
Endothelium Infection and Dysregulation by SARS-CoV-2: Evidence and Caveats in COVID-19




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Search Strategy
3. Entry Receptors and Tropism of SARS-CoV-2 and Related Human Coronaviruses
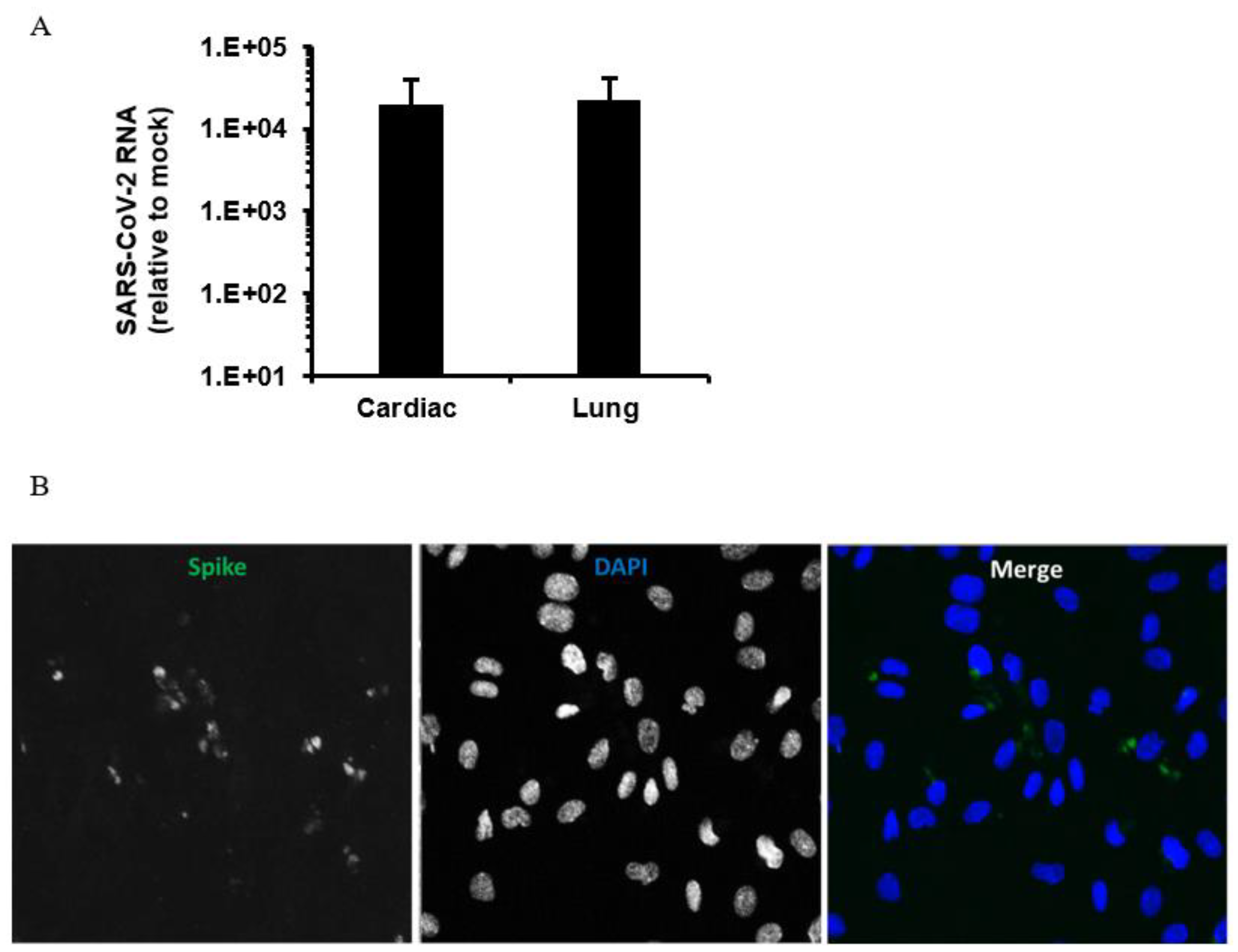
4. Does SARS-CoV-2 Infect Endothelial Cells?
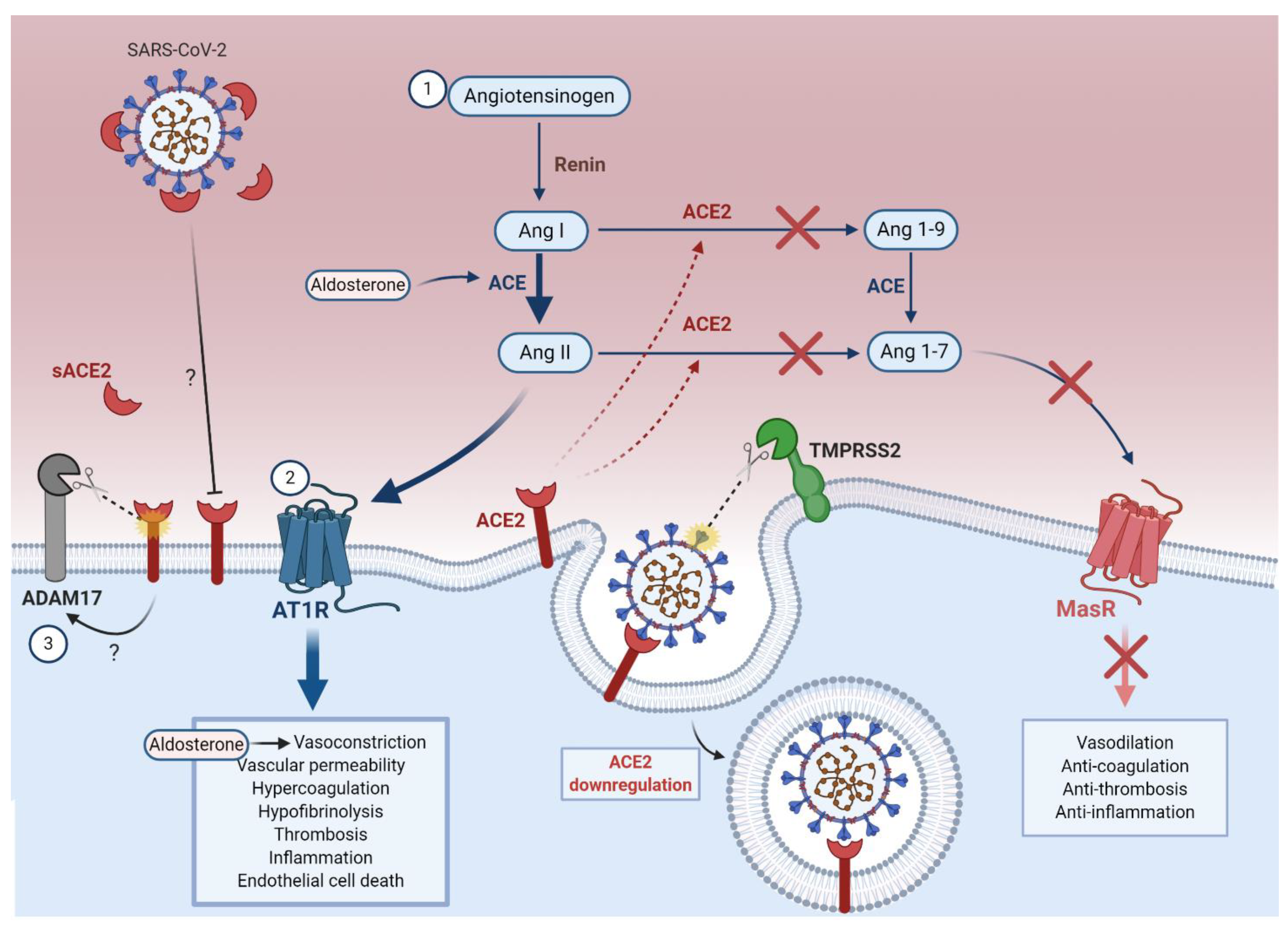
5. Cellular ACE2 Downregulation Leading to Endothelial Dysfunction Is Mediated by SARS-CoV-2
5.1. RAAS Dysfunction
5.1.1. ACE2 Downregulation Increases the Ang II to Ang 1-7 Ratio
5.1.2. Ang II Upregulation Increases the PAI-1 to tPA/uPA Ratio
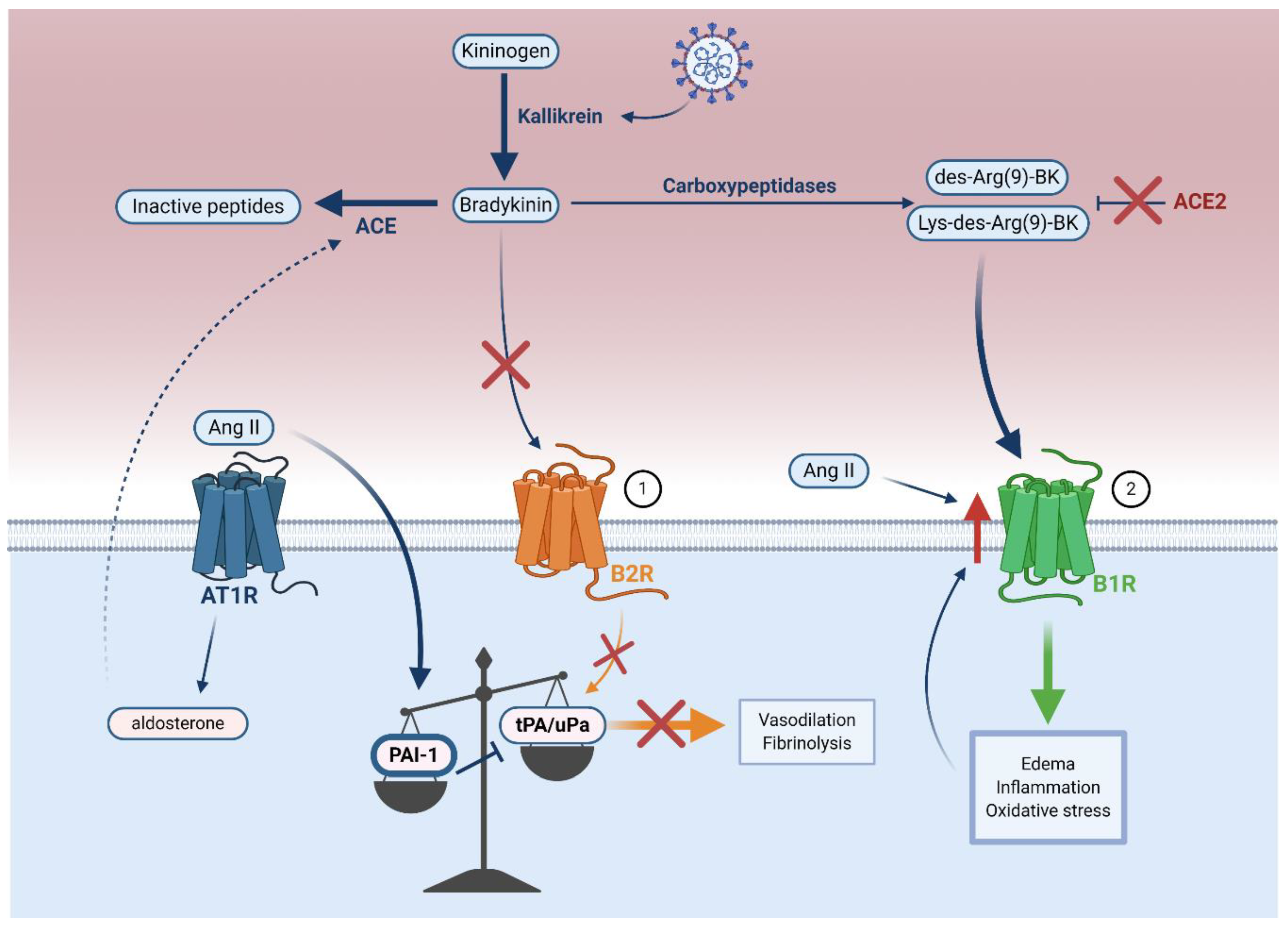
5.2. Bradykinin–Kallikrein Pathway Dysfunction
5.3. ADAM17-Mediated ACE2 Shedding
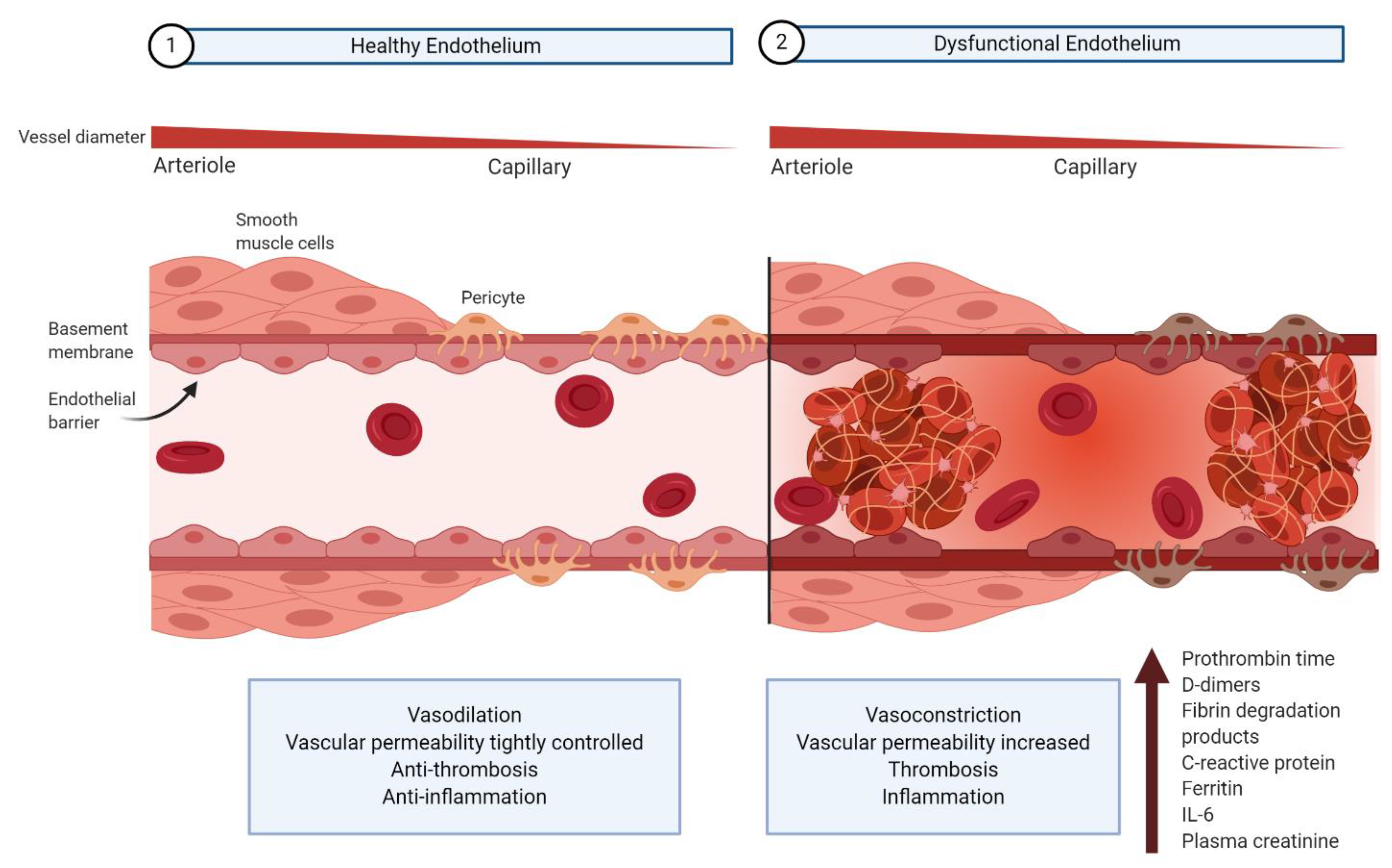
6. Consequences of Endothelium Dysfunction in COVID-19
6.1. Dysfunction of Pericyte–Endothelial Cell Cross-Talk
6.2. Immunothrombosis
6.2.1. Hyperinflammation and Complement Activation
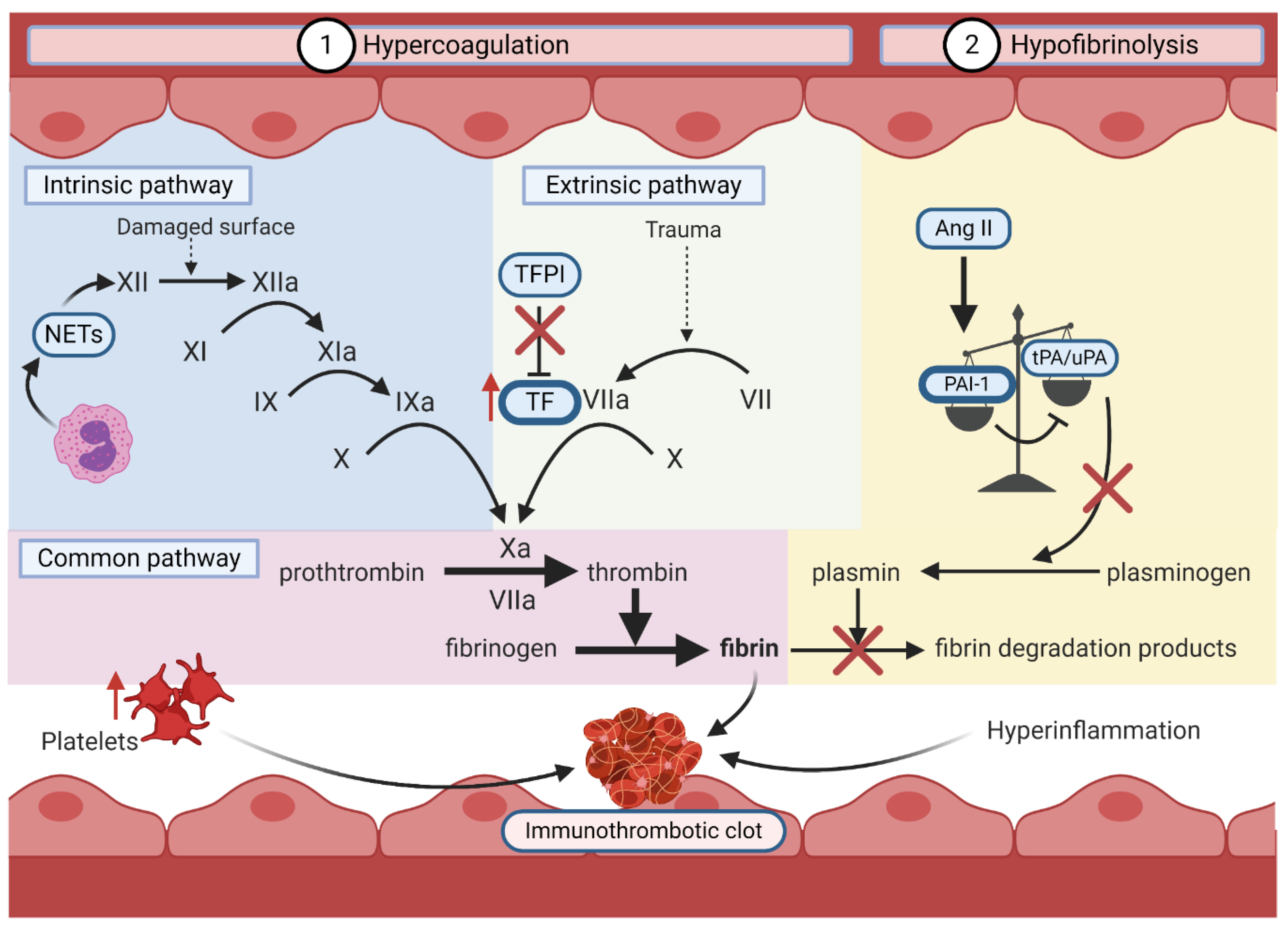
6.2.2. Hypercoagulation and Hypofibrinolysis
6.3. Impaired Antiviral Response
7. Therapeutic Strategies for SARS-CoV-2-Mediated Endothelial Dysfunction
7.1. Limiting SARS-CoV-2 Entry in Endothelial Cells
7.2. RAAS Inhibition
7.3. Immunomodulation
7.4. Inhibition of Thrombosis
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hui, D.S.; Azhar, E.I.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; Mchugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- WHO. Director-General’s Opening Remarks at the Media Briefing on COVID-19—11 March 2020. Available online: https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 28 June 2020).
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Behzad, S.; Aghaghazvini, L.; Radmard, A.R.; Gholamrezanezhad, A. Extrapulmonary manifestations of COVID-19: Radiologic and clinical overview. Clin. Imaging 2020, 66, 35–41. [Google Scholar] [CrossRef]
- Lee, I.-C.; Huo, T.-I.; Huang, Y.-H. Gastrointestinal and liver manifestations in patients with COVID-19. J. Chin. Med. Assoc. 2020. [Google Scholar] [CrossRef]
- Zheng, Y.-Y.; Ma, Y.-T.; Zhang, J.-Y.; Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef]
- Sardu, C.; Gambardella, J.; Morelli, M.B.; Wang, X.; Marfella, R.; Santulli, G. Hypertension, Thrombosis, Kidney Failure, and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence. J. Clin. Med. 2020, 9, 1417. [Google Scholar] [CrossRef]
- Jin, M.; Tong, Q. Rhabdomyolysis as Potential Late Complication Associated with COVID-19. Emerg. Infect. Dis. 2020, 26, 1618–1620. [Google Scholar] [CrossRef]
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020, 19, 767–783. [Google Scholar] [CrossRef]
- Yachou, Y.; El Idrissi, A.; Belapasov, V.; Ait Benali, S. Neuroinvasion, neurotropic, and neuroinflammatory events of SARS-CoV-2: Understanding the neurological manifestations in COVID-19 patients. Neurol. Sci. 2020, 41, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Jung, F.; Krüger-Genge, A.; Franke, R.P.; Hufert, F.; Küpper, J.-H. COVID-19 and the endothelium. Clin. Hemorheol. Microcirc. 2020, 1–5. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.D.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef]
- Pons, S.; Fodil, S.; Azoulay, E.; Zafrani, L. The vascular endothelium: The cornerstone of organ dysfunction in severe SARS-CoV-2 infection. Crit. Care 2020, 24, 353. [Google Scholar] [CrossRef]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15. [Google Scholar] [CrossRef]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.-A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Smith, C.; Dichgans, M. Small vessel disease: Mechanisms and clinical implications. Lancet Neurol. 2019, 18, 684–696. [Google Scholar] [CrossRef]
- Geevarghese, A.; Herman, I.M. Pericyte-Endothelial Cross-Talk: Implications and Opportunities for Advanced Cellular Therapies. Transl. Res. J. Lab. Clin. Med. 2014, 163, 296–306. [Google Scholar] [CrossRef]
- McGonagle, D.; O’Donnell, J.S.; Sharif, K.; Emery, P.; Bridgewood, C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020, 2, e437–e445. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Smadja, D.M.; Guerin, C.L.; Chocron, R.; Yatim, N.; Boussier, J.; Gendron, N.; Khider, L.; Hadjadj, J.; Goudot, G.; Debuc, B.; et al. Angiopoietin-2 as a marker of endothelial activation is a good predictor factor for intensive care unit admission of COVID-19 patients. Angiogenesis 2020, 1–10. [Google Scholar] [CrossRef]
- Ok, F.; Erdogan, O.; Durmus, E.; Carkci, S.; Canik, A. Predictive values of blood urea nitrogen/creatinine ratio and other routine blood parameters on disease severity and survival of COVID-19 patients. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; Barnaby, D.P.; Becker, L.B.; Chelico, J.D.; Cohen, S.L.; et al. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef]
- Desai, R.; Gandhi, Z.; Singh, S.; Sachdeva, S.; Manaktala, P.; Savani, S.; Desai, V.; Sachdeva, R.; Kumar, G. Prevalence of Pulmonary Embolism in COVID-19: A Pooled Analysis. Sn Compr. Clin. Med. 2020, 1–4. [Google Scholar] [CrossRef]
- NIH. What’s New in the Guidelines. Available online: https://www.covid19treatmentguidelines.nih.gov/whats-new/ (accessed on 1 November 2020).
- Spinner, C.D.; Gottlieb, R.L.; Criner, G.J.; Arribas López, J.R.; Cattelan, A.M.; Soriano Viladomiu, A.; Ogbuagu, O.; Malhotra, P.; Mullane, K.M.; Castagna, A.; et al. Effect of Remdesivir vs Standard Care on Clinical Status at 11 Days in Patients With Moderate COVID-19: A Randomized Clinical Trial. JAMA 2020, 324, 1048–1057. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Cohen, J.; Kupferschmidt, K. The ‘very, very Bad Look’ of Remdesivir, the First FDA-Approved COVID-19 Drug. Available online: https://www.sciencemag.org/news/2020/10/very-very-bad-look-remdesivir-first-fda-approved-covid-19-drug (accessed on 8 November 2020).
- WHO Solidarity Trial Consortium; Pan, H.; Peto, R.; Henao-Restrepo, A.-M.; Preziosi, M.-P.; Sathiyamoorthy, V.; Abdool Karim, Q.; Alejandria, M.M.; Hernández García, C.; Kieny, M.-P.; et al. Repurposed Antiviral Drugs for Covid-19—Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- RECOVERY Collaborative Group; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with Covid-19—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Bosch, B.J.; Zee, R.; van der Haan, C.A.M.; de Rottier, P.J.M. The Coronavirus Spike Protein is a Class I Virus Fusion Protein: Structural and Functional Characterization of the Fusion Core Complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef]
- Yeager, C.L.; Ashmun, R.A.; Williams, R.K.; Cardellichio, C.B.; Shapiro, L.H.; Look, A.T.; Holmes, K.V. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 1992, 357, 420–422. [Google Scholar] [CrossRef]
- Huang, X.; Dong, W.; Milewska, A.; Golda, A.; Qi, Y.; Zhu, Q.K.; Marasco, W.A.; Baric, R.S.; Sims, A.C.; Pyrc, K.; et al. Human Coronavirus HKU1 Spike Protein Uses O-Acetylated Sialic Acid as an Attachment Receptor Determinant and Employs Hemagglutinin-Esterase Protein as a Receptor-Destroying Enzyme. J. Virol. 2015, 89, 7202–7213. [Google Scholar] [CrossRef]
- Vlasak, R.; Luytjes, W.; Spaan, W.; Palese, P. Human and bovine coronaviruses recognize sialic acid-containing receptors similar to those of influenza C viruses. Proc. Natl. Acad. Sci. USA 1988, 85, 4526–4529. [Google Scholar] [CrossRef]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.W.; Müller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.A.; Zaki, A.; Fouchier, R.A.M.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Hofmann, H.; Pyrc, K.; van der Hoek, L.; Geier, M.; Berkhout, B.; Pöhlmann, S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. USA 2005, 102, 7988–7993. [Google Scholar] [CrossRef]
- To, K.; Tong, J.H.; Chan, P.K.; Au, F.W.; Chim, S.S.; Allen Chan, K.; Cheung, J.L.; Liu, E.Y.; Tse, G.M.; Lo, A.W.; et al. Tissue and cellular tropism of the coronavirus associated with severe acute respiratory syndrome: An in-situ hybridization study of fatal cases. J. Pathol. 2004, 202, 157–163. [Google Scholar] [CrossRef]
- To, K.F.; Lo, A.W. Exploring the pathogenesis of severe acute respiratory syndrome (SARS): The tissue distribution of the coronavirus (SARS-CoV) and its putative receptor, angiotensin-converting enzyme 2 (ACE2). J. Pathol. 2004, 203, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Gong, E.; Zhang, B.; Zheng, J.; Gao, Z.; Zhong, Y.; Zou, W.; Zhan, J.; Wang, S.; Xie, Z.; et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 2005, 202, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Wang, H.; Shen, H.; Li, Z.; Geng, J.; Han, H.; Cai, J.; Li, X.; Kang, W.; Weng, D.; et al. The clinical pathology of severe acute respiratory syndrome (SARS): A report from China. J. Pathol. 2003, 200, 282–289. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, B.; Xu, J.; Chang, Q.; McNutt, M.A.; Korteweg, C.; Gong, E.; Gu, J. Molecular Pathology in the Lungs of Severe Acute Respiratory Syndrome Patients. Am. J. Pathol. 2007, 170, 538–545. [Google Scholar] [CrossRef]
- Zhang, Q.; Ding, Y.; Hou, J.; He, L.; Huang, Z.; Wang, H.; Cai, J.; Zhang, J.; Zhang, W.; Geng, J.; et al. Detection of severe acute respiratory syndrome (SARS)-associated coronavirus RNA in autopsy tissues with in situ hybridization. Acad. J. First Med. Coll. PLA 2003, 23, 1125–1127. [Google Scholar]
- Glowacka, I.; Bertram, S.; Müller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 Activates the Severe Acute Respiratory Syndrome Coronavirus Spike Protein for Membrane Fusion and Reduces Viral Control by the Humoral Immune Response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient Activation of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein by the Transmembrane Protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Fan, X.; Cao, D.; Kong, L.; Zhang, X. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat. Commun. 2020, 11, 3618. [Google Scholar] [CrossRef]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 1–5. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Yao, H.; Song, Y.; Chen, Y.; Wu, N.; Xu, J.; Sun, C.; Zhang, J.; Weng, T.; Zhang, Z.; Wu, Z.; et al. Molecular Architecture of the SARS-CoV-2 Virus. Cell 2020, 183, 730–738.e13. [Google Scholar] [CrossRef] [PubMed]
- Turoňová, B.; Sikora, M.; Schürmann, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; von Bülow, S.; Gecht, M.; Bagola, K.; Hörner, C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef]
- Chu, H.; Chan, J.F.-W.; Yuen, T.T.-T.; Shuai, H.; Yuan, S.; Wang, Y.; Hu, B.; Yip, C.C.-Y.; Tsang, J.O.-L.; Huang, X.; et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: An observational study. Lancet Microbe 2020, 1, e14–e23. [Google Scholar] [CrossRef]
- Nicin, L.; Abplanalp, W.T.; Mellentin, H.; Kattih, B.; Tombor, L.; John, D.; Schmitto, J.D.; Heineke, J.; Emrich, F.; Arsalan, M.; et al. Cell type-specific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur. Heart J. 2020, 41, 1804–1806. [Google Scholar] [CrossRef]
- Bojkova, D.; McGreig, J.E.; McLaughlin, K.-M.; Masterson, S.G.; Widera, M.; Krähling, V.; Ciesek, S.; Wass, M.N.; Michaelis, M.; Cinatl, J. SARS-CoV-2 and SARS-CoV differ in their cell tropism and drug sensitivity profiles. bioRxiv 2020. [Google Scholar] [CrossRef]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef] [PubMed]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Robson, B. Bioinformatics studies on a function of the SARS-CoV-2 spike glycoprotein as the binding of host sialic acid glycans. Comput. Biol. Med. 2020, 122, 103849. [Google Scholar] [CrossRef]
- Saltos, F.A.; Saltos, A.D.A. Entry of SARS-CoV2 through the Basal Surface of Alveolar Endothelial Cells—A Proposed Mechanism Mediated by CD147 in COVID-19. Preprints 2020. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.-Y.; Geng, J.-J.; et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.-E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Paniz-Mondolfi, A.; Bryce, C.; Grimes, Z.; Gordon, R.E.; Reidy, J.; Lednicky, J.; Sordillo, E.M.; Fowkes, M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J. Med. Virol. 2020, 92, 699–702. [Google Scholar] [CrossRef]
- Colmenero, I.; Santonja, C.; Alonso-Riaño, M.; Noguera-Morel, L.; Hernández-Martín, A.; Andina, D.; Wiesner, T.; Rodríguez-Peralto, J.L.; Requena, L.; Torrelo, A. SARS-CoV-2 endothelial infection causes COVID-19 chilblains: Histopathological, immunohistochemical and ultraestructural study of 7 paediatric cases. Br. J. Dermatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bradley, B.T.; Maioli, H.; Johnston, R.; Chaudhry, I.; Fink, S.L.; Xu, H.; Najafian, B.; Deutsch, G.; Lacy, M.; Williams, T.; et al. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: A case series. Lancet 2020, 396, 320–332. [Google Scholar] [CrossRef]
- Puelles, V.G.; Lütgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, C.S.; Tatti, K.M.; Ksiazek, T.G.; Rollin, P.E.; Comer, J.A.; Lee, W.W.; Rota, P.A.; Bankamp, B.; Bellini, W.J.; Zaki, S.R. Ultrastructural Characterization of SARS Coronavirus. Emerg. Infect. Dis. 2004, 10, 320–326. [Google Scholar] [CrossRef]
- Bromfield, M.; McQuillan, R.; John, R.; Avila-Casado, C. The significance of tubuloreticular inclusions as a marker of systemic stimulation by interferons in a case of focal and segmental glomerulosclerosis associated with cytomegalovirus (CMV) infection. Clin. Kidney J. 2014, 7, 174–178. [Google Scholar] [CrossRef]
- Damsky, W.; Peterson, D.; King, B. When interferon tiptoes through COVID-19: Pernio-like lesions and their prognostic implications during SARS-CoV-2 infection. J. Am. Acad. Dermatol. 2020, 83, e269–e270. [Google Scholar] [CrossRef]
- Hui, K.P.Y.; Cheung, M.-C.; Perera, R.A.P.M.; Ng, K.-C.; Bui, C.H.T.; Ho, J.C.W.; Ng, M.M.T.; Kuok, D.I.T.; Shih, K.C.; Tsao, S.-W.; et al. Tropism, replication competence, and innate immune responses of the coronavirus SARS-CoV-2 in human respiratory tract and conjunctiva: An analysis in ex-vivo and in-vitro cultures. Lancet Respir. Med. 2020, 8, 687–695. [Google Scholar] [CrossRef]
- Schaefer, I.-M.; Padera, R.F.; Solomon, I.H.; Kanjilal, S.; Hammer, M.M.; Hornick, J.L.; Sholl, L.M. In situ detection of SARS-CoV-2 in lungs and airways of patients with COVID-19. Mod. Pathol. 2020, 1–11. [Google Scholar] [CrossRef]
- Yang, L.; Han, Y.; Nilsson-Payant, B.E.; Gupta, V.; Wang, P.; Duan, X.; Tang, X.; Zhu, J.; Zhao, Z.; Jaffré, F.; et al. A Human Pluripotent Stem Cell-based Platform to Study SARS-CoV-2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem Cell 2020, 27, 125–136.e7. [Google Scholar] [CrossRef]
- Goldsmith, C.S.; Miller, S.E.; Martines, R.B.; Bullock, H.A.; Zaki, S.R. Electron microscopy of SARS-CoV-2: A challenging task. Lancet Lond. Engl. 2020, 395, e99. [Google Scholar] [CrossRef]
- Roufosse, C.; Curtis, E.; Moran, L.; Hollinshead, M.; Cook, T.; Hanley, B.; Horsfield, C.; Neil, D. Electron microscopic investigations in COVID-19: Not all crowns are coronas. Kidney Int. 2020, 98, 505–506. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.E.; Brealey, J.K. Visualization of putative coronavirus in kidney. Kidney Int. 2020, 98, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Calomeni, E.; Satoskar, A.; Ayoub, I.; Brodsky, S.; Rovin, B.H.; Nadasdy, T. Multivesicular bodies mimicking SARS-CoV-2 in patients without COVID-19. Kidney Int. 2020, 98, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Dittmayer, C.; Meinhardt, J.; Radbruch, H.; Radke, J.; Heppner, B.I.; Heppner, F.L.; Stenzel, W.; Holland, G.; Laue, M. Why misinterpretation of electron micrographs in SARS-CoV-2-infected tissue goes viral. Lancet 2020, 396, e64–e65. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.; Mehra, M.R.; Scholkmann, F.; Schüpbach, R.; Ruschitzka, F.; et al. Electron microscopy of SARS-CoV-2: A challenging task—Authors’ reply. Lancet 2020, 395, e100. [Google Scholar] [CrossRef]
- Takayama, K. In Vitro and Animal Models for SARS-CoV-2 research. Trends Pharmacol. Sci. 2020, 41, 513–517. [Google Scholar] [CrossRef]
- Cagno, V. SARS-CoV-2 cellular tropism. Lancet Microbe 2020, 1, e2–e3. [Google Scholar] [CrossRef]
- Zhao, J.; Zhou, H.; Huang, W.; Zhou, J.; Qiu, M.; Deng, Z.; Chen, L.; Weng, Y.; Cai, L.; Gu, Y.; et al. Cell morphological analysis of SARS-CoV-2 infection by transmission electron microscopy. J. Thorac. Dis. 2020, 12, 4368–4373. [Google Scholar] [CrossRef]
- Kissling, S.; Rotman, S.; Gerber, C.; Halfon, M.; Lamoth, F.; Comte, D.; Lhopitallier, L.; Sadallah, S.; Fakhouri, F. Collapsing glomerulopathy in a COVID-19 patient. Kidney Int. 2020, 98, 228–231. [Google Scholar] [CrossRef]
- Su, H.; Yang, M.; Wan, C.; Yi, L.-X.; Tang, F.; Zhu, H.-Y.; Yi, F.; Yang, H.-C.; Fogo, A.B.; Nie, X.; et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.L.; Quade, B.; Deshpande, V.; Mino-Kenudson, M.; Ting, D.T.; Desai, N.; Dygulska, B.; Heyman, T.; Salafia, C.; Shen, D.; et al. SARS-CoV-2 can infect the placenta and is not associated with specific placental histopathology: A series of 19 placentas from COVID-19-positive mothers. Mod. Pathol. 2020, 1–12. [Google Scholar] [CrossRef]
- Ko, C.J.; Harigopal, M.; Gehlhausen, J.R.; Bosenberg, M.; McNiff, J.M.; Damsky, W. Discordant anti-SARS-CoV-2 spike protein and RNA staining in cutaneous perniotic lesions suggests endothelial deposition of cleaved spike protein. J. Cutan. Pathol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Henry, B.M.; Vikse, J.; Benoit, S.; Favaloro, E.J.; Lippi, G. Hyperinflammation and derangement of renin-angiotensin-aldosterone system in COVID-19: A novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin. Chim. Acta Int. J. Clin. Chem. 2020, 507, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Limonta, D.; Dyna-Dagman, L.; Branton, W.; Makio, T.; Wozniak, R.W.; Power, C.; Hobman, T.C. Nodosome inhibition as a novel broad-spectrum antiviral strategy against arboviruses and SARS-CoV-2. bioRxiv. 2020. [Google Scholar] [CrossRef]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H.; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446.e14. [Google Scholar] [CrossRef]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharm. 2017, 94, 317–325. [Google Scholar] [CrossRef]
- Li, D.; Yang, B.; Philips, M.I.; Mehta, J.L. Proapoptotic effects of ANG II in human coronary artery endothelial cells: Role of AT1receptor and PKC activation. Am. J. Physiol.-Heart Circ. Physiol. 1999, 276, H786–H792. [Google Scholar] [CrossRef]
- López-Farré, A.; Sánchez de Miguel, L.; Montón, M.; Jiménez, A.; Lopez-Bloya, A.; Gómez, J.; Núñez, A.; Rico, L.; Casado, S. Angiotensin II AT(1) receptor antagonists and platelet activation. Nephrol. Dial. Transpl. 2001, 16 Suppl 1, 45–49. [Google Scholar] [CrossRef]
- Keidar, S.; Kaplan, M.; Gamliel-Lazarovich, A. ACE2 of the heart: From angiotensin I to angiotensin (1–7). Cardiovasc. Res. 2007, 73, 463–469. [Google Scholar] [CrossRef]
- Zaman Tarikuz, A.K.M.; Fujii, S.; Sawa, H.; Goto, D.; Ishimori, N.; Watano, K.; Kaneko, T.; Furumoto, T.; Sugawara, T.; Sakuma, I.; et al. Angiotensin-Converting Enzyme Inhibition Attenuates Hypofibrinolysis and Reduces Cardiac Perivascular Fibrosis in Genetically Obese Diabetic Mice. Circulation 2001, 103, 3123–3128. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues Prestes, T.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simoes-E-Silva, A.C. The Anti-Inflammatory Potential of ACE2/Angiotensin-(1-7)/Mas Receptor Axis: Evidence from Basic and Clinical Research. Curr. Drug Targets 2017, 18, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Silhol, F.; Sarlon, G.; Deharo, J.-C.; Vaïsse, B. Downregulation of ACE2 induces overstimulation of the renin–angiotensin system in COVID-19: Should we block the renin–angiotensin system? Hypertens. Res. 2020, 43, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Kwaan, H.C. Coronavirus Disease 2019: The Role of the Fibrinolytic System from Transmission to Organ Injury and Sequelae. Semin. Thromb. Hemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.; Brosnihan, K.B.; Jacobsen, D.W.; DiCorleto, P.E.; Ferrario, C.M. Production of angiotensin-(1-7) by human vascular endothelium. Hypertension 1992. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Penninger, J.M. The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp. Physiol. 2008, 93, 543–548. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Reddy, R.; Asante, I.; Liu, S.; Parikh, P.; Liebler, J.; Borok, Z.; Rodgers, K.; Baydur, A.; Louie, S.G. Circulating angiotensin peptides levels in Acute Respiratory Distress Syndrome correlate with clinical outcomes: A pilot study. PLoS ONE 2019, 14, e0213096. [Google Scholar] [CrossRef]
- Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.-J.; Xie, J.; Liu, Y.-M.; Zhao, Y.-C.; Huang, X.; Lin, L.; et al. Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers with Mortality Among Patients with Hypertension Hospitalized With COVID-19. Circ. Res. 2020, 126, 1671–1681. [Google Scholar] [CrossRef]
- Khan, A.T.-A.; Khalid, Z.; Zahid, H.; Yousaf, M.A.; Shakoori, A.R. A computational and bioinformatic analysis of ACE2: An elucidation of its dual role in COVID-19 pathology and finding its associated partners as potential therapeutic targets. J. Biomol. Struct. Dyn. 2020, 1–17. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.; Bridget Tallant, E. Ann Diz Debra, I.; Gallagher Patricia, E. Effect of Angiotensin-Converting Enzyme Inhibition and Angiotensin II Receptor Blockers on Cardiac Angiotensin-Converting Enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef]
- Bryce, C.; Grimes, Z.; Pujadas, E.; Ahuja, S.; Beasley, M.B.; Albrecht, R.; Hernandez, T.; Stock, A.; Zhao, Z.; Rasheed, M.A.; et al. Pathophysiology of SARS-CoV-2: Targeting of endothelial cells renders a complex disease with thrombotic microangiopathy and aberrant immune response. The Mount Sinai COVID-19 autopsy experience. medRxiv 2020. [Google Scholar] [CrossRef]
- Sukhanov, S.; Semprun-Prieto, L.; Yoshida, T.; Tabony, A.M.; Higashi, Y.; Galvez, S.; Delafontaine, P. Angiotensin II, Oxidative Stress and Skeletal Muscle Wasting. Am. J. Med. Sci. 2011, 342, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Stangl, K.; Stangl, V. The ubiquitin-proteasome pathway and endothelial (dys)function. Cardiovasc. Res. 2010, 85, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.E.; Lazos, S.A.; Tong, K. Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. J. Clin. Investig. 1995, 95, 995–1001. [Google Scholar] [CrossRef]
- Brown, N.J.; Nakamura, S.; Ma, L.; Nakamura, I.; Donnert, E.; Freeman, M.; Vaughan, D.E.; Fogo, A.B. Aldosterone modulates plasminogen activator inhibitor-1 and glomerulosclerosis in vivo. Kidney Int. 2000, 58, 1219–1227. [Google Scholar] [CrossRef]
- Wright, F.L.; Vogler, T.O.; Moore, E.E.; Moore, H.B.; Wohlauer, M.V.; Urban, S.; Nydam, T.L.; Moore, P.K.; McIntyre, R.C. Fibrinolysis Shutdown Correlation with Thromboembolic Events in Severe COVID-19 Infection. J. Am. Coll. Surg. 2020, 231, 193–203.e1. [Google Scholar] [CrossRef]
- Nougier, C.; Benoit, R.; Simon, M.; Desmurs-Clavel, H.; Marcotte, G.; Argaud, L.; David, J.S.; Bonnet, A.; Negrier, C.; Dargaud, Y. Hypofibrinolytic state and high thrombin generation may play a major role in SARS-COV2 associated thrombosis. J. Thromb. Haemost. 2020, 18, 2215–2219. [Google Scholar] [CrossRef]
- van de Veerdonk, F.L.; Netea, M.G.; van Deuren, M.; van der Meer, J.W.; de Mast, Q.; Brüggemann, R.J.; van der Hoeven, H. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. eLife 2020, 9, e57555. [Google Scholar] [CrossRef]
- Garvin, M.R.; Alvarez, C.; Miller, J.I.; Prates, E.T.; Walker, A.M.; Amos, B.K.; Mast, A.E.; Justice, A.; Aronow, B.; Jacobson, D. A mechanistic model and therapeutic interventions for COVID-19 involving a RAS-mediated bradykinin storm. eLife 2020, 9. [Google Scholar] [CrossRef]
- Terzuoli, E.; Corti, F.; Nannelli, G.; Giachetti, A.; Donnini, S.; Ziche, M. Bradykinin B2 Receptor Contributes to Inflammatory Responses in Human Endothelial Cells by the Transactivation of the Fibroblast Growth Factor Receptor FGFR-1. Int. J. Mol. Sci. 2018, 19, 2638. [Google Scholar] [CrossRef] [PubMed]
- Brown Nancy, J.; Gainer James, V.; Stein, C.M.; Vaughan Douglas, E. Bradykinin Stimulates Tissue Plasminogen Activator Release in Human Vasculature. Hypertension 1999, 33, 1431–1435. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Warnock, M.; Harbaugh, A.; Yalavarthi, S.; Gockman, K.; Zuo, M.; Madison, J.A.; Knight, J.S.; Kanthi, Y.; Lawrence, D.A. Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. medRxiv 2020. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Karp Leaf, R.S.; Dzik, W.H.; Carlson, J.C.T.; Fogerty, A.E.; Waheed, A.; Goodarzi, K.; Bendapudi, P.K.; Bornikova, L.; Gupta, S.; et al. COVID-19 and coagulation: Bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood 2020, 136, 489–500. [Google Scholar] [CrossRef]
- White, D.; MacDonald, S.; Edwards, T.; Bridgeman, C.; Hayman, M.; Sharp, M.; Cox-Morton, S.; Duff, E.; Mahajan, S.; Moore, C.; et al. Evaluation of COVID-19 coagulopathy; laboratory characterization using thrombin generation and nonconventional haemostasis assays. Int. J. Lab. Hematol. 2020. [Google Scholar] [CrossRef]
- Stoll, D.; Yokota, R.; Sanches Aragão, D.; Casarini, D.E. Both aldosterone and spironolactone can modulate the intracellular ACE/ANG II/AT1 and ACE2/ANG (1-7)/MAS receptor axes in human mesangial cells. Physiol. Rep. 2019, 7, e14105. [Google Scholar] [CrossRef]
- Matsumoto, T.; Minai, K.; Horie, H.; Ohira, N.; Takashima, H.; Tarutani, Y.; Yasuda, Y.; Ozawa, T.; Matsuo, S.; Kinoshita, M.; et al. Angiotensin-converting enzyme inhibition but not angiotensin II type 1 receptor antagonism augments coronary release of tissue plasminogen activator in hypertensive patients. J. Am. Coll. Cardiol. 2003, 41, 1373–1379. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Roche, J.A.; Roche, R. A hypothesized role for dysregulated bradykinin signaling in COVID-19 respiratory complications. Faseb J. 2020, 34, 7265–7269. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor Necrosis Factor-α Convertase (ADAM17) Mediates Regulated Ectodomain Shedding of the Severe-acute Respiratory Syndrome-Coronavirus (SARS-CoV) Receptor, Angiotensin-converting Enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [PubMed]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Sfera, A.; Osorio, C.; Jafri, N.; Diaz, E.L.; Campo Maldonado, J.E. Intoxication With Endogenous Angiotensin II: A COVID-19 Hypothesis. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef]
- Xu, J.; Xu, X.; Jiang, L.; Dua, K.; Hansbro, P.M.; Liu, G. SARS-CoV-2 induces transcriptional signatures in human lung epithelial cells that promote lung fibrosis. Respir. Res. 2020, 21. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef]
- Zoufaly, A.; Poglitsch, M.; Aberle, J.H.; Hoepler, W.; Seitz, T.; Traugott, M.; Grieb, A.; Pawelka, E.; Laferl, H.; Wenisch, C.; et al. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir. Med. 2020, 8, 1154–1158. [Google Scholar] [CrossRef]
- Glasgow, A.; Glasgow, J.; Limonta, D.; Solomon, P.; Lui, I.; Zhang, Y.; Nix, M.A.; Rettko, N.J.; Zha, S.; Yamin, R.; et al. Engineered ACE2 receptor traps potently neutralize SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020. [Google Scholar] [CrossRef]
- Guo, S.; Som, A.T.; Arai, K.; Lo, E.H. Effects of angiotensin-II on brain endothelial cell permeability via PPARalpha regulation of para- and trans-cellular pathways. Brain Res. 2019, 1722, 146353. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Mäe, M.A.; Sun, Y.; Muhl, L.; Nahar, K.; Liébanas, E.V.; Fagerlund, M.J.; Oldner, A.; Liu, J.; Genové, G.; et al. Pericyte-specific vascular expression of SARS-CoV-2 receptor ACE2—Implications for microvascular inflammation and hypercoagulopathy in COVID-19 patients. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cardot-Leccia, N.; Hubiche, T.; Dellamonica, J.; Burel-Vandenbos, F.; Passeron, T. Pericyte alteration sheds light on micro-vasculopathy in COVID-19 infection. Intensive Care Med. 2020, 1–2. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, S.E.; Wilson, C.L.; Bond, N.G.; Kaur, A.; Alvarez, X.; Midkiff, C.C.; Schnapp, L.M. Pericytes as novel targets for HIV/SIV infection in the lung. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2020, 319, L848–L853. [Google Scholar] [CrossRef]
- Hellström, M.; Gerhardt, H.; Kalén, M.; Li, X.; Eriksson, U.; Wolburg, H.; Betsholtz, C. Lack of Pericytes Leads to Endothelial Hyperplasia and Abnormal Vascular Morphogenesis. J. Cell Biol. 2001, 153, 543–554. [Google Scholar] [CrossRef]
- Otani, A.; Takagi, H.; Oh, H.; Koyama, S.; Honda, Y. Angiotensin II induces expression of the Tie2 receptor ligand, angiopoietin-2, in bovine retinal endothelial cells. Diabetes 2001, 50, 867–875. [Google Scholar] [CrossRef]
- Ramlall, V.; Thangaraj, P.M.; Meydan, C.; Foox, J.; Butler, D.; Kim, J.; May, B.; De Freitas, J.K.; Glicksberg, B.S.; Mason, C.E.; et al. Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat. Med. 2020, 26, 1609–1615. [Google Scholar] [CrossRef]
- Aid, M.; Busman-Sahay, K.; Vidal, S.J.; Maliga, Z.; Bondoc, S.; Starke, C.; Terry, M.; Jacobson, C.A.; Wrijil, L.; Ducat, S.; et al. Vascular Disease and Thrombosis in SARS-CoV-2-Infected Rhesus Macaques. Cell 2020. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.A.M.P.J.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020. [Google Scholar] [CrossRef] [PubMed]
- Lokugamage, K.G.; Hage, A.; de Vries, M.; Valero-Jimenez, A.M.; Schindewolf, C.; Dittmann, M.; Rajsbaum, R.; Menachery, V.D. Type I interferon susceptibility distinguishes SARS-CoV-2 from SARS-CoV. bioRxiv 2020. [Google Scholar] [CrossRef]
- Prendecki, M.; Clarke, C.; Medjeral-Thomas, N.; McAdoo, S.P.; Sandhu, E.; Peters, J.E.; Thomas, D.C.; Willicombe, M.; Botto, M.; Pickering, M.C. Temporal changes in complement activation in haemodialysis patients with COVID-19 as a predictor of disease progression. Clin. Kidney J. 2020, 13, 889–896. [Google Scholar] [CrossRef]
- Cugno, M.; Meroni, P.L.; Gualtierotti, R.; Griffini, S.; Grovetti, E.; Torri, A.; Lonati, P.; Grossi, C.; Borghi, M.O.; Novembrino, C.; et al. Complement activation and endothelial perturbation parallel COVID-19 severity and activity. J. Autoimmun. 2020, 102560. [Google Scholar] [CrossRef]
- Eriksson, O.; Hultström, M.; Persson, B.; Lipcsey, M.; Ekdahl, K.N.; Nilsson, B.; Frithiof, R. Mannose-Binding Lectin is Associated with Thrombosis and Coagulopathy in Critically Ill COVID-19 Patients. Thromb. Haemost. 2020. [Google Scholar] [CrossRef]
- Gao, T.; Hu, M.; Zhang, X.; Li, H.; Zhu, L.; Liu, H.; Dong, Q.; Zhang, Z.; Wang, Z.; Hu, Y.; et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv 2020. [Google Scholar] [CrossRef]
- Yu, J.; Yuan, X.; Chen, H.; Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood 2020, 136, 2080–2089. [Google Scholar] [CrossRef]
- Foley, J.H.; Walton, B.L.; Aleman, M.M.; O’Byrne, A.M.; Lei, V.; Harrasser, M.; Foley, K.A.; Wolberg, A.S.; Conway, E.M. Complement Activation in Arterial and Venous Thrombosis is Mediated by Plasmin. EBioMedicine 2016, 5, 175–182. [Google Scholar] [CrossRef]
- Mastellos, D.C.; Pires da Silva, B.G.P.; Fonseca, B.A.L.; Fonseca, N.P.; Auxiliadora-Martins, M.; Mastaglio, S.; Ruggeri, A.; Sironi, M.; Radermacher, P.; Chrysanthopoulou, A.; et al. Complement C3 vs C5 inhibition in severe COVID-19: Early clinical findings reveal differential biological efficacy. Clin. Immunol. Orlando FLA 2020, 220, 108598. [Google Scholar] [CrossRef]
- de Nooijer, A.H.; Grondman, I.; Janssen, N.A.F.; Netea, M.G.; Willems, L.; van de Veerdonk, F.L.; Giamarellos-Bourboulis, E.J.; Toonen, E.J.M.; Joosten, L.A.B. Complement activation in the disease course of COVID-19 and its effects on clinical outcomes. J. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Risitano, A.M.; Mastellos, D.C.; Huber-Lang, M.; Yancopoulou, D.; Garlanda, C.; Ciceri, F.; Lambris, J.D. Complement as a target in COVID-19? Nat. Rev. Immunol. 2020, 20, 343–344. [Google Scholar] [CrossRef] [PubMed]
- Holter, J.C.; Pischke, S.E.; Boer, E.; de Lind, A.; Jenum, S.; Holten, A.R.; Tonby, K.; Barratt-Due, A.; Sokolova, M.; Schjalm, C.; et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Natl. Acad. Sci. USA 2020, 117, 25018–25025. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, R.; Usama, S.M.; Babiker, H.M. Physiology, Coagulation Pathways. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil Extracellular Traps (NETs) Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Frantzeskaki, F.; Armaganidis, A.; Orfanos, S.E. Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks between Inflammation and Coagulation. Respiration 2017, 93, 212–225. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Beristain-Covarrubias, N.; Perez-Toledo, M.; Thomas, M.R.; Henderson, I.R.; Watson, S.P.; Cunningham, A.F. Understanding Infection-Induced Thrombosis: Lessons Learned From Animal Models. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Long, W.; Tu, M.; Chen, S.; Huang, Y.; Wang, S.; Zhou, W.; Chen, D.; Zhou, L.; Wang, M.; et al. Lymphocyte subset (CD4+, CD8+) counts reflect the severity of infection and predict the clinical outcomes in patients with COVID-19. J. Infect. 2020. [Google Scholar] [CrossRef]
- Liu, R.; Wang, Y.; Li, J.; Han, H.; Xia, Z.; Liu, F.; Wu, K.; Yang, L.; Liu, X.; Zhu, C. Decreased T cell populations contribute to the increased severity of COVID-19. Clin. Chim. Acta Int. J. Clin. Chem. 2020, 508, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Wang, Q.; Zhang, D.; Ding, J.; Huang, Q.; Tang, Y.-Q.; Wang, Q.; Miao, H. Lymphopenia predicts disease severity of COVID-19: A descriptive and predictive study. Signal. Transduct. Target. Ther. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Davanzo, G.G.; Codo, A.C.; Brunetti, N.S.; Boldrini, V.O.; Knittel, T.L.; Monterio, L.B.; de Moraes, D.; Ferrari, A.J.R.; de Souza, G.F.; Muraro, S.P.; et al. SARS-CoV-2 Uses CD4 to Infect T Helper Lymphocytes. medRxiv 2020. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.-E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef]
- Tamburini, B.A.; Burchill, M.A.; Kedl, R.M. Antigen capture and archiving by lymphatic endothelial cells following vaccination or viral infection. Nat. Commun. 2014, 5, 3989. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Zhou, Y.; Fu, B.; Zheng, X.; Wang, D.; Zhao, C.; Qi, Y.; Sun, R.; Tian, Z.; Xu, X.; Wei, H. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl. Sci. Rev. 2020, 7, 998–1002. [Google Scholar] [CrossRef]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef]
- Zhang, D.; Guo, R.; Lei, L.; Liu, H.; Wang, Y.; Wang, Y.; Qian, H.; Dai, T.; Zhang, T.; Lai, Y.; et al. COVID-19 infection induces readily detectable morphologic and inflammation-related phenotypic changes in peripheral blood monocytes. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef]
- Fahlberg, M.D.; Blair, R.V.; Doyle-Meyers, L.A.; Midkiff, C.C.; Zenere, G.; Russell-Lodrigue, K.E.; Monjure, C.J.; Haupt, E.H.; Penney, T.P.; Lehmicke, G.; et al. Cellular events of acute, resolving or progressive COVID-19 in SARS-CoV-2 infected non-human primates. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Yilla, M.; Harcourt, B.H.; Hickman, C.J.; McGrew, M.; Tamin, A.; Goldsmith, C.S.; Bellini, W.J.; Anderson, L.J. SARS-coronavirus replication in human peripheral monocytes/macrophages. Virus Res. 2005, 107, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Boumaza, A.; Gay, L.; Mezouar, S.; Diallo, A.B.; Michel, M.; Desnues, B.; Raoult, D.; Scola, B.L.; Halfon, P.; Vitte, J.; et al. Monocytes and macrophages, targets of SARS-CoV-2: The clue for Covid-19 immunoparalysis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, Y.; Li, K.; Meyerholz, D.K.; Allamargot, C.; Perlman, S. SARS-CoV-2-induced immune activation and death of monocyte-derived human macrophages and dendritic cells. J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kazancioglu, S.; Yilmaz, F.M.; Bastug, A.; Sakallı, A.; Ozbay, B.O.; Buyuktarakci, C.; Bodur, H.; Yilmaz, G. Lymphocyte Subset Alteration and Monocyte CD4 Expression Reduction in Patients with Severe COVID-19. Viral Immunol. 2020. [Google Scholar] [CrossRef]
- FDA. Coronavirus Treatment Acceleration Program (CTAP). Available online: https://www.fda.gov/drugs/coronavirus-covid-19-drugs/coronavirus-treatment-acceleration-program-ctap (accessed on 8 December 2020).
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef]
- Linsky, T.W.; Vergara, R.; Codina, N.; Nelson, J.W.; Walker, M.J.; Su, W.; Barnes, C.O.; Hsiang, T.-Y.; Esser-Nobis, K.; Yu, K.; et al. De novo design of potent and resilient hACE2 decoys to neutralize SARS-CoV-2. Science 2020, 370, 1208–1214. [Google Scholar] [CrossRef]
- Baum, A.; Ajithdoss, D.; Copin, R.; Zhou, A.; Lanza, K.; Negron, N.; Ni, M.; Wei, Y.; Mohammadi, K.; Musser, B.; et al. REGN-COV2 antibodies prevent and treat SARS-CoV-2 infection in rhesus macaques and hamsters. Science 2020. [Google Scholar] [CrossRef]
- Chen, P.; Nirula, A.; Heller, B.; Gottlieb, R.L.; Boscia, J.; Morris, J.; Huhn, G.; Cardona, J.; Mocherla, B.; Stosor, V.; et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Commissioner, O. Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibodies for Treatment of COVID-19. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibodies-treatment-covid-19 (accessed on 8 December 2020).
- Meng, J.; Xiao, G.; Zhang, J.; He, X.; Ou, M.; Bi, J.; Yang, R.; Di, W.; Wang, Z.; Li, Z.; et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg. Microbes Infect. 2020, 9, 757–760. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, B.; Li, Y.; Zhang, L.; Wang, Y.; Yang, S.; Xiao, X.; Qin, Q. The Use of Renin-Angiotensin-Aldosterone System (RAAS) Inhibitors is Associated with a Lower Risk of Mortality in Hypertensive COVID-19 Patients: A Systematic Review and Meta-analysis. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- RAAS inhibitors are not associated with mortality in COVID-19 patients: Findings from an observational multicenter study in Italy and a meta-analysis of 19 studies. Vascul. Pharmacol. 2020. [CrossRef]
- Fu, W.; Liu, Y.; Liu, L.; Hu, H.; Cheng, X.; Liu, P.; Song, Z.; Zha, L.; Bai, S.; Xu, T.; et al. An open-label, randomized trial of the combination of IFN-κ plus TFF2 with standard care in the treatment of patients with moderate COVID-19. EClinicalMedicine 2020, 27. [Google Scholar] [CrossRef] [PubMed]
- Hung, I.F.-N.; Lung, K.-C.; Tso, E.Y.-K.; Liu, R.; Chung, T.W.-H.; Chu, M.-Y.; Ng, Y.-Y.; Lo, J.; Chan, J.; Tam, A.R.; et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: An open-label, randomised, phase 2 trial. Lancet Lond. Engl. 2020, 395, 1695–1704. [Google Scholar] [CrossRef]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef] [PubMed]
- Guaraldi, G.; Meschiari, M.; Cozzi-Lepri, A.; Milic, J.; Tonelli, R.; Menozzi, M.; Franceschini, E.; Cuomo, G.; Orlando, G.; Borghi, V.; et al. Tocilizumab in patients with severe COVID-19: A retrospective cohort study. Lancet Rheumatol. 2020, 2, E474–E484. [Google Scholar] [CrossRef]
- Stone, J.H.; Frigault, M.J.; Serling-Boyd, N.J.; Fernandes, A.D.; Harvey, L.; Foulkes, A.S.; Horick, N.K.; Healy, B.C.; Shah, R.; Bensaci, A.M.; et al. Efficacy of Tocilizumab in Patients Hospitalized with Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Roschewski, M.; Lionakis, M.S.; Sharman, J.P.; Roswarski, J.; Goy, A.; Monticelli, M.A.; Roshon, M.; Wrzesinski, S.H.; Desai, J.V.; Zarakas, M.A.; et al. Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Luo, W.; Li, Y.-X.; Jiang, L.-J.; Chen, Q.; Wang, T.; Ye, D.-W. Targeting JAK-STAT Signaling to Control Cytokine Release Syndrome in COVID-19. Trends Pharmacol. Sci. 2020, 41, 531–543. [Google Scholar] [CrossRef]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Rawling, M.; Savory, E.; Stebbing, J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020, 395, e30–e31. [Google Scholar] [CrossRef]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Commissioner, O. Coronavirus (COVID-19) Update: FDA Authorizes Drug Combination for Treatment of COVID-19. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-drug-combination-treatment-covid-19 (accessed on 8 December 2020).
- La Rosée, F.; Bremer, H.C.; Gehrke, I.; Kehr, A.; Hochhaus, A.; Birndt, S.; Fellhauer, M.; Henkes, M.; Kumle, B.; Russo, S.G.; et al. The Janus kinase 1/2 inhibitor ruxolitinib in COVID-19 with severe systemic hyperinflammation. Leukemia 2020, 34, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wei, J.; Zou, L.; Jiang, T.; Wang, G.; Chen, L.; Huang, L.; Meng, F.; Huang, L.; Wang, N.; et al. Ruxolitinib in treatment of severe coronavirus disease 2019 (COVID-19): A multicenter, single-blind, randomized controlled trial. J. Allergy Clin. Immunol. 2020, 146, 137–146.e3. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef]
- Buijsers, B.; Yanginlar, C.; de Nooijer, A.; Grondman, I.; Maciej-Hulme, M.L.; Jonkman, I.; Janssen, N.A.F.; Rother, N.; de Graaf, M.; Pickkers, P.; et al. Increased Plasma Heparanase Activity in COVID-19 Patients. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Conzelmann, C.; Müller, J.A.; Perkhofer, L.; Sparrer, K.M.; Zelikin, A.N.; Münch, J.; Kleger, A. Inhaled and systemic heparin as a repurposed direct antiviral drug for prevention and treatment of COVID-19. Clin. Med. Lond. Engl. 2020. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernard, I.; Limonta, D.; Mahal, L.K.; Hobman, T.C. Endothelium Infection and Dysregulation by SARS-CoV-2: Evidence and Caveats in COVID-19. Viruses 2021, 13, 29. https://doi.org/10.3390/v13010029
Bernard I, Limonta D, Mahal LK, Hobman TC. Endothelium Infection and Dysregulation by SARS-CoV-2: Evidence and Caveats in COVID-19. Viruses. 2021; 13(1):29. https://doi.org/10.3390/v13010029
Chicago/Turabian StyleBernard, Isabelle, Daniel Limonta, Lara K. Mahal, and Tom C. Hobman. 2021. "Endothelium Infection and Dysregulation by SARS-CoV-2: Evidence and Caveats in COVID-19" Viruses 13, no. 1: 29. https://doi.org/10.3390/v13010029
APA StyleBernard, I., Limonta, D., Mahal, L. K., & Hobman, T. C. (2021). Endothelium Infection and Dysregulation by SARS-CoV-2: Evidence and Caveats in COVID-19. Viruses, 13(1), 29. https://doi.org/10.3390/v13010029