Immune Checkpoints in Viral Infections


 ,
,
Abstract
1. Introduction
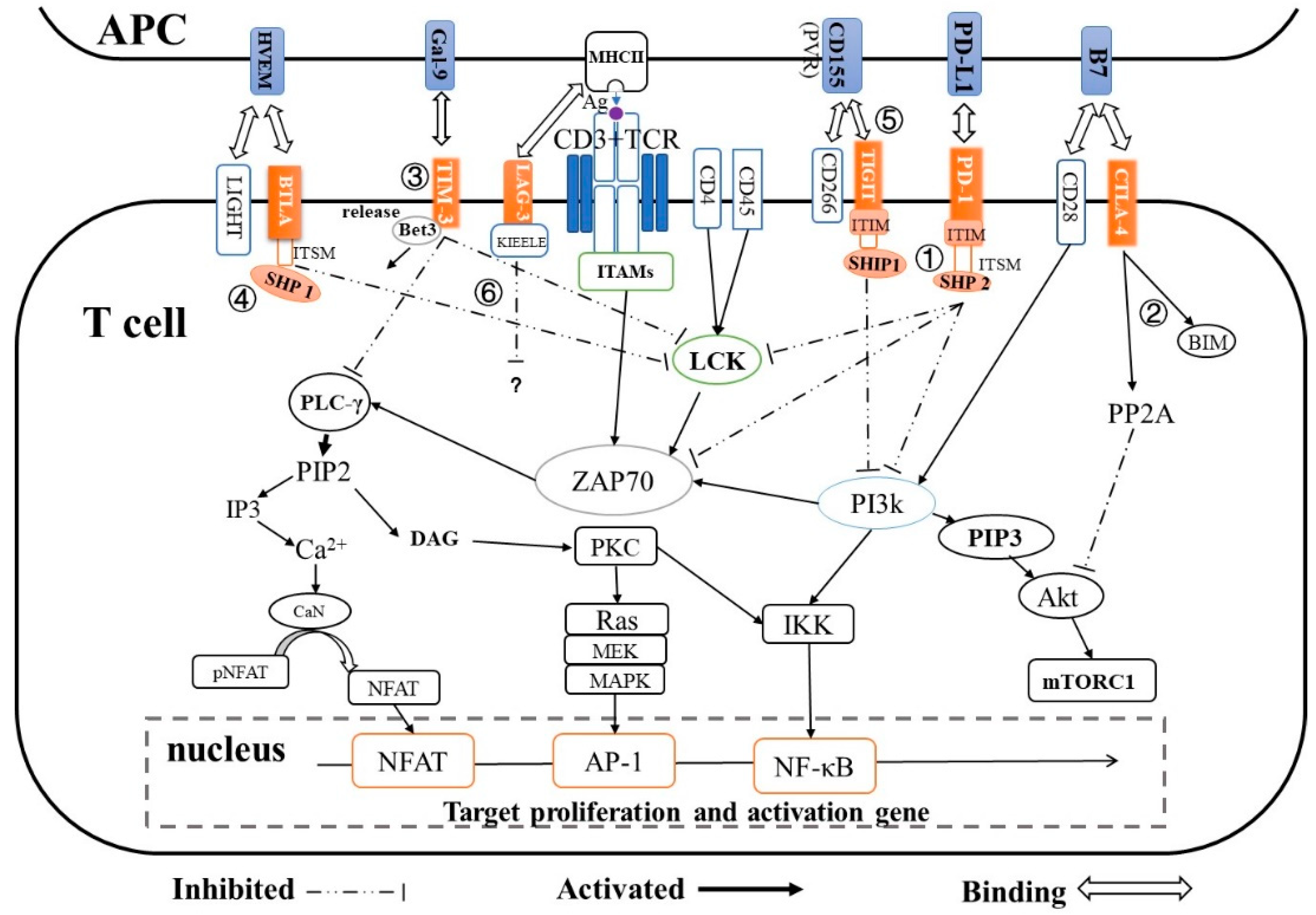
2. Immune Checkpoints and Their T-cell Inactivation Pathways
3. Immune Checkpoints in HIV
3.1. HIV-Specific T-cell Exhaustion Accompanies Immune Checkpoint Upregulation

{kind=link}
| Virus | Manifestations | Checkpoints Involved | Functions | Ref. |
|---|---|---|---|---|
| HIV | Establishment of HIV latency reservoirs | PD-1, CTLA-4, LAG-3, TIGIT | Participate in HIV infection and assist to virus escape from immune clearance | [43,46,47,48,49] |
| CD4+ T-cell dysfunction | PD-1, CTLA-4, LAG-3, TIM-3 | Cause many CD4 T-cells dysfunction and loss | [46,50,51,52,53] | |
| CD8+ T-cell exhaustion | PD-1, TIM-3, LAG-3, TIGIT | Lead to CD8+ T-cell functional impact such as decreased IL-2 secretion and T-cell proliferation | [42,46,54,55] | |
| Improved Treg proliferation | TIM-3, CTLA-4, PD-1 | Decrease the HIV-specific immune responses, contributing to virus persistence | [56,57,58,59] | |
| HBV | CD8+ T-cell exhaustion | PD-1, CTLA-4, Tim-3, TIGIT, LAG-3 | Lead to CD8+ T-cell functional impact such as decreased IL-2 secretion and T-cell proliferation | [60,61,62,63] |
| Upregulation on CD4+ T-cell | PD-1, CTLA-4, Tim-3 | Increase Treg and regulate Th1/Th2 cytokine secretion | [64,65,66,67] | |
| Altered cytokine secretion | See Table 4 | See Table 4 | ||
| Limited liver injury | LAG-3 | Suppress T-cell function and mitigate liver injury | [68] | |
| HCV | CD8+ T-cell exhaustion | PD-1, TIGIT, Tim3, CTLA-4 | cause CD8+ T-cell functional defect such as decreased IL-2 secretion and T-cell proliferation | [69,70,71,72] |
| Up-regulation on CD4+ T-cell | CTLA-4, TIM-3 | Improve viral replication and persistence | [67,73] | |
| Altered cytokine secretion | See Table 4 | See Table 4 | ||
| Influenza | Decrease in CD8+ T-cell response | PD-1, Tim-3 | Reduce the number of virus-specific CD8+ T-cell and cause T-cell dysfunction | [74,75,76,77,78,79,80,81,82] |
| Up-regulation on both CD4+ and CD8+ T-cells from patients with influenza-associated encephalopathy | CTLA-4 | Involved in influenza virus-associated encephalopathy | ||
| SARS-CoV-2 | Up-regulation on T-cells and NK cells from COVID-19 patients. Patient deteriorated from prodromal to symptomatic | PD-1, TIM-3 | Mediate T-cell exhaustion and T-cell lymphopenia | [83,84,85] |
| HSV-1 | CD8+ T-cell exhaustion | PD-1, LAG-3 | Cause T-cell exhaustion and assist viral latency infection | [86,87,88] |
| Reactivation of latency virus | PD-1, CTLA-4, TIM3 | Mediate T-cell exhaustion and make them lost the control of spontaneous HSV-1 reactivation | [89] | |
| EBV | Up-regulated expression | PD-1, CTLA-4, TIM3, 2B4 | Provide T-cell dysfunction status to increase EBV escape and EBV latent infections | [90,91,92,93] |
| Immune escape | PD-1 | |||
| Ebola virus | Upregulation on both CD4+ and CD8+ T-cells; Associated with high viremia and poor outcome | CTLA-4, PD-1 | Mediate immune suppression | [94] |
3.2. The Upregulation of Immune Checkpoint Proteins Helps to Establish a Latent HIV Reservoir
4. Immune Checkpoints in Hepatitis Virus
4.1. Hepatitis Virus Infection Upregulates Immune Checkpoint Proteins on T-cells
4.2. Immune Checkpoints Affect Chronic Hepatitis Virus Infection through Cytokine Regulation
5. Immune Checkpoints in Influenza
6. Immune Checkpoints in COVID-19
7. Immune Checkpoints in Other Viruses
8. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Schweitzer, A.; Horn, J.; Mikolajczyk, R.T.; Krause, G.; Ott, J.J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet (Lond. Engl.) 2015, 386, 1546–1555. [Google Scholar] [CrossRef]
- DALYs, G.B.D.; Collaborators, H. Global, regional, and national disability-adjusted life-years (DALYs) for 333 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet (Lond. Engl.) 2017, 390, 1260–1344. [Google Scholar]
- Angelosanto, J.M.; Blackburn, S.D.; Crawford, A.; Wherry, E.J. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J. Virol. 2012, 86, 8161–8170. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Kamphorst, A.O.; Im, S.J.; Kissick, H.T.; Pillai, R.N.; Ramalingam, S.S.; Araki, K.; Ahmed, R. CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Ann. Rev. Med. 2018, 69, 301–318. [Google Scholar] [CrossRef] [PubMed]
- Alegre, M.L.; Frauwirth, K.A.; Thompson, C.B. T-cell regulation by CD28 and CTLA-4. Nat. Rev. Immunol. 2001, 1, 220–228. [Google Scholar] [CrossRef]
- Chen, L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar] [CrossRef]
- Korman, A.J.; Peggs, K.S.; Allison, J.P. Checkpoint blockade in cancer immunotherapy. Adv. Immunol. 2006, 90, 297–339. [Google Scholar]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Rangachari, M.; Kuchroo, V.K. Tim-3: A co-receptor with diverse roles in T cell exhaustion and tolerance. Semin. Immunol. 2019, 42, 101302. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Cohn, L.B.; Chomont, N.; Deeks, S.G. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe 2020, 27, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Kasamon, Y.L.; de Claro, R.A.; Wang, Y.; Shen, Y.L.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Nivolumab for the Treatment of Relapsed or Progressive Classical Hodgkin Lymphoma. Oncologist 2017, 22, 585–591. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef]
- Popovic, A.; Jaffee, E.M.; Zaidi, N. Emerging strategies for combination checkpoint modulators in cancer immunotherapy. J. Clin. Investig. 2018, 128, 3209–3218. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef]
- Thibult, M.-L.; Mamessier, E.; Gertner-Dardenne, J.; Pastor, S.; Just-Landi, S.; Xerri, L.; Chetaille, B.; Olive, D. PD-1 is a novel regulator of human B-cell activation. Int. Immunol. 2013, 25, 129–137. [Google Scholar] [CrossRef]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef]
- Sheppard, K.-A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Rota, G.; Niogret, C.; Dang, A.T.; Barros, C.R.; Fonta, N.P.; Alfei, F.; Morgado, L.; Zehn, D.; Birchmeier, W.; Vivier, E.; et al. Shp-2 Is Dispensable for Establishing T Cell Exhaustion and for PD-1 Signaling In Vivo. Cell Rep. 2018, 23, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, A.; Gharibi, T.; Marofi, F.; Babaloo, Z.; Baradaran, B. CTLA-4: From mechanism to autoimmune therapy. Int. Immunopharmacol. 2020, 80, 106221. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Kaur, S.; Hou, T.Z.; Jeffery, L.E.; Poulter, N.S.; Briggs, Z.; Kenefeck, R.; Willox, A.K.; Royle, S.J.; Rappoport, J.Z.; et al. Constitutive clathrin-mediated endocytosis of CTLA-4 persists during T cell activation. J. Biol. Chem. 2012, 287, 9429–9440. [Google Scholar] [CrossRef]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef]
- Chuang, E.; Fisher, T.S.; Morgan, R.W.; Robbins, M.D.; Duerr, J.M.; Vander Heiden, M.G.; Gardner, J.P.; Hambor, J.E.; Neveu, M.J.; Thompson, C.B. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity 2000, 13, 313–322. [Google Scholar] [CrossRef]
- Fraser, J.H.; Rincón, M.; McCoy, K.D.; Le Gros, G. CTLA4 ligation attenuates AP-1, NFAT and NF-kappaB activity in activated T cells. Eur. J. Immunol. 1999, 29, 838–844. [Google Scholar] [CrossRef]
- Olsson, C.; Riesbeck, K.; Dohlsten, M.; Michaëlsson, E.; Riebeck, K. CTLA-4 ligation suppresses CD28-induced NF-kappaB and AP-1 activity in mouse T cell blasts. J. Biol. Chem. 1999, 274, 14400–14405. [Google Scholar] [CrossRef]
- Schurich, A.; Khanna, P.; Lopes, A.R.; Han, K.J.; Peppa, D.; Micco, L.; Nebbia, G.; Kennedy, P.T.F.; Geretti, A.-M.; Dusheiko, G.; et al. Role of the coinhibitory receptor cytotoxic T lymphocyte antigen-4 on apoptosis-Prone CD8 T cells in persistent hepatitis B virus infection. Hepatology 2011, 53, 1494–1503. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Diff. 2013, 20, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, M.; Zhu, C.; Sakuishi, K.; Xiao, S.; Karman, J.; Chen, A.; Angin, M.; Wakeham, A.; Greenfield, E.A.; Sobel, R.A.; et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat. Med. 2012, 18, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Tomkowicz, B.; Walsh, E.; Cotty, A.; Verona, R.; Sabins, N.; Kaplan, F.; Santulli-Marotto, S.; Chin, C.-N.; Mooney, J.; Lingham, R.B.; et al. TIM-3 Suppresses Anti-CD3/CD28-Induced TCR Activation and IL-2 Expression through the NFAT Signaling Pathway. PLoS ONE 2015, 10, e0140694. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Woo, M.-Y.; Chwae, Y.-J.; Kwon, M.-H.; Kim, K.; Park, S. Down-regulation of interleukin-2 production by CD4(+) T cells expressing TIM-3 through suppression of NFAT dephosphorylation and AP-1 transcription. Immunobiology 2012, 217, 986–995. [Google Scholar] [CrossRef]
- Joller, N.; Hafler, J.P.; Brynedal, B.; Kassam, N.; Spoerl, S.; Levin, S.D.; Sharpe, A.H.; Kuchroo, V.K. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol. 2011, 186, 1338–1342. [Google Scholar] [CrossRef]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V.; et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef]
- Maeda, T.K.; Sugiura, D.; Okazaki, I.-M.; Maruhashi, T.; Okazaki, T. Atypical motifs in the cytoplasmic region of the inhibitory immune co-receptor LAG-3 inhibit T cell activation. J. Biol. Chem. 2019, 294, 6017–6026. [Google Scholar] [CrossRef]
- Cai, G.; Freeman, G.J. The CD160, BTLA, LIGHT/HVEM pathway: A bidirectional switch regulating T-cell activation. Immunol. Rev. 2009, 229, 244–258. [Google Scholar] [CrossRef]
- Xu, X.; Hou, B.; Fulzele, A.; Masubuchi, T.; Zhao, Y.; Wu, Z.; Hu, Y.; Jiang, Y.; Ma, Y.; Wang, H.; et al. PD-1 and BTLA regulate T cell signaling differentially and only partially through SHP1 and SHP2. J. Cell Biol. 2020, 219, 6. [Google Scholar] [CrossRef]
- Gavrieli, M.; Murphy, K.M. Association of Grb-2 and PI3K p85 with phosphotyrosile peptides derived from BTLA. Biochem. Biophys. Res. Commun. 2006, 345, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Banga, R.; Procopio, F.A.; Noto, A.; Pollakis, G.; Cavassini, M.; Ohmiti, K.; Corpataux, J.M.; de Leval, L.; Pantaleo, G.; Perreau, M. PD-1(+) and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat. Med. 2016, 22, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Pardons, M.; Baxter, A.E.; Massanella, M.; Pagliuzza, A.; Fromentin, R.; Dufour, C.; Leyre, L.; Routy, J.P.; Kaufmann, D.E.; Chomont, N. Single-cell characterization and quantification of translation-competent viral reservoirs in treated and untreated HIV infection. PLoS Pathog. 2019, 15, e1007619. [Google Scholar] [CrossRef]
- Tian, X.; Zhang, A.; Qiu, C.; Wang, W.; Yang, Y.; Qiu, C.; Liu, A.; Zhu, L.; Yuan, S.; Hu, H.; et al. The upregulation of LAG-3 on T cells defines a subpopulation with functional exhaustion and correlates with disease progression in HIV-infected subjects. J. Immunol. 2015, 194, 3873–3882. [Google Scholar] [CrossRef]
- Noto, A.; Procopio, F.A.; Banga, R.; Suffiotti, M.; Corpataux, J.M.; Cavassini, M.; Riva, A.; Fenwick, C.; Gottardo, R.; Perreau, M.; et al. CD32 and PD-1 Lymph Node CD4 T Cells Support Persistent HIV-1 Transcription in Treated Aviremic Individuals. J. Virol. 2018, 92, 20. [Google Scholar] [CrossRef]
- Riley, J.L.; Schlienger, K.; Blair, P.J.; Carreno, B.; Craighead, N.; Kim, D.; Carroll, R.G.; June, C.H. Modulation of susceptibility to HIV-1 infection by the cytotoxic T lymphocyte antigen 4 costimulatory molecule. J. Exp. Med. 2000, 191, 1987–1997. [Google Scholar] [CrossRef]
- Hatano, H.; Jain, V.; Hunt, P.W.; Lee, T.H.; Sinclair, E.; Do, T.D.; Hoh, R.; Martin, J.N.; McCune, J.M.; Hecht, F.; et al. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. J. Infect. Dis. 2013, 208, 50–56. [Google Scholar] [CrossRef]
- Leng, Q.; Bentwich, Z.; Magen, E.; Kalinkovich, A.; Borkow, G. CTLA-4 upregulation during HIV infection: Association with anergy and possible target for therapeutic intervention. AIDS (Lond. Engl.) 2002, 16, 519–529. [Google Scholar] [CrossRef]
- Kaufmann, D.E.; Kavanagh, D.G.; Pereyra, F.; Zaunders, J.J.; Mackey, E.W.; Miura, T.; Palmer, S.; Brockman, M.; Rathod, A.; Piechocka-Trocha, A.; et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat. Immunol. 2007, 8, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, D.E.; Walker, B.D. PD-1 and CTLA-4 inhibitory cosignaling pathways in HIV infection and the potential for therapeutic intervention. J. Immunol. 2009, 182, 5891–5897. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; Ndhlovu, L.C.; Barbour, J.D.; Sheth, P.M.; Jha, A.R.; Long, B.R.; Wong, J.C.; Satkunarajah, M.; Schweneker, M.; Chapman, J.M.; et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 2008, 205, 2763–2779. [Google Scholar] [CrossRef]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef]
- Chew, G.M.; Fujita, T.; Webb, G.M.; Burwitz, B.J.; Wu, H.L.; Reed, J.S.; Hammond, K.B.; Clayton, K.L.; Ishii, N.; Abdel-Mohsen, M.; et al. TIGIT Marks Exhausted T Cells, Correlates with Disease Progression, and Serves as a Target for Immune Restoration in HIV and SIV Infection. PLoS Pathog. 2016, 12, e1005349. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.H.; Xue, Y.L.; Wang, Y. [Association of PD-1 expression on CD4+ CD25 nt/hi CD127 lo regulatory T cells with disease progression in HIV-1 infected patients]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi Chin. J. Cell. Mol. Immunol. 2009, 25, 1020–1022. [Google Scholar]
- Khaitan, A.; Kravietz, A.; Mwamzuka, M.; Marshed, F.; Ilmet, T.; Said, S.; Ahmed, A.; Borkowsky, W.; Unutmaz, D. FOXP3+Helios+ Regulatory T Cells, Immune Activation, and Advancing Disease in HIV-Infected Children. J. Acquir. Immune Defic. Syndr. (1999) 2016, 72, 474–484. [Google Scholar] [CrossRef][Green Version]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef]
- Koch, K.; Koch, N.; Sandaradura de Silva, U.; Jung, N.; Schulze zur Wiesch, J.; Fätkenheuer, G.; Hartmann, P.; Romerio, F.; Lehmann, C. Increased Frequency of CD49b/LAG-3(+) Type 1 Regulatory T Cells in HIV-Infected Individuals. AIDS Res. Hum. Retrovir. 2015, 31, 1238–1246. [Google Scholar] [CrossRef]
- Zong, L.; Peng, H.; Sun, C.; Li, F.; Zheng, M.; Chen, Y.; Wei, H.; Sun, R.; Tian, Z. Breakdown of adaptive immunotolerance induces hepatocellular carcinoma in HBsAg-tg mice. Nat. Commun. 2019, 10, 221. [Google Scholar] [CrossRef]
- Li, F.J.; Zhang, Y.; Jin, G.X.; Yao, L.; Wu, D.Q. Expression of LAG-3 is coincident with the impaired effector function of HBV-specific CD8(+) T cell in HCC patients. Immunol. Lett. 2013, 150, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, K.; Tao, K.; Chen, L.; Zheng, Q.; Lu, X.; Liu, J.; Shi, L.; Liu, C.; Wang, G.; et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2012, 56, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Shi, Y.; Li, S.; Zhang, Y.; Liu, Y.; Wu, Y.; Chen, Z. Blockade of Tim-3 signaling restores the virus-specific CD8⁺ T-cell response in patients with chronic hepatitis B. Eur. J. Immunol. 2012, 42, 1180–1191. [Google Scholar] [CrossRef]
- Dong, Y.; Li, X.; Zhang, L.; Zhu, Q.; Chen, C.; Bao, J.; Chen, Y. CD4 T cell exhaustion revealed by high PD-1 and LAG-3 expression and the loss of helper T cell function in chronic hepatitis B. BMC Immunol. 2019, 20, 27. [Google Scholar] [CrossRef] [PubMed]
- Nebbia, G.; Peppa, D.; Schurich, A.; Khanna, P.; Singh, H.D.; Cheng, Y.; Rosenberg, W.; Dusheiko, G.; Gilson, R.; ChinAleong, J.; et al. Upregulation of the Tim-3/galectin-9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS ONE 2012, 7, e47648. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, C.; Peng, Q.; Shi, J.; Gu, G. Expression levels of CD28, CTLA-4, PD-1 and Tim-3 as novel indicators of T-cell immune function in patients with chronic hepatitis B virus infection. Biomed. Rep. 2014, 2, 270–274. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yu, Y.; Wu, H.; Tang, Z.; Zang, G. CTLA4 silencing with siRNA promotes deviation of Th1/Th2 in chronic hepatitis B patients. Cell. Mol. Immunol. 2009, 6, 123–127. [Google Scholar] [CrossRef]
- Ye, B.; Li, X.; Dong, Y.; Wang, Y.; Tian, L.; Lin, S.; Liu, X.; Kong, H.; Chen, Y. Increasing LAG-3 expression suppresses T-cell function in chronic hepatitis B: A balance between immunity strength and liver injury extent. Medicine 2017, 96, e5275. [Google Scholar] [CrossRef]
- Nakamoto, N.; Kaplan, D.E.; Coleclough, J.; Li, Y.; Valiga, M.E.; Kaminski, M.; Shaked, A.; Olthoff, K.; Gostick, E.; Price, D.A.; et al. Functional restoration of HCV-specific CD8 T cells by PD-1 blockade is defined by PD-1 expression and compartmentalization. Gastroenterology 2008, 134, 1927–1937.e1–2. [Google Scholar] [CrossRef]
- Ackermann, C.; Smits, M.; Woost, R.; Eberhard, J.M.; Peine, S.; Kummer, S.; Marget, M.; Kuntzen, T.; Kwok, W.W.; Lohse, A.W.; et al. HCV-specific CD4+ T cells of patients with acute and chronic HCV infection display high expression of TIGIT and other co-inhibitory molecules. Sci. Rep. 2019, 9, 10624. [Google Scholar] [CrossRef]
- Golden-Mason, L.; Palmer, B.E.; Kassam, N.; Townshend-Bulson, L.; Livingston, S.; McMahon, B.J.; Castelblanco, N.; Kuchroo, V.; Gretch, D.R.; Rosen, H.R. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J. Virol. 2009, 83, 9122–9130. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.L.; El-Badawy, A. Programmed death-1/programmed death-L1 signaling pathway and its blockade in hepatitis C virus immunotherapy. World J. Hepatol. 2015, 7, 2449–2458. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ponce, C.; Dominguez-Villar, M.; Aguado, E.; Garcia-Cozar, F. CD4+ primary T cells expressing HCV-core protein upregulate Foxp3 and IL-10, suppressing CD4 and CD8 T cells. PLoS ONE 2014, 9, e85191. [Google Scholar] [CrossRef] [PubMed]
- McNally, B.; Ye, F.; Willette, M.; Flaño, E. Local blockade of epithelial PDL-1 in the airways enhances T cell function and viral clearance during influenza virus infection. J. Virol. 2013, 87, 12916–12924. [Google Scholar] [CrossRef]
- Katoh, S.; Ikeda, M.; Shimizu, H.; Mouri, K.; Obase, Y.; Kobashi, Y.; Fukushima, K.; Hirashima, M.; Oka, M. Increased Levels of Plasma Galectin-9 in Patients with Influenza Virus Infection. Tohoku J. Exp. Med. 2014, 232, 263–267. [Google Scholar] [CrossRef]
- Sharma, S.; Sundararajan, A.; Suryawanshi, A.; Kumar, N.; Veiga-Parga, T.; Kuchroo, V.K.; Thomas, P.G.; Sangster, M.Y.; Rouse, B.T. T cell immunoglobulin and mucin protein-3 (Tim-3)/Galectin-9 interaction regulates influenza A virus-specific humoral and CD8 T-cell responses. Proc. Natl. Acad. Sci. USA 2011, 108, 19001–19006. [Google Scholar] [CrossRef]
- Erickson, J.J.; Rogers, M.C.; Hastings, A.K.; Tollefson, S.J.; Williams, J.V. Programmed death-1 impairs secondary effector lung CD8⁺ T cells during respiratory virus reinfection. J. Immunol. 2014, 193, 5108–5117. [Google Scholar] [CrossRef]
- Erickson, J.J.; Gilchuk, P.; Hastings, A.K.; Tollefson, S.J.; Johnson, M.; Downing, M.B.; Boyd, K.L.; Johnson, J.E.; Kim, A.S.; Joyce, S.; et al. Viral acute lower respiratory infections impair CD8+ T cells through PD-1. J. Clin. Investig. 2012, 122, 2967–2982. [Google Scholar] [CrossRef]
- Rutigliano, J.A.; Sharma, S.; Morris, M.Y.; Oguin, T.H.; McClaren, J.L.; Doherty, P.C.; Thomas, P.G. Highly pathological influenza A virus infection is associated with augmented expression of PD-1 by functionally compromised virus-specific CD8+ T cells. J. Virol. 2014, 88, 1636–1651. [Google Scholar] [CrossRef]
- Ayukawa, H.; Matsubara, T.; Kaneko, M.; Hasegawa, M.; Ichiyama, T.; Furukawa, S. Expression of CTLA-4 (CD152) in peripheral blood T cells of children with influenza virus infection including encephalopathy in comparison with respiratory syncytial virus infection. Clin. Exp. Immunol. 2004, 137, 151–155. [Google Scholar] [CrossRef]
- Schorer, M.; Rakebrandt, N.; Lambert, K.; Hunziker, A.; Pallmer, K.; Oxenius, A.; Kipar, A.; Stertz, S.; Joller, N. TIGIT limits immune pathology during viral infections. Nat. Commun. 2020, 11, 1288. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.L.; Roche, M.I.; Sandall, B.; Brass, A.L.; Seed, B.; Xavier, R.J.; Medoff, B.D. Enhanced Tim3 activity improves survival after influenza infection. J. Immunol. 2012, 189, 2879–2889. [Google Scholar] [CrossRef] [PubMed]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Chiappelli, F.; Khakshooy, A.; Greenberg, G. CoViD-19 Immunopathology and Immunotherapy. Bioinformation 2020, 16, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Mott, K.R.; Zandian, M.; Ghiasi, H. Immunization with different viral antigens alters the pattern of T cell exhaustion and latency in herpes simplex virus type 1-infected mice. J. Virol. 2010, 84, 12315–12324. [Google Scholar] [CrossRef]
- Mott, K.R.; Bresee, C.J.; Allen, S.J.; BenMohamed, L.; Wechsler, S.L.; Ghiasi, H. Level of herpes simplex virus type 1 latency correlates with severity of corneal scarring and exhaustion of CD8+ T cells in trigeminal ganglia of latently infected mice. J. Virol. 2009, 83, 2246–2254. [Google Scholar] [CrossRef]
- Roy, S.; Coulon, P.G.; Prakash, S.; Srivastava, R.; Geertsema, R.; Dhanushkodi, N.; Lam, C.; Nguyen, V.; Gorospe, E.; Nguyen, A.M.; et al. Blockade of PD-1 and LAG-3 Immune Checkpoints Combined with Vaccination Restores the Function of Antiviral Tissue-Resident CD8 T Cells and Reduces Ocular Herpes Simplex Infection and Disease in HLA Transgenic Rabbits. J. Virol. 2019, 93, 18. [Google Scholar] [CrossRef]
- Srivastava, R.; Dervillez, X.; Khan, A.A.; Chentoufi, A.A.; Chilukuri, S.; Shukr, N.; Fazli, Y.; Ong, N.N.; Afifi, R.E.; Osorio, N.; et al. The Herpes Simplex Virus Latency-Associated Transcript Gene Is Associated with a Broader Repertoire of Virus-Specific Exhausted CD8+ T Cells Retained within the Trigeminal Ganglia of Latently Infected HLA Transgenic Rabbits. J. Virol. 2016, 90, 3913–3928. [Google Scholar] [CrossRef]
- Larsen, M.; Sauce, D.; Deback, C.; Arnaud, L.; Mathian, A.; Miyara, M.; Boutolleau, D.; Parizot, C.; Dorgham, K.; Papagno, L.; et al. Exhausted cytotoxic control of Epstein-Barr virus in human lupus. PLoS Pathog. 2011, 7, e1002328. [Google Scholar] [CrossRef]
- Ma, S.D.; Xu, X.; Jones, R.; Delecluse, H.J.; Zumwalde, N.A.; Sharma, A.; Gumperz, J.E.; Kenney, S.C. PD-1/CTLA-4 Blockade Inhibits Epstein-Barr Virus-Induced Lymphoma Growth in a Cord Blood Humanized-Mouse Model. PLoS Pathog. 2016, 12, e1005642. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.; Deng, Y.; Holler, A.; Nunez, N.; Azzi, T.; Vanoaica, L.D.; Müller, A.; Zdimerova, H.; Antsiferova, O.; Zbinden, A.; et al. CD8+ T cells retain protective functions despite sustained inhibitory receptor expression during Epstein-Barr virus infection in vivo. PLoS Pathog. 2019, 15, e1007748. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.-W.; Wang, H.; Zhang, W.-W.; Wang, J.-H.; Liu, W.-J.; Xia, Z.-J.; Huang, H.-Q.; Jiang, W.-Q.; Zhang, Y.-J.; Wang, L. PD-L1 is upregulated by EBV-driven LMP1 through NF-κB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J. Hematol. Oncol. 2016, 9, 109. [Google Scholar] [CrossRef] [PubMed]
- Ruibal, P.; Oestereich, L.; Lüdtke, A.; Becker-Ziaja, B.; Wozniak, D.M.; Kerber, R.; Korva, M.; Cabeza-Cabrerizo, M.; Bore, J.A.; Koundouno, F.R.; et al. Unique human immune signature of Ebola virus disease in Guinea. Nature 2016, 533, 100–104. [Google Scholar] [CrossRef]
- Sakhdari, A.; Mujib, S.; Vali, B.; Yue, F.Y.; MacParland, S.; Clayton, K.; Jones, R.B.; Liu, J.; Lee, E.Y.; Benko, E.; et al. Tim-3 negatively regulates cytotoxicity in exhausted CD8+ T cells in HIV infection. PLoS ONE 2012, 7, e40146. [Google Scholar] [CrossRef]
- Seung, E.; Dudek, T.E.; Allen, T.M.; Freeman, G.J.; Luster, A.D.; Tager, A.M. PD-1 blockade in chronically HIV-1-infected humanized mice suppresses viral loads. PLoS ONE 2013, 8, e77780. [Google Scholar] [CrossRef]
- Grabmeier-Pfistershammer, K.; Stecher, C.; Zettl, M.; Rosskopf, S.; Rieger, A.; Zlabinger, G.J.; Steinberger, P. Antibodies targeting BTLA or TIM-3 enhance HIV-1 specific T cell responses in combination with PD-1 blockade. Clin. Immunol. 2017, 183, 167–173. [Google Scholar] [CrossRef]
- Rallón, N.; García, M.; García-Samaniego, J.; Cabello, A.; Álvarez, B.; Restrepo, C.; Nistal, S.; Górgolas, M.; Benito, J.M. Expression of PD-1 and Tim-3 markers of T-cell exhaustion is associated with CD4 dynamics during the course of untreated and treated HIV infection. PLoS ONE 2018, 13, e0193829. [Google Scholar] [CrossRef]
- de Kivit, S.; Lempsink, L.J.; Plants, J.; Martinson, J.; Keshavarzian, A.; Landay, A.L. Modulation of TIM-3 expression on NK and T cell subsets in HIV immunological non-responders. Clin. Immunol. 2015, 156, 28–35. [Google Scholar] [CrossRef]
- Morou, A.; Palmer, B.E.; Kaufmann, D.E. Distinctive features of CD4+ T cell dysfunction in chronic viral infections. Curr. Opin. HIV AIDS 2014, 9, 446–451. [Google Scholar] [CrossRef]
- Fromentin, R.; Bakeman, W.; Lawani, M.B.; Khoury, G.; Hartogensis, W.; DaFonseca, S.; Killian, M.; Epling, L.; Hoh, R.; Sinclair, E.; et al. CD4+ T Cells Expressing PD-1, TIGIT and LAG-3 Contribute to HIV Persistence during ART. PLoS Pathog. 2016, 12, e1005761. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, G.N.; Seclén, E.; Wietgrefe, S.; Liu, S.; Chateau, M.; Pei, H.; Perkey, K.; Marsden, M.D.; Hinkley, S.J.; Paschon, D.E.; et al. Humanized Mouse Model of HIV-1 Latency with Enrichment of Latent Virus in PD-1 and TIGIT CD4 T Cells. J. Virol. 2019, 93, 10. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; Rudensky, A. Roles of Regulatory T Cells in Tissue Pathophysiology and Metabolism. Cell Metab. 2020, 31, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Ciurkiewicz, M.; Herder, V.; Beineke, A. Beneficial and Detrimental Effects of Regulatory T Cells in Neurotropic Virus Infections. Int. J. Mol. Sci. 2020, 21, 1705. [Google Scholar] [CrossRef] [PubMed]
- Rowe, J.H.; Ertelt, J.M.; Way, S.S. Foxp3(+) regulatory T cells, immune stimulation and host defence against infection. Immunology 2012, 136, 1–10. [Google Scholar] [CrossRef]
- Elahi, S.; Niki, T.; Hirashima, M.; Horton, H. Galectin-9 binding to Tim-3 renders activated human CD4+ T cells less susceptible to HIV-1 infection. Blood 2012, 119, 4192–4204. [Google Scholar] [CrossRef]
- Jain, N.; Nguyen, H.; Chambers, C.; Kang, J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc. Natl. Acad. Sci. USA 2010, 107, 1524–1528. [Google Scholar] [CrossRef]
- Nikolova, M.; Wiedemann, A.; Muhtarova, M.; Achkova, D.; Lacabaratz, C.; Lévy, Y. Subset- and Antigen-Specific Effects of Treg on CD8+ T Cell Responses in Chronic HIV Infection. PLoS Pathog. 2016, 12, e1005995. [Google Scholar] [CrossRef]
- Jones, R.B.; Walker, B.D. HIV-specific CD8⁺ T cells and HIV eradication. J. Clin. Investig. 2016, 126, 455–463. [Google Scholar] [CrossRef]
- Dyck, L.; Mills, K.H.G. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur. J. Immunol. 2017, 47, 765–779. [Google Scholar] [CrossRef]
- Freeman, M.L.; Shive, C.L.; Nguyen, T.P.; Younes, S.-A.; Panigrahi, S.; Lederman, M.M. Cytokines and T-Cell Homeostasis in HIV Infection. J. Infect. Dis. 2016, 214 (Suppl. 2), S51–S57. [Google Scholar] [CrossRef] [PubMed]
- Akhmetzyanova, I.; Drabczyk, M.; Neff, C.P.; Gibbert, K.; Dietze, K.K.; Werner, T.; Liu, J.; Chen, L.; Lang, K.S.; Palmer, B.E.; et al. PD-L1 Expression on Retrovirus-Infected Cells Mediates Immune Escape from CD8+ T Cell Killing. PLoS Pathog. 2015, 11, e1005224. [Google Scholar]
- Meyaard, L.; Miedema, F. Immune dysregulation and CD4+ T cell loss in HIV-1 infection. Springer Semin. Immunopathol. 1997, 18, 285–303. [Google Scholar] [CrossRef] [PubMed]
- Doitsh, G.; Galloway, N.L.K.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Muñoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.W.; Badley, A.D. Making sense of how HIV kills infected CD4 T cells: Implications for HIV cure. Mol. Cell Ther. 2014, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Doitsh, G.; Greene, W.C. Dissecting How CD4 T Cells Are Lost During HIV Infection. Cell Host Microbe 2016, 19, 280–291. [Google Scholar] [CrossRef]
- Lee, G.Q.; Lichterfeld, M. Diversity of HIV-1 reservoirs in CD4+ T-cell subpopulations. Curr. Opin. HIV AIDS 2016, 11, 383–387. [Google Scholar] [CrossRef]
- Pierson, T.; McArthur, J.; Siliciano, R.F. Reservoirs for HIV-1: Mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu. Rev. Immunol. 2000, 18, 665–708. [Google Scholar] [CrossRef]
- Kwon, K.J.; Timmons, A.E.; Sengupta, S.; Simonetti, F.R.; Zhang, H.; Hoh, R.; Deeks, S.G.; Siliciano, J.D.; Siliciano, R.F. Different human resting memory CD4 T cell subsets show similar low inducibility of latent HIV-1 proviruses. Sci. Transl. Med. 2020, 12, 528. [Google Scholar] [CrossRef]
- Martínez-Bonet, M.; Puertas, M.C.; Fortuny, C.; Ouchi, D.; Mellado, M.J.; Rojo, P.; Noguera-Julian, A.; Muñoz-Fernández, M.A.; Martinez-Picado, J. Establishment and Replenishment of the Viral Reservoir in Perinatally HIV-1-infected Children Initiating Very Early Antiretroviral Therapy. Clin. Infect. Dis. 2015, 61, 1169–1178. [Google Scholar] [CrossRef]
- Strongin, Z.; Sharaf, R.; VanBelzen, D.J.; Jacobson, J.M.; Connick, E.; Volberding, P.; Skiest, D.J.; Gandhi, R.T.; Kuritzkes, D.R.; O’Doherty, U.; et al. Effect of Short-Term Antiretroviral Therapy Interruption on Levels of Integrated HIV DNA. J. Virol. 2018, 92, 12. [Google Scholar] [CrossRef] [PubMed]
- Horiike, M.; Iwami, S.; Kodama, M.; Sato, A.; Watanabe, Y.; Yasui, M.; Ishida, Y.; Kobayashi, T.; Miura, T.; Igarashi, T. Lymph nodes harbor viral reservoirs that cause rebound of plasma viremia in SIV-infected macaques upon cessation of combined antiretroviral therapy. Virology 2012, 423, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.M.; Zaunders, J.J.; McBride, K.L.; Xu, Y.; Bailey, M.; Suzuki, K.; Cooper, D.A.; Emery, S.; Kelleher, A.D.; Koelsch, K.K. HIV DNA subspecies persist in both activated and resting memory CD4+ T cells during antiretroviral therapy. J. Virol. 2014, 88, 3516–3526. [Google Scholar] [CrossRef] [PubMed]
- Mohan, T.; Bhatnagar, S.; Gupta, D.L.; Rao, D.N. Current understanding of HIV-1 and T-cell adaptive immunity: Progress to date. Microb. Pathog. 2014, 73, 60–69. [Google Scholar] [CrossRef]
- D’Souza, M.; Fontenot, A.P.; Mack, D.G.; Lozupone, C.; Dillon, S.; Meditz, A.; Wilson, C.C.; Connick, E.; Palmer, B.E. Programmed death 1 expression on HIV-specific CD4+ T cells is driven by viral replication and associated with T cell dysfunction. J. Immunol. 2007, 179, 1979–1987. [Google Scholar] [CrossRef]
- Evans, V.A.; van der Sluis, R.M.; Solomon, A.; Dantanarayana, A.; McNeil, C.; Garsia, R.; Palmer, S.; Fromentin, R.; Chomont, N.; Sékaly, R.P.; et al. Programmed cell death-1 contributes to the establishment and maintenance of HIV-1 latency. AIDS (Lond. Engl.) 2018, 32, 1491–1497. [Google Scholar] [CrossRef]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef]
- Perreau, M.; Savoye, A.L.; De Crignis, E.; Corpataux, J.M.; Cubas, R.; Haddad, E.K.; De Leval, L.; Graziosi, C.; Pantaleo, G. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J. Exp. Med. 2013, 210, 143–156. [Google Scholar] [CrossRef]
- Garcia-Bates, T.M.; Palma, M.L.; Shen, C.; Gambotto, A.; Macatangay, B.J.C.; Ferris, R.L.; Rinaldo, C.R.; Mailliard, R.B. Contrasting Roles of the PD-1 Signaling Pathway in Dendritic Cell-Mediated Induction and Regulation of HIV-1-Specific Effector T Cell Functions. J. Virol. 2019, 93, 5. [Google Scholar] [CrossRef]
- McGary, C.S.; Deleage, C.; Harper, J.; Micci, L.; Ribeiro, S.P.; Paganini, S.; Kuri-Cervantes, L.; Benne, C.; Ryan, E.S.; Balderas, R.; et al. CTLA-4(+)PD-1(-) Memory CD4(+) T Cells Critically Contribute to Viral Persistence in Antiretroviral Therapy-Suppressed, SIV-Infected Rhesus Macaques. Immunity 2017, 47, 776–788.e5. [Google Scholar] [CrossRef]
- El-Far, M.; Ancuta, P.; Routy, J.-P.; Zhang, Y.; Bakeman, W.; Bordi, R.; DaFonseca, S.; Said, E.A.; Gosselin, A.; Tep, T.-S.; et al. Nef promotes evasion of human immunodeficiency virus type 1-infected cells from the CTLA-4-mediated inhibition of T-cell activation. J. Gen. Virol. 2015, 96 Pt 6, 1463–1477. [Google Scholar] [CrossRef][Green Version]
- Porichis, F.; Kwon, D.S.; Zupkosky, J.; Tighe, D.P.; McMullen, A.; Brockman, M.A.; Pavlik, D.F.; Rodriguez-Garcia, M.; Pereyra, F.; Freeman, G.J.; et al. Responsiveness of HIV-specific CD4 T cells to PD-1 blockade. Blood 2011, 118, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Wherry, E.J.; Ahmed, R.; Sharpe, A.H. Reinvigorating exhausted HIV-specific T cells via PD-1-PD-1 ligand blockade. J. Exp. Med. 2006, 203, 2223–2227. [Google Scholar] [CrossRef] [PubMed]
- Fromentin, R.; DaFonseca, S.; Costiniuk, C.T.; El-Far, M.; Procopio, F.A.; Hecht, F.M.; Hoh, R.; Deeks, S.G.; Hazuda, D.J.; Lewin, S.R.; et al. PD-1 blockade potentiates HIV latency reversal ex vivo in CD4(+) T cells from ART-suppressed individuals. Nat. Commun. 2019, 10, 814. [Google Scholar] [CrossRef] [PubMed]
- Peeridogaheh, H.; Meshkat, Z.; Habibzadeh, S.; Arzanlou, M.; Shahi, J.M.; Rostami, S.; Gerayli, S.; Teimourpour, R. Current concepts on immunopathogenesis of hepatitis B virus infection. Virus Res. 2018, 245, 29–43. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1. [Google Scholar] [CrossRef]
- Piekarska, A.; Jabłonowska, E.; Garlicki, A.; Sitko, M.; Mazur, W.; Jaroszewicz, J.; Czauz-Andrzejuk, A.; Buczyńska, I.; Simon, K.; Lorenc, B.; et al. Real life results of direct acting antiviral therapy for HCV infection in HIV-HCV-coinfected patients: Epi-Ter2 study. AIDS Care 2019, 32, 762–769. [Google Scholar] [CrossRef]
- Allweiss, L.; Dandri, M. The Role of cccDNA in HBV Maintenance. Viruses 2017, 9, 156. [Google Scholar] [CrossRef]
- Chen, D.S. Toward elimination and eradication of hepatitis B. J. Gastroenterol. Hepatol. 2010, 25, 19–25. [Google Scholar] [CrossRef]
- Mohd-Ismail, N.K.; Lim, Z.; Gunaratne, J.; Tan, Y.-J. Mapping the Interactions of HBV cccDNA with Host Factors. Int. J. Mol. Sci. 2019, 20, 4276. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, J.Y.; Wherry, E.J.; Jin, B.; Xu, B.; Zou, Z.S.; Zhang, S.Y.; Li, B.S.; Wang, H.F.; Wu, H.; et al. Dynamic programmed death 1 expression by virus-specific CD8 T cells correlates with the outcome of acute hepatitis B. Gastroenterology 2008, 134, 1938–1949.e1–3. [Google Scholar] [CrossRef] [PubMed]
- Yong, Y.K.; Saeidi, A.; Tan, H.Y.; Rosmawati, M.; Enström, P.F.; Batran, R.A.; Vasuki, V.; Chattopadhyay, I.; Murugesan, A.; Vignesh, R.; et al. Hyper-Expression of PD-1 Is Associated with the Levels of Exhausted and Dysfunctional Phenotypes of Circulating CD161TCR iVα7.2 Mucosal-Associated Invariant T Cells in Chronic Hepatitis B Virus Infection. Front. Immunol. 2018, 9, 472. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Nie, X.; Li, L.; Hu, L.; Wu, B.; Lin, J.; Jiang, C.; Wang, H.; Wang, X.; Shen, Q. B and T lymphocyte attenuator is highly expressed on intrahepatic T cells during chronic HBV infection and regulates their function. J. Gastroenterol. 2013, 48, 1362–1372. [Google Scholar] [CrossRef] [PubMed]
- Wieland, D.; Hofmann, M.; Thimme, R. Overcoming CD8+ T-Cell Exhaustion in Viral Hepatitis: Lessons from the Mouse Model and Clinical Perspectives. Dig. Dis. 2017, 35, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Golden-Mason, L.; Palmer, B.; Klarquist, J.; Mengshol, J.A.; Castelblanco, N.; Rosen, H.R. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J. Virol. 2007, 81, 9249–9258. [Google Scholar] [CrossRef] [PubMed]
- Raziorrouh, B.; Ulsenheimer, A.; Schraut, W.; Heeg, M.; Kurktschiev, P.; Zachoval, R.; Jung, M.; Thimme, R.; Neumann-Haefelin, C.; Horster, S.; et al. Inhibitory molecules that regulate expansion and restoration of HCV-specific CD4+ T cells in patients with chronic infection. (1528-0012 (Electronic)). Gastroenterol 2011, 141, 1422–1431.e6. [Google Scholar] [CrossRef]
- Tavakolpour, S.; Alavian, S.M.; Sali, S. Manipulation of Regulatory Cells’ Responses to Treatments for Chronic Hepatitis B Virus Infection. Hepat. Mon. 2016, 16, e37927. [Google Scholar] [CrossRef]
- Trehanpati, N.; Vyas, A.K. Immune Regulation by T Regulatory Cells in Hepatitis B Virus-Related Inflammation and Cancer. Scand. J. Immunol. 2017, 85, 175–181. [Google Scholar] [CrossRef]
- Hu, C.C.; Jeng, W.J.; Chen, Y.C.; Fang, J.H.; Huang, C.H.; Teng, W.; Hsieh, Y.C.; Lin, Y.C.; Chien, R.N.; Sheen, I.S.; et al. Memory Regulatory T cells Increase Only In Inflammatory Phase of Chronic Hepatitis B Infection and Related to Galectin-9/Tim-3 interaction. Sci. Rep. 2017, 7, 15280. [Google Scholar] [CrossRef]
- Feng, C.; Cao, L.J.; Song, H.F.; Xu, P.; Chen, H.; Xu, J.C.; Zhu, X.Y.; Zhang, X.G.; Wang, X.F. Expression of PD-L1 on CD4+CD25+Foxp3+ Regulatory T Cells of Patients with Chronic HBV Infection and Its Correlation with Clinical Parameters. Viral Immunol. 2015, 28, 418–424. [Google Scholar] [CrossRef]
- Cao, H.; Zhang, R.; Zhang, W. CTLA-4 interferes with the HBV-specific T cell immune response (Review). Int. J. Mol. Med. 2018, 42, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Dinney, C.M.; Zhao, L.D.; Conrad, C.D.; Duker, J.M.; Karas, R.O.; Hu, Z.; Hamilton, M.A.; Gillis, T.R.; Parker, T.M.; Fan, B.; et al. Regulation of HBV-specific CD8(+) T cell-mediated inflammation is diversified in different clinical presentations of HBV infection. J. Microbiol. 2015, 53, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Kroy, D.C.; Ciuffreda, D.; Cooperrider, J.H.; Tomlinson, M.; Hauck, G.D.; Aneja, J.; Berger, C.; Wolski, D.; Carrington, M.; Wherry, E.J.; et al. Liver environment and HCV replication affect human T-cell phenotype and expression of inhibitory receptors. Gastroenterology 2014, 146, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef]
- Wang, Q.; Pan, W.; Liu, Y.; Luo, J.; Zhu, D.; Lu, Y.; Feng, X.; Yang, X.; Dittmer, U.; Lu, M.; et al. Hepatitis B Virus-Specific CD8+ T Cells Maintain Functional Exhaustion after Antigen Reexposure in an Acute Activation Immune Environment. Front. Immunol. 2018, 9, 219. [Google Scholar] [CrossRef]
- Balsitis, S.; Gali, V.; Mason, P.J.; Chaniewski, S.; Levine, S.M.; Wichroski, M.J.; Feulner, M.; Song, Y.; Granaldi, K.; Loy, J.K.; et al. Safety and efficacy of anti-PD-L1 therapy in the woodchuck model of HBV infection. PLoS ONE 2018, 13, e0190058. [Google Scholar] [CrossRef]
- Xia, Y.; Protzer, U. Control of Hepatitis B Virus by Cytokines. Viruses 2017, 9, 18. [Google Scholar] [CrossRef]
- Xia, Y.; Stadler, D.; Lucifora, J.; Reisinger, F.; Webb, D.; Hösel, M.; Michler, T.; Wisskirchen, K.; Cheng, X.; Zhang, K.; et al. Interferon-γ and Tumor Necrosis Factor-α Produced by T Cells Reduce the HBV Persistence Form, cccDNA, Without Cytolysis. Gastroenterology 2016, 150, 194–205. [Google Scholar] [CrossRef]
- Ouaguia, L.; Morales, O.; Mrizak, D.; Ghazal, K.; Boleslawski, E.; Auriault, C.; Pancré, V.; de Launoit, Y.; Conti, F.; Delhem, N. Overexpression of Regulatory T Cells Type 1 (Tr1) Specific Markers in a Patient with HCV-Induced Hepatocellular Carcinoma. ISRN Hepatol. 2013, 2013, 928485. [Google Scholar] [CrossRef]
- Li, X.; Liu, X.; Tian, L.; Chen, Y. Cytokine-Mediated Immunopathogenesis of Hepatitis B Virus Infections. Clin. Rev. Allergy Immunol. 2016, 50, 41–54. [Google Scholar] [CrossRef]
- Speletas, M.; Argentou, N.; Germanidis, G.; Vasiliadis, T.; Mantzoukis, K.; Patsiaoura, K.; Nikolaidis, P.; Karanikas, V.; Ritis, K.; Germenis, A.E. Foxp3 expression in liver correlates with the degree but not the cause of inflammation. Med. Inflamm. 2011, 2011, 827565. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Su, Z.J.; Yu, X.P.; Guo, R.Y.; Ming, D.S.; Huang, L.Y.; Su, M.L.; Deng, Y.; Lin, Z.Z. Changes in the balance between Treg and Th17 cells in patients with chronic hepatitis B. Diagn. Microbiol. Infect. Dis. 2013, 76, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Lechmann, M.; Woitas, R.P.; Langhans, B.; Kaiser, R.; Ihlenfeldt, H.G.; Jung, G.; Sauerbruch, T.; Spengler, U. Decreased frequency of HCV core-specific peripheral blood mononuclear cells with type 1 cytokine secretion in chronic hepatitis C. J. Hepatol. 1999, 31, 971–978. [Google Scholar] [CrossRef]
- Chen, N.; Liu, Y.; Guo, Y.; Chen, Y.; Liu, X.; Liu, M. Lymphocyte activation gene 3 negatively regulates the function of intrahepatic hepatitis C virus-specific CD8+ T cells. J. Gastroenterol. Hepatol. 2015, 30, 1788–1795. [Google Scholar] [CrossRef]
- Maier, H.; Isogawa, M.; Freeman, G.J.; Chisari, F.V. PD-1:PD-L1 interactions contribute to the functional suppression of virus-specific CD8+ T lymphocytes in the liver. J. Immunol. 2007, 178, 2714–2720. [Google Scholar] [CrossRef]
- Horiuchi, Y.; Takagi, A.; Kobayashi, N.; Moriya, O.; Nagai, T.; Moriya, K.; Tsutsumi, T.; Koike, K.; Akatsuka, T. Effect of the infectious dose and the presence of hepatitis C virus core gene on mouse intrahepatic CD8 T cells. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2014, 44, E240–E252. [Google Scholar] [CrossRef]
- Cooksley, H.; Riva, A.; Katzarov, K.; Hadzhiolova-Lebeau, T.; Pavlova, S.; Simonova, M.; Williams, R.; Chokshi, S. Differential Expression of Immune Inhibitory Checkpoint Signatures on Antiviral and Inflammatory T Cell Populations in Chronic Hepatitis B. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2018, 38, 273–282. [Google Scholar] [CrossRef]
- Wang, W.; Tong, Z.; Zhong, J.; Zhang, L.; Zhang, H.; Su, Y.; Gao, B.; Zhang, C. Identification of IL-10-secreting CD8CD28PD-1 regulatory T cells associated with chronic hepatitis C virus infection. Immunol. Lett. 2018, 202, 16–22. [Google Scholar] [CrossRef]
- Xiao, W.; Jiang, L.F.; Deng, X.Z.; Zhu, D.Y.; Pei, J.P.; Xu, M.L.; Li, B.J.; Wang, C.J.; Zhang, J.H.; Zhang, Q.; et al. PD-1/PD-L1 signal pathway participates in HCV F protein-induced T cell dysfunction in chronic HCV infection. Immunol. Res. 2016, 64, 412–423. [Google Scholar] [CrossRef]
- Townsend, E.C.; Zhang, G.Y.; Ali, R.; Firke, M.; Moon, M.S.; Han, M.A.T.; Fram, B.; Glenn, J.S.; Kleiner, D.E.; Koh, C.; et al. The balance of type 1 and type 2 immune responses in the contexts of hepatitis B infection and hepatitis D infection. J. Gastroenterol. Hepatol. 2019, 34, 764–775. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Wahl, S.M. Engagement of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) induces transforming growth factor beta (TGF-beta) production by murine CD4(+) T cells. J. Exp. Med. 1998, 188, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.J.; Ma, C.J.; Wang, J.M.; Wu, X.Y.; Niki, T.; Hirashima, M.; Moorman, J.P.; Yao, Z.Q. HCV-infected hepatocytes drive CD4+ CD25+ Foxp3+ regulatory T-cell development through the Tim-3/Gal-9 pathway. Eur. J. Immunol. 2013, 43, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Ma, C.J.; Li, G.Y.; Wu, X.Y.; Thayer, P.; Greer, P.; Smith, A.M.; High, K.P.; Moorman, J.P.; Yao, Z.Q. Tim-3 alters the balance of IL-12/IL-23 and drives TH17 cells: Role in hepatitis B vaccine failure during hepatitis C infection. Vaccine 2013, 31, 2238–2245. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Shi, L.; Ma, C.J.; Ji, X.J.; Ying, R.S.; Wu, X.Y.; Wang, K.S.; Li, G.; Moorman, J.P.; Yao, Z.Q. Differential regulation of interleukin-12 (IL-12)/IL-23 by Tim-3 drives T(H)17 cell development during hepatitis C virus infection. J. Virol. 2013, 87, 4372–4383. [Google Scholar] [CrossRef]
- Chigbu, D.I.; Loonawat, R.; Sehgal, M.; Patel, D.; Jain, P. Hepatitis C Virus Infection: Host⁻Virus Interaction and Mechanisms of Viral Persistence. Cells 2019, 8, 376. [Google Scholar] [CrossRef]
- Ma, C.J.; Ni, L.; Zhang, Y.; Zhang, C.L.; Wu, X.Y.; Atia, A.N.; Thayer, P.; Moorman, J.P.; Yao, Z.Q. PD-1 negatively regulates interleukin-12 expression by limiting STAT-1 phosphorylation in monocytes/macrophages during chronic hepatitis C virus infection. Immunology 2011, 132, 421–431. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Chowdhury, M.A.; Hossain, N.; Kashem, M.A.; Shahid, M.A.; Alam, A. Immune response in COVID-19: A review. J. Infect. Public Health 2020. [Google Scholar] [CrossRef]
- Citarella, F.; Russano, M.; Pantano, F.; Dell’Aquila, E.; Vincenzi, B.; Tonini, G.; Santini, D. Facing SARS-CoV-2 outbreak in immunotherapy era. Future Oncol. 2020, 16, 1475–1485. [Google Scholar] [CrossRef]
- Guihot, A.; Litvinova, E.; Autran, B.; Debré, P.; Vieillard, V. Cell-Mediated Immune Responses to COVID-19 Infection. Front. Immunol. 2020, 11, 1662. [Google Scholar] [CrossRef]
- Gambichler, T.; Reuther, J.; Scheel, C.; Becker, J. On the use of immune checkpoint inhibitors in patients with viral infections including COVID-19. J. ImmunoTher. Cancer 2020, 8, e001145. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Demaria, O.; Carvelli, J.; Batista, L.; Thibult, M.-L.; Morel, A.; André, P.; Morel, Y.; Vély, F.; Vivier, E. Identification of druggable inhibitory immune checkpoints on Natural Killer cells in COVID-19. Cell. Mol. Immunol. 2020, 1–3. [Google Scholar] [CrossRef]
- Lee, L.Y.W.; Cazier, J.B.; Starkey, T.; Turnbull, C.D.; Kerr, R.; Middleton, G. COVID-19 mortality in patients with cancer on chemotherapy or other anticancer treatments: A prospective cohort study. Lancet 2020, 395, 1919–1926. [Google Scholar] [CrossRef]
- Luo, J.; Rizvi, H.; Egger, J.; Preeshagul, I.; Wolchok, J.; Hellmann, M. Impact of PD-1 Blockade on Severity of COVID-19 in Patients with Lung Cancers. Cancer Discov. 2020, CD-20. [Google Scholar] [CrossRef] [PubMed]
- Robilotti, E.; Babady, N.; Mead, P.; Rolling, T.; Perez-Johnston, R.; Bernardes, M.; Bogler, Y.; Caldararo, M.; Figueroa, C.; Glickman, M.; et al. Determinants of COVID-19 disease severity in patients with cancer. Nat. Med. 2020, 26, 1218–1223. [Google Scholar] [CrossRef]
- Suresh, K.; Voong, K.; Shankar, B.; Forde, P.; Ettinger, D.; Marrone, K.; Kelly, R.; Hann, C.; Levy, B.; Feliciano, J.; et al. Pneumonitis in Non-Small Cell Lung Cancer Patients Receiving Immune Checkpoint Immunotherapy: Incidence and Risk Factors. J. Thorac. Oncol. 2018, 13, 1930–1939. [Google Scholar] [CrossRef]
- Esau, D. Viral Causes of Lymphoma: The History of Epstein-Barr Virus and Human T-Lymphotropic Virus 1. Virology (Auckl) 2017, 8, 1178122X17731772. [Google Scholar] [CrossRef]
- Ressing, M.E.; van Gent, M.; Gram, A.M.; Hooykaas, M.J.G.; Piersma, S.J.; Wiertz, E.J.H.J. Immune Evasion by Epstein-Barr Virus. Curr. Top Microbiol. Immunol. 2015, 391, 355–381. [Google Scholar]
- Kennedy, P.G.E.; Rovnak, J.; Badani, H.; Cohrs, R.J. A comparison of herpes simplex virus type 1 and varicella-zoster virus latency and reactivation. J. Gen. Virol. 2015, 96 Pt 7, 1581–1602. [Google Scholar] [CrossRef]
- Lobo, A.-M.; Agelidis, A.M.; Shukla, D. Pathogenesis of herpes simplex keratitis: The host cell response and ocular surface sequelae to infection and inflammation. Ocul. Surf. 2019, 17, 40–49. [Google Scholar] [CrossRef] [PubMed]

| Immune Checkpoint | Super Family | Ligand | Expression | T-cell Inactivation Pathway | Ref. |
|---|---|---|---|---|---|
| PD-1 | CD28 | PD-L1/L2 | T-cells, B-cells, natural killer T-cells, DCs and activated monocytes |
| [20,21,22] |
| CTLA-4 | CD28 | B7 (CD80/CD86) | Active T-cells | CTLA-4 → PP2A → Akt → mTORc1 → regulates T-cell proliferation and differentiation | [25,26,27] |
| TIM-3 | TIM | Galcetin-9 | Th1, Tc1, Treg cells |
| [32] |
| TIGIT | CD28 | CD155 on DCs | T-cells and NK cells | TIGIT → SHIP1 → PI3K → AKT/PKC/IKK → NF-κB | [31,35] |
| LAG-3 | Immunoglobulin | MHC class Ⅱ | Activated T-cells Treg cells, B-cells, DCs and NK cells | LAG- KIEELE motif /FXXL motif/ C terminus EX repeat → unknown | [38] |
| BTLA/CD160 | CD28 | HVEM | T-cells and B-cells, activated T-cells |
| [39,41] |
| Infection | Target | NCT ID | Research Title/Objective | Drug | Status |
|---|---|---|---|---|---|
| HIV | PD-1 | NCT03239899 | PD-1 Inhibition to Determine CNS Reservoir of HIV-Infection | Pembrolizumab | Phase Ⅰ |
| PD-1 | NCT03787095 | Safety and Immunotherapeutic Activity of an Anti-PD-1 Antibody (Cemiplimab) in HIV-1-infected Participants on Suppressive cART | Cemiplimab | Phase Ⅰ | |
| IMC | NCT03354936 | Assess the safety of the use of immune checkpoint inhibitors in HIV infected patients | Nivolumab, Pembrolizumab | Recruiting | |
| PD-1 | NCT03367754 | A Single Dose of Pembrolizumab in HIV-Infected People | Pembrolizumab | Phase Ⅰ | |
| HBV | PD-L1 | NCT03899428 | Immune Checkpoint Therapy vs Target Therapy in Reducing Serum HBsAg Levels in Patients with HBsAg+ Advanced Stage HCC | Durvalumab | Phase Ⅱ |
| HCV | PD-1 | NCT00703469 | A Study of MDX-1106 to Treat Patients with Hepatitis C Infection (MDX1106-02) | MDX-1106 (PD-1 ab) | Phase Ⅰ |
| Influenza | PD-1 | NCT03061955 | Safety and Efficacy of Concurrent Administration of Influenza Vaccine in Patients Undergoing Anti-PD-1 Immunotherapy (Nivolumab, Pembrolizumab) | Nivolumab, Pembrolizumab | Completed |
| PD-1/CTLA-4 | NCT03590808 | Influenza Vaccination in Patients Receiving Immune Checkpoint Inhibitor | Pembrolizumab, Nivolumab, Ipilimumab | Completed | |
| HPV | CTLA-4 | NCT01693783 | Ipilimumab in Treating Patients with Metastatic or Recurrent Human Papilloma Virus-Related Cervical Cancer | Ipilimumab | Phase Ⅱ |
| EBV | PD-1 | NCT03755440 | PD-1 Antibody in EBV Positive Metastatic Gastric Cancer Patients | SHR-1210 (PD-1 ab) | Phase Ⅱ |
| PD-1 | NCT02488759 | An Investigational Immuno-Therapy Study to Investigate the Safety and Effectiveness of Nivolumab, and Nivolumab Combination Therapy in Virus-associated Tumors (CheckMate358) | Nivolumab | Phase Ⅰ |
| Chronic Virus | Cytokines Secretion during Viral Infection | Associated Immune Checkpoint | Effects | Ref. | |
|---|---|---|---|---|---|
| HBV | TH-1/CD8+T | IFN-γ ↓ | CTLA-4, PD-1, Tim-3, LAG-3, BTLA | Activate multiple immune responses Enhance Th1 responses Inhibit cccDNA replication and accumulation | [62,63,64,68,100,143,151,152,166] |
| IL-2 ↓ | CTLA-4, BTLA, PD-1, Tim-3, LAG-3 | Induce differentiation and effector function of T-cell, NK and LAK | [62,64,67,143] | ||
| TNF-α ↓ | PD-1, Tim-3, LAG-3 | Lead to direct clearance of virus-infection cells | [64,152,166] | ||
| IL-12 ↓ | PD-1 | Impair host defense and viral clearance | [176] | ||
| Treg | IL-10, TGF-β ↑ | CTLA-4, PD-1, Tim-3 | suppress effector T-cells and regulate liver fibrosis | [171,172,173] | |
| TH-2 | IL-4, IL-5, IL-10 ↓ | LAG-3, PD-1 | Downregulate the activation of B- and T-cell proliferation Suppress TH1 response | [64] | |
| TH-17 | IL-17 ↑ | TIM-3 | Promote the exacerbation of liver inflammation and sustain the proinflammatory response | [167] | |
| HCV | TH-1/CD8+ T-cell | IFN-γ ↓ | LAG-3, PD-1, Tim-3 | Promote Th1 immune response, suppress Th2 and Th17 responses | [71,164] |
| IL-2 ↓ | PD-1, CTLA-4 | Inactivate multiple immune-cell subsets, including T-cells, NK cells, B-cells, monocytes, macrophages, and neutrophils | [145] | ||
| TNF-α ↓ | LAG-3 | Decrease the direct clearance of virus-infection cells | [164] | ||
| IL-12 ↓ | Tim-3 | Decrease the stimulation of TH1 responses that are essential for host defense and pathogen clearance | [173] | ||
| Treg | IL-10, TGF-β ↑ | PD-1, Tim-3, CTLA-4 | Limit the secretion of proinflammatory cytokines and suppress effector T-cells | [73,146,168] | |
| TH-2 | IL-4, IL-5, IL-10 ↑ | PD-1 | Promote the activation of B and T-cell proliferation and limit the secretion of proinflammatory cytokines | [169] | |
| TH-17 | IL-17 ↑ | Tim-3 | Promote the exacerbation of liver inflammation and injury | [173,174] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, H.; Liu, G.; Zhong, J.; Zheng, K.; Xiao, H.; Li, C.; Song, X.; Li, Y.; Xu, C.; Wu, H.; et al. Immune Checkpoints in Viral Infections. Viruses 2020, 12, 1051. https://doi.org/10.3390/v12091051
Cai H, Liu G, Zhong J, Zheng K, Xiao H, Li C, Song X, Li Y, Xu C, Wu H, et al. Immune Checkpoints in Viral Infections. Viruses. 2020; 12(9):1051. https://doi.org/10.3390/v12091051
Chicago/Turabian StyleCai, Huiming, Ge Liu, Jianfeng Zhong, Kai Zheng, Haitao Xiao, Chenyang Li, Xun Song, Ying Li, Chenshu Xu, Haiqiang Wu, and et al. 2020. "Immune Checkpoints in Viral Infections" Viruses 12, no. 9: 1051. https://doi.org/10.3390/v12091051
APA StyleCai, H., Liu, G., Zhong, J., Zheng, K., Xiao, H., Li, C., Song, X., Li, Y., Xu, C., Wu, H., He, Z., & Zhu, Q. (2020). Immune Checkpoints in Viral Infections. Viruses, 12(9), 1051. https://doi.org/10.3390/v12091051