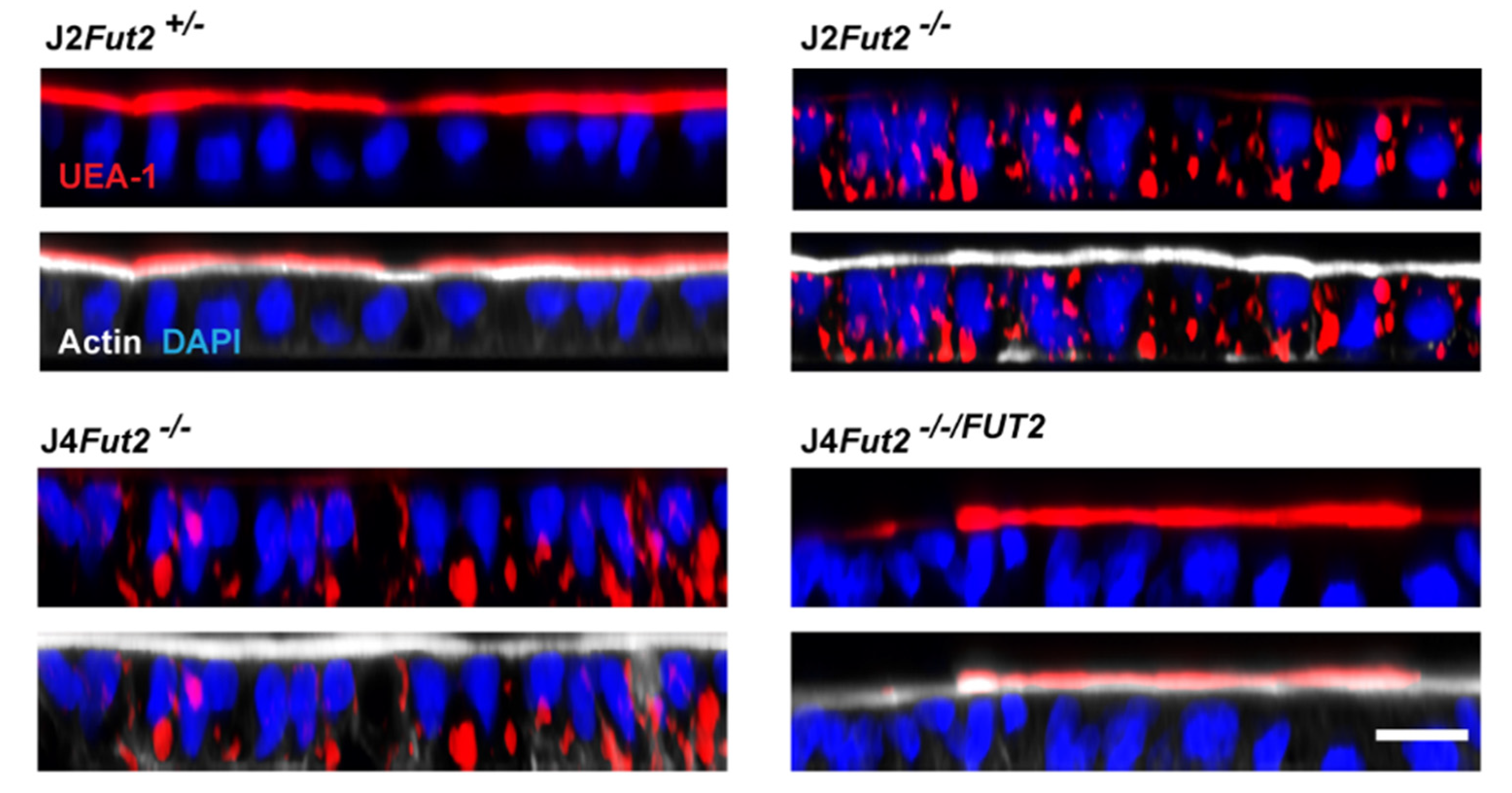
5.2. Genetic Manipulation of HIEs Reveals FUT2 Is Necessary and Sufficient for HIE Infection by Most HuNoV Strains
5.3. Progress towards Identification of the Fucosylated Glycan Receptor for HuNoVs
6. HBGA Interactions and Immune Response to HuNoVs
7. Glycan Interactions for Other Viral Pathogens
8. Conclusions and Outstanding Questions

Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paulson, J.C.; Colley, K.J. Glycosyltransferases: Structure, localization, and control of cell type-specific glycosylation. J. Biol. Chem. 1989, 264, 17615–17618. [Google Scholar] [CrossRef]
- Gagneux, P.; Aebi, M.; Varki, A. Evolution of Glycan Diversity. In Essentials of Glycobiology; [Internet]; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2015–2017; pp. 253–264. [Google Scholar] [CrossRef]
- Rini, J.M.; Esko, J.D. Glycosyltransferases and Glycan-Processing Enzymes. In Essentials of Glycobiology; [Internet]; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2015–2017; pp. 65–75. [Google Scholar] [CrossRef]
- Ruvoen-Clouet, N.; Magalhaes, A.; Marcos-Silva, L.; Breiman, A.; Figueiredo, C.; David, L.; Le Pendu, J. Increase in Genogroup II.4 Norovirus Host Spectrum by CagA-Positive Helicobacter pylori Infection. J. Infect. Dis. 2014, 210, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Qu, D.; Wang, G.; Yu, L.; Tian, F.; Chen, W.; Zhai, Q. The effects of diet and gut microbiota on the regulation of intestinal mucin glycosylation. Carbohydr. Polym. 2021, 258, 117651. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.; McGuckin, M.; Wesselingh, S.; Rogers, G.B. Infection’s Sweet Tooth: How Glycans Mediate Infection and Disease Susceptibility. Trends Microbiol. 2017, 26, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Ramani, S.; Hu, L.; Prasad, B.V.; Estes, M.K. Diversity in Rotavirus–Host Glycan Interactions: A “Sweet” Spectrum. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 263–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Crawford, S.E.; Czako, R.; Cortes-Penfield, N.W.; Smith, D.F.; Le Pendu, J.; Estes, M.K.; Prasad, B.V.V. Cell attachment protein VP8* of a human rotavirus specifically interacts with A-type histo-blood group antigen. Nature 2012, 485, 256–259. [Google Scholar] [CrossRef]
- Marionneau, S.; Ruvoën, N.; Le Moullac–Vaidye, B.; Clement, M.; Cailleau–Thomas, A.; Ruiz–Palacois, G.; Huang, P.; Jiang, X.; Le Pendu, J. Norwalk virus binds to histo-blood group antigens present on gastroduodenal epithelial cells of secretor individuals. Gastroenterology 2002, 122, 1967–1977. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.C.; Moe, C.L.; Marionneau, S.; Ruvoen, N.; Jiang, X.; Lindblad, L.; Stewart, P.W.; LePendu, J.; Baric, R.S. Human susceptibility and resistance to Norwalk virus infection. Nat. Med. 2003, 9, 548–553. [Google Scholar] [CrossRef]
- Ravn, V.; Dabelsteen, E. Tissue distribution of histo-blood group antigens. Review article. APMIS 2000, 108, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, B.; Kindberg, E.; Buesa, J.; Rydell, G.E.; Lidón, M.F.; Montava, R.; Abu Mallouh, R.; Grahn, A.; Rodriguez-Díaz, J.; Bellido, J.; et al. The G428A Nonsense Mutation in FUT2 Provides Strong but Not Absolute Protection against Symptomatic GII.4 Norovirus Infection. PLoS ONE 2009, 4, e5593. [Google Scholar] [CrossRef]
- Ferrer-Admetlla, A.; Sikora, M.; Laayouni, H.; Esteve-Codina, A.; Roubinet, F.; Blancher, A.; Calafell, F.; Bertranpetit, J.; Casals, F. A Natural History of FUT2 Polymorphism in Humans. Mol. Biol. Evol. 2009, 26, 1993–2003. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.J.; Rouquier, S.; Giorgi, D.; Lennon, G.G.; Lowe, J.B. Sequence and Expression of a Candidate for the Human Secretor Blood Group α(1,2)Fucosyltransferase Gene (FUT2). Homozygosity for an enzyme-inactivating nonsense mutation commonly correlates with the non-secretor phenotype. J. Biol. Chem. 1995, 270, 4640–4649. [Google Scholar] [CrossRef] [Green Version]
- Koda, Y.; Soejima, M.; Liu, Y.; Kimura, H. Molecular basis for secretor type alpha(1,2)-fucosyltransferase gene deficiency in a Japanese population: A fusion gene generated by unequal crossover responsible for the enzyme deficiency. Am. J. Hum. Genet. 1996, 59, 343–350. [Google Scholar]
- Costache, M.; Cailleau, A.; Fernández-Mateos, P.; Oriol, R.; Mollicone, R. Advances in molecular genetics of α-2- and α-3/4-fucosyltransferases. Transfus. Clin. Et Biol. 1997, 4, 367–382. [Google Scholar] [CrossRef]
- Möller, M.; Jöud, M.; Storry, J.R.; Olsson, M.L. Erythrogene: A database for in-depth analysis of the extensive variation in 36 blood group systems in the 1000 Genomes Project. Blood Adv. 2016, 1, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutson, A.M.; Airaud, F.; LePendu, J.; Estes, M.K.; Atmar, R.L. Norwalk virus infection associates with secretor status genotyped from sera. J. Med. Virol. 2005, 77, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Lopman, B.A.; Trivedi, T.; Vicuña, Y.; Costantini, V.; Collins, N.; Gregoricus, N.; Parashar, U.; Sandoval, C.; Broncano, N.; Vaca, M.; et al. Norovirus Infection and Disease in an Ecuadorian Birth Cohort: Association of Certain Norovirus Genotypes with Host FUT2 Secretor Status. J. Infect. Dis. 2014, 211, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Nordgren, J.; Svensson, L. Genetic Susceptibility to Human Norovirus Infection: An Update. Viruses 2019, 11, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frenck, R.; Bernstein, D.I.; Xia, M.; Huang, P.; Zhong, W.; Parker, S.; Dickey, M.; McNeal, M.; Jiang, X. Predicting Susceptibility to Norovirus GII.4 by Use of a Challenge Model Involving Humans. J. Infect. Dis. 2012, 206, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Stanley, P.; Cummings, R.D. Structures Common to Different Glycans. In Essentials of Glycobiology; [Internet]; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2015–2017; pp. 161–178. [Google Scholar] [CrossRef]
- Hu, L.; Sankaran, B.; Laucirica, D.R.; Patil, K.; Salmen, W.; Ferreon, A.C.M.; Tsoi, P.S.; Lasanajak, Y.; Smith, D.F.; Ramani, S.; et al. Glycan recognition in globally dominant human rotaviruses. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kapikian, A.Z.; Wyatt, R.G.; Dolin, R.; Thornhill, T.S.; Kalica, A.R.; Chanock, R.M. Visualization by Immune Electron Microscopy of a 27-nm Particle Associated with Acute Infectious Nonbacterial Gastroenteritis. J. Virol. 1972, 10, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Belliot, G.; Lopman, B.; Ambert-Balay, K.; Pothier, P. The burden of norovirus gastroenteritis: An important foodborne and healthcare-related infection. Clin. Microbiol. Infect. 2014, 20, 724–730. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.M.; Hall, A.J.; Robinson, E.A.; Verhoef, L.; Premkumar, P.; Parashar, U.D.; Koopmans, M.; Lopman, A.B. Global prevalence of norovirus in cases of gastroenteritis: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Atmar, R.L.; Ramani, S.; Estes, M.K. Human noroviruses: Recent advances in a 50-year history. Curr. Opin. Infect. Dis. 2018, 31, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.C.; Vinje, J.; Szilagyi, P.G.; Edwards, K.M.; Staat, M.A.; Weinberg, G.; Hall, C.B.; Chappell, J.; Bernstein, D.I.; Curns, A.T.; et al. Norovirus and Medically Attended Gastroenteritis in U.S. Children. N. Engl. J. Med. 2013, 368, 1121–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, H.L.; Neill, F.H.; Estes, M.K.; Munoz, F.M.; Cameron, A.; DuPont, H.L.; Atmar, R.L. Noroviruses: The Most Common Pediatric Viral Enteric Pathogen at a Large University Hospital After Introduction of Rotavirus Vaccination. J. Pediatr. Infect. Dis. Soc. 2012, 2, 57–60. [Google Scholar] [CrossRef] [Green Version]
- Bagci, S.; Eis-Hübinger, A.M.; Yassin, A.F.; Simon, A.; Bartmann, P.; Franz, A.R.; Mueller, A. Clinical characteristics of viral intestinal infection in preterm and term neonates. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 1079–1084. [Google Scholar] [CrossRef]
- Brown, L.-A.K.; Clark, I.; Brown, J.; Breuer, J.; Lowe, D.M. Norovirus infection in primary immune deficiency. Rev. Med Virol. 2017, 27, e1926. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, T.K.; Desai, R.; Hall, A.J.; Patel, M.; Parashar, U.D.; Lopman, B.A. Clinical characteristics of norovirus-associated deaths: A systematic literature review. Am. J. Infect. Control. 2013, 41, 654–657. [Google Scholar] [CrossRef]
- Bartsch, S.M.; Lopman, B.A.; Ozawa, S.; Hall, A.J.; Lee, B.Y. Global Economic Burden of Norovirus Gastroenteritis. PLoS ONE 2016, 11, e0151219. [Google Scholar] [CrossRef] [Green Version]
- Vinjé, J.; Estes, M.K.; Esteves, P.; Green, K.Y.; Katayama, K.; Knowles, N.J.; L’Homme, Y.; Martella, V.; Vennema, H.; White, P.; et al. ICTV Virus Taxonomy Profile: Caliciviridae. J. Gen. Virol. 2019, 100, 1469–1470. [Google Scholar] [CrossRef]
- Chhabra, P.; De Graaf, M.; Parra, G.I.; Chan, M.C.-W.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393–1406. [Google Scholar] [CrossRef]
- Parra, I.G. Emergence of norovirus strains: A tale of two genes. Virus Evol. 2019, 5, vez048. [Google Scholar] [CrossRef] [PubMed]
- Rydell, E.G.; Dahlin, A.B.; Höök, F.; Larson, G. QCM-D studies of human norovirus VLPs binding to glycosphingolipids in supported lipid bilayers reveal strain-specific characteristics. Glycobiology 2009, 19, 1176–1184. [Google Scholar] [CrossRef] [Green Version]
- Kendra, J.A.; Tohma, K.; Ford-Siltz, L.A.; Lepore, C.J.; Parra, G.I. Antigenic cartography reveals complexities of genetic determinants that lead to antigenic differences among pandemic GII.4 noroviruses. Proc. Natl. Acad. Sci. USA 2021, 118, e2015874118. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Farkas, T.; Marionneau, S.; Zhong, W.; Ruvoën-Clouet, N.; Morrow, A.L.; Altaye, M.; Pickering, L.K.; Newburg, D.S.; LePendu, J.; et al. Noroviruses Bind to Human ABO, Lewis, and Secretor Histo–Blood Group Antigens: Identification of 4 Distinct Strain-Specific Patterns. J. Infect. Dis. 2003, 188, 19–31. [Google Scholar] [CrossRef]
- Huang, P.; Farkas, T.; Zhong, W.; Tan, M.; Thornton, S.; Morrow, A.L.; Jiang, X. Norovirus and Histo-Blood Group Antigens: Demonstration of a Wide Spectrum of Strain Specificities and Classification of Two Major Binding Groups among Multiple Binding Patterns. J. Virol. 2005, 79, 6714–6722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheetham, S.; Souza, M.; McGregor, R.; Meulia, T.; Wang, Q.; Saif, L.J. Binding Patterns of Human Norovirus-Like Particles to Buccal and Intestinal Tissues of Gnotobiotic Pigs in Relation to A/H Histo-Blood Group Antigen Expression. J. Virol. 2007, 81, 3535–3544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creutznacher, R.; Maass, T.; Ogrissek, P.; Wallmann, G.; Feldmann, C.; Peters, H.; Lingemann, M.; Taube, S.; Peters, T.; Mallagaray, A. NMR Experiments Shed New Light on Glycan Recognition by Human and Murine Norovirus Capsid Proteins. Viruses 2021, 13, 416. [Google Scholar] [CrossRef]
- Creutznacher, R.; Schulze, E.; Wallmann, G.; Peters, T.; Stein, M.; Mallagaray, A. Chemical-Shift Perturbations Reflect Bile Acid Binding to Norovirus Coat Protein: Recognition Comes in Different Flavors. ChemBioChem 2019, 21, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Fiege, B.; Rademacher, C.; Cartmell, J.; Kitov, P.I.; Parra, F.; Peters, T. Molecular Details of the Recognition of Blood Group Antigens by a Human Norovirus as Determined by STD NMR Spectroscopy. Angew. Chem. Int. Ed. 2011, 51, 928–932. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, G.; Salmen, W.; Ettayebi, K.; Hu, L.; Sankaran, B.; Estes, M.K.; Prasad, B.V.V.; Crowe, J.E. Broadly cross-reactive human antibodies that inhibit genogroup I and II noroviruses. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Shanker, S.; Choi, J.-M.; Sankaran, B.; Atmar, R.L.; Estes, M.K.; Prasad, B.V.V. Structural Analysis of Histo-Blood Group Antigen Binding Specificity in a Norovirus GII.4 Epidemic Variant: Implications for Epochal Evolution. J. Virol. 2011, 85, 8635–8645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.; Neill, F.H.; Blutt, S.E.; Zeng, X.-L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef]
- Haga, K.; Ettayebi, K.; Tenge, V.R.; Karandikar, U.C.; Lewis, M.A.; Lin, S.-C.; Neill, F.H.; Ayyar, B.V.; Zeng, X.-L.; Larson, G.; et al. Genetic Manipulation of Human Intestinal Enteroids Demonstrates the Necessity of a Functional Fucosyltransferase 2 Gene for Secretor-Dependent Human Norovirus Infection. mBio 2020, 11, e00251-20. [Google Scholar] [CrossRef] [PubMed]
- Ettayebi, K.; Tenge, V.R.; Cortes-Penfield, N.W.; Crawford, S.E.; Neill, F.H.; Zeng, X.-L.; Yu, X.; Ayyar, B.V.; Burrin, D.; Ramani, S.; et al. New Insights and Enhanced Human Norovirus Cultivation in Human Intestinal Enteroids. mSphere 2021, 6, e01136-20. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Vicente, N.; Vila-Vicent, S.; Allen, D.; Gozalbo-Rovira, R.; Iturriza-Gómara, M.; Buesa, J.; Rodríguez-Díaz, J. Characterization of a Novel Conformational GII.4 Norovirus Epitope: Implications for Norovirus-Host Interactions. J. Virol. 2016, 90, 7703–7714. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2012, 41, D36–D42. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Chhabra, P.; Diez-Valcarce, M.; Barclay, L.; Cannon, J.L.; Vinjé, J. Human Calicivirus Typing tool: A web-based tool for genotyping human norovirus and sapovirus sequences. J. Clin. Virol. 2020, 134, 104718. [Google Scholar] [CrossRef]
- Kroneman, A.; Vennema, H.; Deforche, K.; Avoort, H.; Peñaranda, S.; Oberste, M.; Vinje, J.; Koopmans, M. An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 2011, 51, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, M.; Graham, D.Y.; Estes, M.K. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J. Virol. 1992, 66, 6527–6532. [Google Scholar] [CrossRef] [Green Version]
- Green, K.Y.; Lew, J.F.; Jiang, X.; Kapikian, A.Z.; Estes, M.K. Comparison of the reactivities of baculovirus-expressed recombinant Norwalk virus capsid antigen with those of the native Norwalk virus antigen in serologic assays and some epidemiologic observations. J. Clin. Microbiol. 1993, 31, 2185–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, B.V.V.; Hardy, M.E.; Dokland, T.; Bella, J.; Rossmann, M.G.; Estes, M.K. X-ray Crystallographic Structure of the Norwalk Virus Capsid. Science 1999, 286, 287–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrino, T.A.; Schreiber, D.S.; Trier, J.S.; Kapikian, A.Z.; Blacklow, N.R. Clinical Immunity in Acute Gastroenteritis Caused by Norwalk Agent. N. Engl. J. Med. 1977, 297, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Ruvoën-Clouet, N.; Ganière, J.P.; André-Fontaine, G.; Blanchard, D.; Le Pendu, J. Binding of Rabbit Hemorrhagic Disease Virus to Antigens of the ABH Histo-Blood Group Family. J. Virol. 2000, 74, 11950–11954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutson, A.M.; Atmar, R.L.; Graham, D.Y.; Estes, M.K. Norwalk Virus Infection and Disease Is Associated with ABO Histo–Blood Group Type. J. Infect. Dis. 2002, 185, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Hutson, A.M.; Atmar, R.L.; Marcus, D.M.; Estes, M.K. Norwalk Virus-Like Particle Hemagglutination by Binding to H Histo-Blood Group Antigens. J. Virol. 2003, 77, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Thorven, M.; Grahn, A.; Hedlund, K.-O.; Johansson, H.; Wahlfrid, C.; Larson, G.; Svensson, L. A Homozygous Nonsense Mutation (428G→A) in the Human Secretor (FUT2) Gene Provides Resistance to Symptomatic Norovirus (GGII) Infections. J. Virol. 2005, 79, 15351–15355. [Google Scholar] [CrossRef] [Green Version]
- Larsson, M.M.; Rydell, G.E.P.; Grahn, A.; Rodriguez-Diaz, J.; Åkerlind, B.; Hutson, A.M.; Estes, M.K.; Larson, G.; Svensson, L. Antibody Prevalence and Titer to Norovirus (Genogroup II) Correlate with Secretor(FUT2)but Not with ABO Phenotype or Lewis(FUT3)Genotype. J. Infect. Dis. 2006, 194, 1422–1427. [Google Scholar] [CrossRef]
- Tian, P.; Brandl, M.; Mandrell, R. Porcine gastric mucin binds to recombinant norovirus particles and competitively inhibits their binding to histo-blood group antigens and Caco-2 cells. Lett. Appl. Microbiol. 2005, 41, 315–320. [Google Scholar] [CrossRef]
- Haynes, J.; Perry, V.; Benson, E.; Meeks, A.; Watts, G.; Watkins, H.; Braun, R. In Depth Breadth Analyses of Human Blockade Responses to Norovirus and Response to Vaccination. Viruses 2019, 11, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.-M.; Hutson, A.M.; Estes, M.K.; Prasad, B.V.V. Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proc. Natl. Acad. Sci. USA 2008, 105, 9175–9180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansman, G.S.; Biertümpfel, C.; Georgiev, I.; McLellan, J.S.; Chen, L.; Zhou, T.; Katayama, K.; Kwong, P.D. Crystal Structures of GII.10 and GII.12 Norovirus Protruding Domains in Complex with Histo-Blood Group Antigens Reveal Details for a Potential Site of Vulnerability. J. Virol. 2011, 85, 6687–6701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koromyslova, A.; Tripathi, S.; Morozov, V.; Schroten, H.; Hansman, G.S. Human norovirus inhibition by a human milk oligosaccharide. Virology 2017, 508, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Shanker, S.; Czako, R.; Sankaran, B.; Atmar, R.L.; Estes, M.K.; Prasad, B.V.V. Structural Analysis of Determinants of Histo-Blood Group Antigen Binding Specificity in Genogroup I Noroviruses. J. Virol. 2014, 88, 6168–6180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taube, S.; Mallagaray, A.; Peters, T. Norovirus, glycans and attachment. Curr. Opin. Virol. 2018, 31, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Etzold, S.; Bode, L. Glycan-dependent viral infection in infants and the role of human milk oligosaccharides. Curr. Opin. Virol. 2014, 7, 101–107. [Google Scholar] [CrossRef]
- Ruiz-Palacios, G.M.; Cervantes, L.E.; Ramos, P.; Chavez-Munguia, B.; Newburg, D.S. Campylobacter jejuni Binds Intestinal H(O) Antigen (Fucα1, 2Galβ1, 4GlcNAc), and Fucosyloligosaccharides of Human Milk Inhibit Its Binding and Infection. J. Biol. Chem. 2003, 278, 14112–14120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichert, S.; Koromyslova, A.; Singh, B.K.; Hansman, S.; Jennewein, S.; Schroten, H.; Hansman, G. Structural Basis for Norovirus Inhibition by Human Milk Oligosaccharides. J. Virol. 2016, 90, 4843–4848. [Google Scholar] [CrossRef] [Green Version]
- White, L.J.; Ball, J.M.; Hardy, E.M.; Tanaka, T.N.; Kitamoto, N.; Estes, M.K. Attachment and entry of recombinant Norwalk virus capsids to cultured human and animal cell lines. J. Virol. 1996, 70, 6589–6597. [Google Scholar] [CrossRef] [Green Version]
- Duizer, E.; Schwab, K.J.; Neill, F.H.; Atmar, R.L.; Koopmans, M.P.G.; Estes, M.K. Laboratory efforts to cultivate noroviruses. J. Gen. Virol. 2004, 85, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Guix, S.; Asanaka, M.; Katayama, K.; Crawford, S.E.; Neill, F.H.; Atmar, R.L.; Estes, M.K. Norwalk Virus RNA Is Infectious in Mammalian Cells. J. Virol. 2007, 81, 12238–12248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varki, A.; Kannagi, R.; Toole, B.; Stanley, P. Glycosylation Changes in Cancer. In Essentials of Glycobiology; [Internet]; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2015–2017; pp. 597–609. [Google Scholar] [CrossRef]
- Pearce, O.M.T.; Läubli, H. Sialic acids in cancer biology and immunity. Glycobiology 2015, 26, 111–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, J.M.H.; Thomsson, K.A.; Rodríguez-Piñeiro, A.M.; Karlsson, H.; Hansson, G.C. Studies of mucus in mouse stomach, small intestine, and colon. III. Gastrointestinal Muc5ac and Muc2 mucin O-glycan patterns reveal a regiospecific distribution. Am. J. Physiol. Liver Physiol. 2013, 305, G357–G363. [Google Scholar] [CrossRef] [Green Version]
- Van Dycke, J.; Ny, A.; Conceição-Neto, N.; Maes, J.; Hosmillo, M.; Cuvry, A.; Goodfellow, I.; Nogueira, T.C.; Verbeken, E.; Matthijnssens, J.; et al. A robust human norovirus replication model in zebrafish larvae. PLoS Pathog. 2019, 15, e1008009. [Google Scholar] [CrossRef] [Green Version]
- Yamakawa, N.; Vanbeselaere, J.; Chang, L.-Y.; Yu, S.-Y.; Ducrocq, L.; Harduin-Lepers, A.; Kurata, J.; Aoki-Kinoshita, K.F.; Sato, C.; Khoo, K.-H.; et al. Systems glycomics of adult zebrafish identifies organ-specific sialylation and glycosylation patterns. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.; Ferrante, M.; Vries, R.G.; van Es, J.H.; Brink, S.V.D.; van Houdt, W.; Pronk, A.; van Gorp, J.; Siersema, P.D.; et al. Long-term Expansion of Epithelial Organoids From Human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Estes, M.K.; Ettayebi, K.; Tenge, V.R.; Murakami, K.; Karandikar, U.; Lin, S.-C.; Ayyar, B.V.; Cortes-Penfield, N.W.; Haga, K.; Neill, F.H.; et al. Human Norovirus Cultivation in Nontransformed Stem Cell-Derived Human Intestinal Enteroid Cultures: Success and Challenges. Viruses 2019, 11, 638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rimkute, I.; Thorsteinsson, K.; Henricsson, M.; Tenge, V.R.; Yu, X.; Lin, S.-C.; Haga, K.; Atmar, R.L.; Lycke, N.; Nilsson, J.; et al. Histo-blood group antigens of glycosphingolipids predict susceptibility of human intestinal enteroids to norovirus infection. J. Biol. Chem. 2020, 295, 15974–15987. [Google Scholar] [CrossRef]
- Murakami, K.; Tenge, V.R.; Karandikar, U.C.; Lin, S.-C.; Ramani, S.; Ettayebi, K.; Crawford, S.E.; Zeng, X.-L.; Neill, F.H.; Ayyar, B.V.; et al. Bile acids and ceramide overcome the entry restriction for GII.3 human norovirus replication in human intestinal enteroids. Proc. Natl. Acad. Sci. USA 2020, 117, 1700–1710. [Google Scholar] [CrossRef]
- Kilic, T.; Koromyslova, A.; Hansman, G.S. Structural Basis for Human Norovirus Capsid Binding to Bile Acids. J. Virol. 2019, 93, e01581-18. [Google Scholar] [CrossRef] [Green Version]
- Tenge, V.R.; Murakami, K.; Salmen, W.; Lin, S.C.; Crawford, S.E.; Neill, F.H.; Prasad, B.V.V.; Atmar, R.L.; Estes, M.K. Bile Goes Viral. Viruses 2021, 13, 998. [Google Scholar] [CrossRef] [PubMed]
- Mallory, M.L.; Lindesmith, L.C.; Brewer-Jensen, P.D.; Graham, R.L.; Baric, R.S. Bile Facilitates Human Norovirus Interactions with Diverse Histoblood Group Antigens, Compensating for Capsid Microvariation Observed in 2016–2017 GII.2 Strains. Viruses 2020, 12, 989. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.C.; Brewer-Jensen, P.D.; Mallory, M.L.; Jensen, K.; Yount, B.L.; Costantini, V.; Collins, M.H.; Edwards, C.E.; Sheahan, T.P.; Vinjé, J.; et al. Virus–Host Interactions Between Nonsecretors and Human Norovirus. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 245–267. [Google Scholar] [CrossRef] [PubMed]
- Ayouni, S.; Estienney, M.; Sdiri-Loulizi, K.; Ambert-Balay, K.; de Rougemont, A.; Aho, S.; Hammami, S.; Aouni, M.; Guédiche, M.; Pothier, P.; et al. Relationship between GII.3 norovirus infections and blood group antigens in young children in Tunisia. Clin. Microbiol. Infect. 2015, 21, 874.e1–874.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-C.; Qu, L.; Ettayebi, K.; Crawford, S.E.; Blutt, S.E.; Robertson, M.J.; Zeng, X.-L.; Tenge, V.R.; Ayyar, B.V.; Karandikar, U.C.; et al. Human norovirus exhibits strain-specific sensitivity to host interferon pathways in human intestinal enteroids. Proc. Natl. Acad. Sci. USA 2020, 117, 23782–23793. [Google Scholar] [CrossRef]
- Tan, K.-P.; Ho, M.-Y.; Cho, H.-C.; Yu, J.; Hung, J.-T.; Yu, A. Fucosylation of LAMP-1 and LAMP-2 by FUT1 correlates with lysosomal positioning and autophagic flux of breast cancer cells. Cell Death Dis. 2016, 7, e2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickard, J.M.; Maurice, C.F.; Kinnebrew, M.A.; Abt, M.C.; Schenten, D.; Golovkina, T.V.; Bogatyrev, S.R.; Ismagilov, R.F.; Pamer, E.G.; Turnbaugh, P.; et al. Rapid fucosylation of intestinal epithelium sustains host–commensal symbiosis in sickness. Nature 2014, 514, 638–641. [Google Scholar] [CrossRef] [Green Version]
- Pickard, J.M.; Chervonsky, A.V. Intestinal Fucose as a Mediator of Host–Microbe Symbiosis. J. Immunol. 2015, 194, 5588–5593. [Google Scholar] [CrossRef] [Green Version]
- Breimer, E.M.; Hansson, G.C.; Karlsson, K.-A.; Larson, G.; Leffler, H. Glycosphingolipid composition of epithelial cells isolated along the villus axis of small intestine of a single human individual. Glycobiology 2012, 22, 1721–1730. [Google Scholar] [CrossRef] [Green Version]
- Rydell, E.G.; Nilsson, J.; Rodriguez-Diaz, J.; Ruvoën-Clouet, N.; Svensson, L.; Le Pendu, J.; Larson, G. Human noroviruses recognize sialyl Lewis x neoglycoprotein. Glycobiology 2008, 19, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Bally, M.; Rydell, G.E.; Zahn, R.; Nasir, W.; Eggeling, C.; Breimer, M.E.; Svensson, L.; Höök, F.; Larson, G. Norovirus GII.4 Virus-like Particles Recognize Galactosylceramides in Domains of Planar Supported Lipid Bilayers. Angew. Chem. Int. Ed. 2012, 51, 12020–12024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasir, W.; Frank, M.; Koppisetty, C.A.K.; Larson, G.; Nyholm, P.-G. Lewis histo-blood group α1,3/α1,4 fucose residues may both mediate binding to GII.4 noroviruses. Glycobiology 2012, 22, 1163–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.-S.; Hosmillo, M.; Alfajaro, M.M.; Kim, J.-Y.; Park, J.-G.; Son, K.-Y.; Ryu, E.-H.; Sorgeloos, F.; Kwon, H.-J.; Park, S.-J.; et al. Both α2,3- and α2,6-Linked Sialic Acids on O-Linked Glycoproteins Act as Functional Receptors for Porcine Sapovirus. PLoS Pathog. 2014, 10, e1004172. [Google Scholar] [CrossRef] [Green Version]
- Stuart, A.D.; Brown, T.D.K. α2,6-Linked sialic acid acts as a receptor for Feline calicivirus. J. Gen. Virol. 2007, 88, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Wei, C.; Huang, P.; Fan, Q.; Quigley, C.; Xia, M.; Fang, H.; Zhang, X.; Zhong, W.; Klassen, J.S.; et al. Tulane virus recognizes sialic acids as cellular receptors. Sci. Rep. 2015, 5, 11784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Huang, P.; Zou, L.; Lowary, T.L.; Tan, M.; Jiang, X. Tulane Virus Recognizes the A Type 3 and B Histo-Blood Group Antigens. J. Virol. 2015, 89, 1419–1427. [Google Scholar] [CrossRef] [Green Version]
- Ossiboff, R.J.; Parker, J.S.L. Identification of Regions and Residues in Feline Junctional Adhesion Molecule Required for Feline Calicivirus Binding and Infection. J. Virol. 2007, 81, 13608–13621. [Google Scholar] [CrossRef] [Green Version]
- Farkas, T.; Yang, K.; Le Pendu, J.; Baines, J.D.; Cardin, R.D. The Coxsackievirus and Adenovirus Receptor, a Required Host Factor for Recovirus Infection, Is a Putative Enteric Calicivirus Receptor. J. Virol. 2019, 93, e00869-19. [Google Scholar] [CrossRef]
- Alfajaro, M.M.; Cho, E.-H.; Kim, D.-S.; Kim, J.-Y.; Park, J.-G.; Soliman, M.; Baek, Y.-B.; Park, C.-H.; Kang, M.-I.; Park, S.-I.; et al. Early Porcine Sapovirus Infection Disrupts Tight Junctions and Uses Occludin as a Coreceptor. J. Virol. 2019, 93, e01773-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orchard, R.C.; Wilen, C.B.; Doench, J.; Baldridge, M.; McCune, B.T.; Lee, Y.-C.; Lee, S.; Pruett-Miller, S.M.; Nelson, C.A.; Fremont, D.H.; et al. Discovery of a proteinaceous cellular receptor for a norovirus. Science 2016, 353, 933–936. [Google Scholar] [CrossRef] [Green Version]
- Haga, K.; Fujimoto, A.; Takai-Todaka, R.; Miki, M.; Doan, Y.H.; Murakami, K.; Yokoyama, M.; Murata, K.; Nakanishi, A.; Katayama, K. Functional receptor molecules CD300lf and CD300ld within the CD300 family enable murine noroviruses to infect cells. Proc. Natl. Acad. Sci. USA 2016, 113, E6248–E6255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graziano, V.R.; Walker, F.C.; Kennedy, E.A.; Wei, J.; Ettayebi, K.; Strine, M.S.; Filler, R.B.; Hassan, E.; Hsieh, L.L.; Kim, A.S.; et al. CD300lf is the primary physiologic receptor of murine norovirus but not human norovirus. PLoS Pathog. 2020, 16, e1008242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furlong, K.; Biering, S.B.; Choi, J.; Wilen, C.B.; Orchard, R.C.; Wobus, C.E.; Nelson, C.A.; Fremont, D.H.; Baldridge, M.T.; Randall, G.; et al. CD300LF Polymorphisms of Inbred Mouse Strains Confer Resistance to Murine Norovirus Infection in a Cell Type-Dependent Manner. J. Virol. 2020, 94, e00837-20. [Google Scholar] [CrossRef]
- Ramani, S.; Estes, M.K.; Atmar, R.L. Correlates of Protection against Norovirus Infection and Disease—Where Are We Now, Where Do We Go? PLoS Pathog. 2016, 12, e1005334. [Google Scholar] [CrossRef] [Green Version]
- Harrington, P.R.; Lindesmith, L.; Yount, B.; Moe, C.L.; Baric, R.S. Binding of Norwalk Virus-Like Particles to ABH Histo-Blood Group Antigens Is Blocked by Antisera from Infected Human Volunteers or Experimentally Vaccinated Mice. J. Virol. 2002, 76, 12335–12343. [Google Scholar] [CrossRef] [Green Version]
- Reeck, A.; Kavanagh, O.; Estes, M.K.; Opekun, A.R.; Gilger, M.A.; Graham, D.Y.; Atmar, R.L. Serological Correlate of Protection against Norovirus-Induced Gastroenteritis. J. Infect. Dis. 2010, 202, 1212–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atmar, R.L.; Ettayebi, K.; Ayyar, B.V.; Neill, F.H.; Braun, R.P.; Ramani, S.; Estes, M.K. Comparison of Microneutralization and Histo-Blood Group Antigen–Blocking Assays for Functional Norovirus Antibody Detection. J. Infect. Dis. 2020, 221, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Atmar, R.L.; Bernstein, D.I.; Lyon, G.M.; Treanor, J.J.; Al-Ibrahim, M.S.; Graham, D.Y.; Vinje, J.; Jiang, X.; Gregoricus, N.; Frenck, R.W.; et al. Serological Correlates of Protection against a GII.4 Norovirus. Clin. Vaccine Immunol. 2015, 22, 923–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanker, S.; Czako, R.; Sapparapu, G.; Alvarado, G.; Viskovska, M.; Sankaran, B.; Atmar, R.L.; Crowe, J.E.; Estes, M.K.; Prasad, B.V.V. Structural basis for norovirus neutralization by an HBGA blocking human IgA antibody. Proc. Natl. Acad. Sci. USA 2016, 113, E5830–E5837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapparapu, G.; Czako, R.; Alvarado, G.; Shanker, S.; Prasad, B.; Atmar, R.L.; Estes, M.K.; Crowe, J.E., Jr. Frequent Use of the IgA Isotype in Human B Cells Encoding Potent Norovirus-Specific Monoclonal Antibodies That Block HBGA Binding. PLoS Pathog. 2016, 12, e1005719. [Google Scholar] [CrossRef] [PubMed]
- Ruoff, K.; Kilic, T.; Devant, J.; Koromyslova, A.; Ringel, A.; Hempelmann, A.; Geiss, C.; Graf, J.; Haas, M.; Roggenbach, I.; et al. Structural Basis of Nanobodies Targeting the Prototype Norovirus. J. Virol. 2019, 93, e02005-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindesmith, L.C.; Donaldson, E.F.; LoBue, A.D.; Cannon, J.L.; Zheng, D.-P.; Vinje, J.; Baric, R.S. Mechanisms of GII.4 Norovirus Persistence in Human Populations. PLoS Med. 2008, 5, e0050031. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.L.; Lindesmith, L.C.; Donaldson, E.F.; Saxe, L.; Baric, R.S.; Vinje, J. Herd Immunity to GII.4 Noroviruses Is Supported by Outbreak Patient Sera. J. Virol. 2009, 83, 5363–5374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rougemont, A.; Ruvoen-Clouet, N.; Simon, B.; Estienney, M.; Elie-Caille, C.; Aho, S.; Pothier, P.; Le Pendu, J.; Boireau, W.; Belliot, G. Qualitative and Quantitative Analysis of the Binding of GII.4 Norovirus Variants onto Human Blood Group Antigens. J. Virol. 2011, 85, 4057–4070. [Google Scholar] [CrossRef] [Green Version]
- Golinelli, D.; Boetto, E.; Maietti, E.; Fantini, M.P. The association between ABO blood group and SARS-CoV-2 infection: A meta-analysis. PLoS ONE 2020, 15, e0239508. [Google Scholar] [CrossRef]
- Le Pendu, J.; Breiman, A.; Rocher, J.; Dion, M.; Ruvoën-Clouet, N. ABO Blood Types and COVID-19: Spurious, Anecdotal, or Truly Important Relationships? A Reasoned Review of Available Data. Viruses 2021, 13, 160. [Google Scholar] [CrossRef]
- Settembre, E.C.; Chen, J.Z.; Dormitzer, P.R.; Grigorieff, N.; Harrison, S.C. Atomic model of an infectious rotavirus particle. EMBO J. 2010, 30, 408–416. [Google Scholar] [CrossRef]
- Sharma, S.; Hagbom, M.; Svensson, L.; Nordgren, J. The Impact of Human Genetic Polymorphisms on Rotavirus Susceptibility, Epidemiology, and Vaccine Take. Viruses 2020, 12, 324. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Huang, P.; Tan, M.; Liu, Y.; Biesiada, J.; Meller, J.; Castello, A.A.; Jiang, B.; Jiang, X. Rotavirus VP8*: Phylogeny, Host Range, and Interaction with Histo-Blood Group Antigens. J. Virol. 2012, 86, 9899–9910. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, S.; Woodruff, A.L.; Xia, M.; Tan, M.; Kennedy, M.A.; Jiang, X. Structural basis of glycan specificity of P[19] VP8*: Implications for rotavirus zoonosis and evolution. PLoS Pathog. 2017, 13, e1006707. [Google Scholar] [CrossRef] [Green Version]
- Ramani, S.; Cortes-Penfield, N.W.; Hu, L.; Crawford, S.E.; Czako, R.; Smith, D.F.; Kang, G.; Ramig, R.F.; Le Pendu, J.; Prasad, B.V.V.; et al. The VP8 * Domain of Neonatal Rotavirus Strain G10P[11] Binds to Type II Precursor Glycans. J. Virol. 2013, 87, 7255–7264. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Huang, P.; Jiang, B.; Tan, M.; Morrow, A.L.; Jiang, X. Poly-LacNAc as an Age-Specific Ligand for Rotavirus P[11] in Neonates and Infants. PLoS ONE 2013, 8, e78113. [Google Scholar] [CrossRef] [Green Version]
- Gozalbo-Rovira, R.; Ciges-Tomas, J.R.; Vila-Vicent, S.; Buesa, J.; Santiso-Bellón, C.; Monedero, V.; Yebra, M.; Marina, A.; Rodríguez-Díaz, J. Unraveling the role of the secretor antigen in human rotavirus attachment to histo-blood group antigens. PLoS Pathog. 2019, 15, e1007865. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Dang, L.; Li, D.; Qi, J.; Wang, M.; Chai, W.; Zhang, Q.; Wang, H.; Bai, R.; Tan, M.; et al. Structural Basis of Glycan Recognition in Globally Predominant Human P[8] Rotavirus. Virol. Sin. 2019, 35, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ahmed, L.U.; Stuckert, M.R.; McGinnis, K.R.; Liu, Y.; Tan, M.; Huang, P.; Zhong, W.; Zhao, D.; Jiang, X.; et al. Molecular basis of P[II] major human rotavirus VP8* domain recognition of histo-blood group antigens. PLoS Pathog. 2020, 16, e1008386. [Google Scholar] [CrossRef] [Green Version]
- Kambhampati, A.; Payne, D.C.; Costantini, V.; Lopman, B.A. Host Genetic Susceptibility to Enteric Viruses: A Systematic Review and Metaanalysis. Clin. Infect. Dis. 2015, 62, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramani, S.; Giri, S. Influence of histo blood group antigen expression on susceptibility to enteric viruses and vaccines. Curr. Opin. Infect. Dis. 2019, 32, 445–452. [Google Scholar] [CrossRef]
- Saxena, K.; Blutt, S.E.; Ettayebi, K.; Zeng, X.-L.; Broughman, J.R.; Crawford, S.E.; Karandikar, U.C.; Sastri, N.P.; Conner, M.E.; Opekun, A.R.; et al. Human Intestinal Enteroids: A New Model To Study Human Rotavirus Infection, Host Restriction, and Pathophysiology. J. Virol. 2016, 90, 43–56. [Google Scholar] [CrossRef] [Green Version]
- Barbé, L.; Le Moullac-Vaidye, B.; Echasserieau, K.; Bernardeau, K.; Carton, T.; Bovin, N.; Nordgren, J.; Svensson, L.; Ruvoën-Clouet, N.; Le Pendu, J. Histo-blood group antigen-binding specificities of human rotaviruses are associated with gastroenteritis but not with in vitro infection. Sci. Rep. 2018, 8, 12961. [Google Scholar] [CrossRef]
- Wu, S.-C.; Arthur, C.M.; Wang, J.; Verkerke, H.; Josephson, C.D.; Kalman, D.; Roback, J.D.; Cummings, R.D.; Stowell, S.R. The SARS-CoV-2 receptor-binding domain preferentially recognizes blood group A. Blood Adv. 2021, 5, 1305–1309. [Google Scholar] [CrossRef] [PubMed]
- Ryzhikov, A.B.; Onkhonova, G.S.; Imatdinov, I.R.; Gavrilova, E.V.; Maksyutov, R.A.; Gordeeva, E.A.; Pazynina, G.V.; Ryzhov, I.M.; Shilova, N.V.; Bovin, N.V. Recombinant SARS-CoV-2 S Protein Binds to Glycans of the Lactosamine Family in vitro. Biochem. (Moscow) 2021, 86, 243–247. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.e15. [Google Scholar] [CrossRef] [PubMed]
- De Pasquale, V.; Quiccione, M.; Tafuri, S.; Avallone, L.; Pavone, L. Heparan Sulfate Proteoglycans in Viral Infection and Treatment: A Special Focus on SARS-CoV-2. Int. J. Mol. Sci. 2021, 22, 6574. [Google Scholar] [CrossRef]
- Bally, M.; Block, S.; Höök, F.; Larson, G.; Parveen, N.; Rydell, G.E. Physicochemical tools for studying virus interactions with targeted cell membranes in a molecular and spatiotemporally resolved context. Anal. Bioanal. Chem. 2021, 1–22. [Google Scholar] [CrossRef]
- Costantini, V.; Morantz, E.K.; Browne, H.; Ettayebi, K.; Zeng, X.-L.; Atmar, R.L.; Estes, M.K.; Vinje, J. Human Norovirus Replication in Human Intestinal Enteroids as Model to Evaluate Virus Inactivation. Emerg. Infect. Dis. 2018, 24, 1453–1464. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Description in Manuscript | Manuscript Reference | Type | Accession Number 1 | RIVM Norovirus Typing Tool | Human Calicivirus Typing Tool |
|---|---|---|---|---|---|
| Dijon | [37] | VLP | AF472623 | GII.4, US95/96 | GII.4 |
| GII.4 011617 | [38] | Stool virus isolate | MN782359 | GII.4[P16] Sydney_2012 | GII.4[P16] |
| Grimsby (GrV) | [39,40] | VLP | AJ004864 | GII.4, US95/96 | GII.4 |
| HS66 | [41] | VLP | EU105469 | GII.4, US95/96 | GII.4 |
| MD145 | [41] | VLP | AY032605 | GII.4[P4], Camberwell 1994 | GII.4[P12] |
| MI001 | [42] | P domain | KC631814 | GII.4[P4], Yerseke_2006a | GII.4[P4] |
| PDB 4WZT | Figure 2 | P domain | JX459908 | GII.4[P31], Sydney_2012 | GII.4[P31] |
| Saga | [42,43] | P domain | AB447457 | GII.4[P4], Den_Haag_2006b | GII.4[P4] |
| Spanish isolate Ast6139/01/Sp | [44] | VLP | AJ583672 | GII.4[P4], Farmington_Hills_2002 | GII.4[P4] |
| Sydney_2012 | [45] | VLP | JX459908 | GII.4[P31], Sydney_2012 | GII.4[P31] |
| TCH05 | [46] | P domain | JF827296 | GII.4, Hunter _2004 | GII.4 |
| TCH12-580 | [47,48,49] | Stool virus isolate | Unpublished | GII.4[P31], Sydney_2012 | GII.4[P31] |
| VA387 | [39,40,50] | VLP | AY038600 | GII.4[P4], US95/96 | GII.4[P4] |
| VA387 | [42] | P domain | AY038600 | GII.4[P4], US95/96 | GII.4[P4] |
| Saliva Binding Pattern | |||||||
|---|---|---|---|---|---|---|---|
| Secretors | |||||||
| Binding Group | VLP Name | Genotype (Variant) | A | B | H | Non-Secretors | Reference |
| 1 | VA387 | GII.4 (US95/96) | Y | Y | Y | N | [39,40] |
| Grimsby (GrV) | GII.4 (US95/96) | Y | Y | Y | N | [40] | |
| 2 | MOH | GII.5 | Y | Y | N | N | [39,40] |
| Mexico (MxV) | GII.3 | Y | Y | N | N | [40] | |
| Parris Island (PiV) | GII.3 | Y | Y | N | N | [40] | |
| 3 | BUDS | GII.2 | Y | N | N | N | [40] |
| 4 | Norwalk | GI.1 | Y | N | Y | N | [39,40] |
| C59 | GI.2 | Y | N | Y | N | [40] | |
| 5 | Boxer | GI.1 | Y | N | Y | Y | [40] |
| VA207 | GII.9 | Y | N | Y | Y | [39,40] | |
| 6 | Operation Iraqi Freedom 031998 (OIF) | GII.21 | N | N | Y | Y | [40] |
| 7 | Desert Shield virus (DSV) | GI.3 | N | N | N | N | [40] |
| VA115 | GI.3 | N | N | N | N | [40] | |
| Hawaii virus (HV) | GII.1 | N | N | N | N | [40] | |
| HIE Line | Modification | Secretor Status | HBGA Expression | GI.1 Infection | GII.3 Infection | GII.4 Infection | GII.17 Infection |
|---|---|---|---|---|---|---|---|
| J2Fut2+/− | Parental | Positive | B Leb | Yes | Yes | Yes | Yes |
| J2Fut2−/− | FUT2 KO | Negative | Lea | No | Yes | No | No |
| J4Fut2−/− | Parental | Negative | Lea | No | No | No | No |
| J4Fut2−/−/FUT2 | FUT2 KI | Positive | Leb | Yes | Yes | Yes | Yes |
| Fucosyltransferase | Activity | J2 Expression (cpm) |
|---|---|---|
| FUT1 | Alpha(1,2) | 112 |
| FUT2 | Alpha(1,2) | 6587 |
| FUT3 | Alpha(1,3/1,4) | 9276 |
| FUT4 | Alpha(1,3) | 3411 |
| FUT5 | Alpha(1,3) | ND |
| FUT6 | Alpha(1,3) | 2485 |
| FUT7 | Alpha(1,3) | ND |
| FUT8 | Alpha(1,6) | 2534 |
| FUT9 | Alpha(1,3) | 47 |
| FUT10 | Alpha(1,3) putative | 715 |
| FUT11 | Alpha(1,3) putative | 1568 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tenge, V.R.; Hu, L.; Prasad, B.V.V.; Larson, G.; Atmar, R.L.; Estes, M.K.; Ramani, S. Glycan Recognition in Human Norovirus Infections. Viruses 2021, 13, 2066. https://doi.org/10.3390/v13102066
Tenge VR, Hu L, Prasad BVV, Larson G, Atmar RL, Estes MK, Ramani S. Glycan Recognition in Human Norovirus Infections. Viruses. 2021; 13(10):2066. https://doi.org/10.3390/v13102066
Chicago/Turabian StyleTenge, Victoria R., Liya Hu, B. V. Venkataram Prasad, Göran Larson, Robert L. Atmar, Mary K. Estes, and Sasirekha Ramani. 2021. "Glycan Recognition in Human Norovirus Infections" Viruses 13, no. 10: 2066. https://doi.org/10.3390/v13102066
APA StyleTenge, V. R., Hu, L., Prasad, B. V. V., Larson, G., Atmar, R. L., Estes, M. K., & Ramani, S. (2021). Glycan Recognition in Human Norovirus Infections. Viruses, 13(10), 2066. https://doi.org/10.3390/v13102066