
Utilizing the VirIdAl Pipeline to Search for Viruses in the Metagenomic Data of Bat Samples


 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
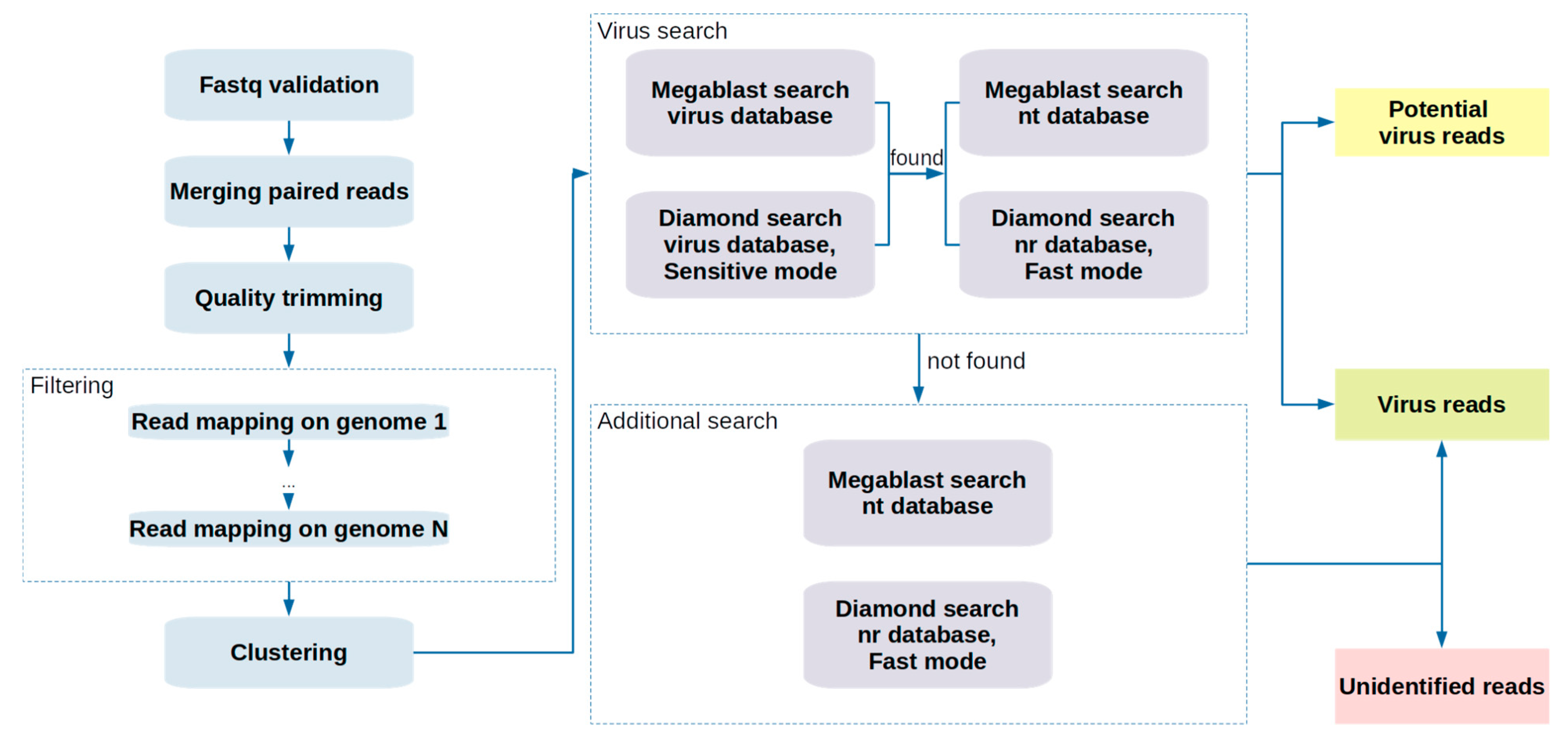
2.1. Pipeline Description
2.2. Computation Details
2.3. Sample Collection, Storage, and Library Preparation
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anthony, S.J.; Epstein, J.H.; Murray, K.A.; Navarrete-Macias, I.; Zambrana-Torrelio, C.M.; Solovyov, A.; Ojeda-Flores, R.; Arrigo, N.C.; Islam, A.; Ali Khan, S.; et al. A Strategy to Estimate Unknown Viral Diversity in Mammals. MBio 2013, 4, e00598-13. [Google Scholar] [CrossRef]
- Woolhouse, M.; Scott, F.; Hudson, Z.; Howey, R.; Chase-Topping, M. Human Viruses: Discovery and Emergence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2864–2871. [Google Scholar] [CrossRef]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global Trends in Emerging Infectious Diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Sørensen, M.D.; Sørensen, B.; Gonzalez-Dosal, R.; Melchjorsen, C.J.; Weibel, J.; Wang, J.; Jun, C.W.; Huanming, Y.; Kristensen, P. Severe Acute Respiratory Syndrome (SARS): Development of Diagnostics and Antivirals. Ann. N. Y. Acad. Sci. 2006, 1067, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Stadler, K.; Masignani, V.; Eickmann, M.; Becker, S.; Abrignani, S.; Klenk, H.-D.; Rappuoli, R. SARS—Beginning to Understand a New Virus. Nat. Rev. Microbiol. 2003, 1, 209–218. [Google Scholar] [CrossRef]
- Park, D.J.; Dudas, G.; Wohl, S.; Goba, A.; Whitmer, S.L.M.; Andersen, K.G.; Sealfon, R.S.; Ladner, J.T.; Kugelman, J.R.; Matranga, C.B.; et al. Ebola Virus Epidemiology, Transmission, and Evolution during Seven Months in Sierra Leone. Cell 2015, 161, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, N.; ECDC Internal Response Team; Catchpole, M. Novel Coronavirus Associated with Severe Respiratory Disease: Case Definition and Public Health Measures. Euro Surveill. 2012, 17. [Google Scholar] [CrossRef]
- Corman, V.M.; Eckerle, I.; Bleicker, T.; Zaki, A.; Landt, O.; Eschbach-Bludau, M.; van Boheemen, S.; Gopal, R.; Ballhause, M.; Bestebroer, T.M.; et al. Detection of a Novel Human Coronavirus by Real-Time Reverse-Transcription Polymerase Chain Reaction. Euro Surveill. 2012, 17. [Google Scholar] [CrossRef]
- Metsky, H.C.; Matranga, C.B.; Wohl, S.; Schaffner, S.F.; Freije, C.A.; Winnicki, S.M.; West, K.; Qu, J.; Baniecki, M.L.; Gladden-Young, A.; et al. Zika Virus Evolution and Spread in the Americas. Nature 2017, 546, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Segreto, R.; Deigin, Y. The Genetic Structure of SARS-CoV-2 Does Not Rule out a Laboratory Origin: SARS-CoV-2 Chimeric Structure and Furin Cleavage Site Might Be the Result of Genetic Manipulation. Bioessays 2021, 43, e2000240. [Google Scholar] [CrossRef] [PubMed]
- Burki, T. The Origin of SARS-CoV-2. Lancet Infect. Dis. 2020, 20, 1018–1019. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The Proximal Origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Piplani, S.; Singh, P.K.; Winkler, D.A.; Petrovsky, N. In Silico Comparison of SARS-CoV-2 Spike Protein-ACE2 Binding Affinities across Species and Implications for Virus Origin. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing Technologies—The next Generation. Nat. Rev. Genet. 2009, 11, 31–46. [Google Scholar] [CrossRef]
- Adams, I.P.; Glover, R.H.; Monger, W.A.; Mumford, R.; Jackeviciene, E.; Navalinskiene, M.; Samuitiene, M.; Boonham, N. Next-Generation Sequencing and Metagenomic Analysis: A Universal Diagnostic Tool in Plant Virology. Mol. Plant Pathol. 2009, 10, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Radford, A.D.; Chapman, D.; Dixon, L.; Chantrey, J.; Darby, A.C.; Hall, N. Application of next-Generation Sequencing Technologies in Virology. J. Gen. Virol. 2012, 93, 1853–1868. [Google Scholar] [CrossRef]
- Jansen, S.A.; Nijhuis, W.; Leavis, H.L.; Riezebos-Brilman, A.; Lindemans, C.A.; Schuurman, R. Broad Virus Detection and Variant Discovery in Fecal Samples of Hematopoietic Transplant Recipients Using Targeted Sequence Capture Metagenomics. Front. Microbiol. 2020, 11, 560179. [Google Scholar] [CrossRef]
- Chiu, C.Y. Viral Pathogen Discovery. Curr. Opin. Microbiol. 2013, 16, 468–478. [Google Scholar] [CrossRef]
- Giallonardo, F.D.; Töpfer, A.; Rey, M.; Prabhakaran, S.; Duport, Y.; Leemann, C.; Schmutz, S.; Campbell, N.K.; Joos, B.; Lecca, M.R.; et al. Full-Length Haplotype Reconstruction to Infer the Structure of Heterogeneous Virus Populations. Nucleic Acids Res. 2014, 42, e115. [Google Scholar] [CrossRef]
- De Vries, J.J.C.; Brown, J.R.; Couto, N.; Beer, M.; Le Mercier, P.; Sidorov, I.; Papa, A.; Fischer, N.; Oude Munnink, B.B.; Rodriquez, C.; et al. Recommendations for the Introduction of Metagenomic next-Generation Sequencing in Clinical Virology, Part II: Bioinformatic Analysis and Reporting. J. Clin. Virol. 2021, 138, 104812. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, D.; Matsvay, A.; Abramov, I.; Dedkov, V.; Shipulin, G.; Khafizov, K. Current Trends in Diagnostics of Viral Infections of Unknown Etiology. Viruses 2020, 12, 211. [Google Scholar] [CrossRef]
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu. Rev. Pathol. 2019, 14, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Alquezar-Planas, D.E.; Mourier, T.; Bruhn, C.A.W.; Hansen, A.J.; Vitcetz, S.N.; Mørk, S.; Gorodkin, J.; Nielsen, H.A.; Guo, Y.; Sethuraman, A.; et al. Discovery of a Divergent HPIV4 from Respiratory Secretions Using Second and Third Generation Metagenomic Sequencing. Sci. Rep. 2013, 3, 2468. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental Genome Shotgun Sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy-O’Grady, H.; Workentine, M.L. The Challenge and Potential of Metagenomics in the Clinic. Front. Immunol. 2016, 7, 29. [Google Scholar] [CrossRef]
- McLaren, M.R.; Willis, A.D.; Callahan, B.J. Consistent and Correctable Bias in Metagenomic Sequencing Experiments. Elife 2019, 8. [Google Scholar] [CrossRef]
- Boers, S.A.; Jansen, R.; Hays, J.P. Understanding and Overcoming the Pitfalls and Biases of next-Generation Sequencing (NGS) Methods for Use in the Routine Clinical Microbiological Diagnostic Laboratory. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1059–1070. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Liu, T.; Yu, C.-H.; Chiang, T.-Y.; Hwang, C.-C. Effects of GC Bias in next-Generation-Sequencing Data on de Novo Genome Assembly. PLoS ONE 2013, 8, e62856. [Google Scholar] [CrossRef]
- Kustin, T.; Ling, G.; Sharabi, S.; Ram, D.; Friedman, N.; Zuckerman, N.; Bucris, E.D.; Glatman-Freedman, A.; Stern, A.; Mandelboim, M. A Method to Identify Respiratory Virus Infections in Clinical Samples Using next-Generation Sequencing. Sci. Rep. 2019, 9, 2606. [Google Scholar] [CrossRef]
- Choi, S.-H.; Hong, S.-B.; Ko, G.-B.; Lee, Y.; Park, H.J.; Park, S.-Y.; Moon, S.M.; Cho, O.-H.; Park, K.-H.; Chong, Y.P.; et al. Viral Infection in Patients with Severe Pneumonia Requiring Intensive Care Unit Admission. Am. J. Respir. Crit. Care Med. 2012, 186, 325–332. [Google Scholar] [CrossRef]
- Datta, S.; Budhauliya, R.; Das, B.; Chatterjee, S.; Vanlalhmuaka; Veer, V. Next-Generation Sequencing in Clinical Virology: Discovery of New Viruses. World J. Virol. 2015, 4, 265–276. [Google Scholar] [CrossRef]
- Hijano, D.R.; Brazelton de Cardenas, J.; Maron, G.; Garner, C.D.; Ferrolino, J.A.; Dallas, R.H.; Gu, Z.; Hayden, R.T. Clinical Correlation of Influenza and Respiratory Syncytial Virus Load Measured by Digital PCR. PLoS ONE 2019, 14, e0220908. [Google Scholar] [CrossRef]
- Allen, U.D.; Hu, P.; Pereira, S.L.; Robinson, J.L.; Paton, T.A.; Beyene, J.; Khodai-Booran, N.; Dipchand, A.; Hébert, D.; Ng, V.; et al. The Genetic Diversity of Epstein-Barr Virus in the Setting of Transplantation Relative to Non-Transplant Settings: A Feasibility Study. Pediatr. Transplant. 2016, 20, 124–129. [Google Scholar] [CrossRef]
- Matranga, C.B.; Andersen, K.G.; Winnicki, S.; Busby, M.; Gladden, A.D.; Tewhey, R.; Stremlau, M.; Berlin, A.; Gire, S.K.; England, E.; et al. Enhanced Methods for Unbiased Deep Sequencing of Lassa and Ebola RNA Viruses from Clinical and Biological Samples. Genome Biol. 2014, 15, 519. [Google Scholar] [CrossRef] [PubMed]
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M.P.G. Overview of Virus Metagenomic Classification Methods and Their Biological Applications. Front. Microbiol. 2018, 9, 749. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Wu, G.; Lim, E.S.; Droit, L.; Krishnamurthy, S.; Barouch, D.H.; Virgin, H.W.; Wang, D. VirusSeeker, a Computational Pipeline for Virus Discovery and Virome Composition Analysis. Virology 2017, 503, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Andrusch, A.; Dabrowski, P.W.; Klenner, J.; Tausch, S.H.; Kohl, C.; Osman, A.A.; Renard, B.Y.; Nitsche, A. PAIPline: Pathogen Identification in Metagenomic and Clinical next Generation Sequencing Samples. Bioinformatics 2018, 34, i715–i721. [Google Scholar] [CrossRef] [PubMed]
- Plyusnin, I.; Kant, R.; Jääskeläinen, A.J.; Sironen, T.; Holm, L.; Vapalahti, O.; Smura, T. Novel NGS Pipeline for Virus Discovery from a Wide Spectrum of Hosts and Sample Types. Virus Evol. 2020, 6, veaa091. [Google Scholar] [CrossRef]
- Wylie, T.N.; Wylie, K.M. ViroMatch: A Computational Pipeline for the Detection of Viral Sequences from Complex Metagenomic Data. Microbiol. Resour. Announc. 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Nie, K.; Zhang, C.; Zhang, Y.; Wang, J.; Niu, P.; Ma, X. VIP: An Integrated Pipeline for Metagenomics of Virus Identification and Discovery. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Garretto, A.; Hatzopoulos, T.; Putonti, C. virMine: Automated Detection of Viral Sequences from Complex Metagenomic Samples. PeerJ 2019, 7, e6695. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast Metagenomic Sequence Classification Using Exact Alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and Sensitive Classification of Metagenomic Sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A Greedy Algorithm for Aligning DNA Sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Ye, S.H.; Siddle, K.J.; Park, D.J.; Sabeti, P.C. Benchmarking Metagenomics Tools for Taxonomic Classification. Cell 2019, 178, 779–794. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in Homology Search: HMMER3 and Convergent Evolution of Coiled-Coil Regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining Viral Signal from Microbial Genomic Data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Antipov, D.; Raiko, M.; Lapidus, A.; Pevzner, P.A. Metaviral SPAdes: Assembly of Viruses from Metagenomic Data. Bioinformatics 2020, 36, 4126–4129. [Google Scholar] [CrossRef]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A Novel K-Mer Based Tool for Identifying Viral Sequences from Assembled Metagenomic Data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A Multi-Classifier, Expert-Guided Approach to Detect Diverse DNA and RNA Viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Amgarten, D.; Braga, L.P.P.; da Silva, A.M.; Setubal, J.C. MARVEL, a Tool for Prediction of Bacteriophage Sequences in Metagenomic Bins. Front. Genet. 2018, 9, 304. [Google Scholar] [CrossRef] [PubMed]
- Auslander, N.; Gussow, A.B.; Benler, S.; Wolf, Y.I.; Koonin, E.V. Seeker: Alignment-Free Identification of Bacteriophage Genomes by Deep Learning. Nucleic Acids Res. 2020, 48, e121. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Song, K.; Deng, C.; Ahlgren, N.A.; Fuhrman, J.A.; Li, Y.; Xie, X.; Poplin, R.; Sun, F. Identifying Viruses from Metagenomic Data Using Deep Learning. Quant. Biol. 2020, 8, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Tampuu, A.; Bzhalava, Z.; Dillner, J.; Vicente, R. ViraMiner: Deep Learning on Raw DNA Sequences for Identifying Viral Genomes in Human Samples. PLoS ONE 2019, 14, e0222271. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewicz, J.M.; Seidel, A.; Renard, B.Y. Interpretable Detection of Novel Human Viruses from Genome Sequencing Data. NAR Genom. Bioinform. 2021, 3, lqab004. [Google Scholar] [CrossRef]
- Kivistö, I.; Tidenberg, E.-M.; Lilley, T.; Suominen, K.; Forbes, K.M.; Vapalahti, O.; Huovilainen, A.; Sironen, T. First Report of Coronaviruses in Northern European Bats. Vector Borne Zoonotic Dis. 2020, 20, 155–158. [Google Scholar] [CrossRef]
- Li, B.; Si, H.-R.; Zhu, Y.; Yang, X.-L.; Anderson, D.E.; Shi, Z.-L.; Wang, L.-F.; Zhou, P. Discovery of Bat Coronaviruses through Surveillance and Probe Capture-Based next-Generation Sequencing. mSphere 2020, 5. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Zhang, D.; Lou, X.; Yan, H.; Pan, J.; Mao, H.; Tang, H.; Shu, Y.; Zhao, Y.; Liu, L.; Li, J.; et al. Metagenomic Analysis of Viral Nucleic Acid Extraction Methods in Respiratory Clinical Samples. BMC Genom. 2018, 19, 773. [Google Scholar] [CrossRef] [PubMed]
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important Reservoir Hosts of Emerging Viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef]
- Banerjee, A.; Kulcsar, K.; Misra, V.; Frieman, M.; Mossman, K. Bats and Coronaviruses. Viruses 2019, 11, 41. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Ma, H.; Tan, T.W.; Ban, K.H.K. A Multi-Task CNN Learning Model for Taxonomic Assignment of Human Viruses. BMC Bioinform. 2021, 22, 194. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.J.; Eddy, S.R. Nhmmer: DNA Homology Search with Profile HMMs. Bioinformatics 2013, 29, 2487–2489. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sample ID | Number of Sequences in Clustered Fasta Files | Number of Unidentified Sequences | Percentage of Unidentified Sequences (%) |
|---|---|---|---|
| 2 | 1,496,002 | 892,558 | 59.7 |
| 6 | 738,554 | 363,592 | 49.2 |
| 16 | 653,809 | 119,176 | 18.2 |
| 19 | 906,791 | 404,517 | 44.6 |
| 20 | 538,581 | 145,623 | 27.0 |
| 21 | 764,442 | 428,544 | 56.1 |
| 22 | 777,210 | 272,237 | 35.0 |
| 23 | 1,466,914 | 711,736 | 48.5 |
| 27 | 2,245,717 | 1,498,197 | 66.7 |
| 30 | 651,436 | 197,040 | 30.3 |
| 31 | 830,335 | 313,898 | 37.8 |
| Sample ID | Unidentified Sequences | Sequences with DeePaC-Vir Score > 0.5 | Virus Sequences Identified by HMMER Hmmscan Only, E-Value < 10 |
|---|---|---|---|
| 21 | 428,544 | 63,969 | 177 |
| 22 | 272,237 | 38,413 | 82 |
| 33 | 482,033 | 35,361 | 69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budkina, A.Y.; Korneenko, E.V.; Kotov, I.A.; Kiselev, D.A.; Artyushin, I.V.; Speranskaya, A.S.; Khafizov, K.; Akimkin, V.G. Utilizing the VirIdAl Pipeline to Search for Viruses in the Metagenomic Data of Bat Samples. Viruses 2021, 13, 2006. https://doi.org/10.3390/v13102006
Budkina AY, Korneenko EV, Kotov IA, Kiselev DA, Artyushin IV, Speranskaya AS, Khafizov K, Akimkin VG. Utilizing the VirIdAl Pipeline to Search for Viruses in the Metagenomic Data of Bat Samples. Viruses. 2021; 13(10):2006. https://doi.org/10.3390/v13102006
Chicago/Turabian StyleBudkina, Anna Y., Elena V. Korneenko, Ivan A. Kotov, Daniil A. Kiselev, Ilya V. Artyushin, Anna S. Speranskaya, Kamil Khafizov, and Vasily G. Akimkin. 2021. "Utilizing the VirIdAl Pipeline to Search for Viruses in the Metagenomic Data of Bat Samples" Viruses 13, no. 10: 2006. https://doi.org/10.3390/v13102006
APA StyleBudkina, A. Y., Korneenko, E. V., Kotov, I. A., Kiselev, D. A., Artyushin, I. V., Speranskaya, A. S., Khafizov, K., & Akimkin, V. G. (2021). Utilizing the VirIdAl Pipeline to Search for Viruses in the Metagenomic Data of Bat Samples. Viruses, 13(10), 2006. https://doi.org/10.3390/v13102006