Reversal of T Cell Exhaustion in Chronic HCV Infection


Abstract
1. Introduction
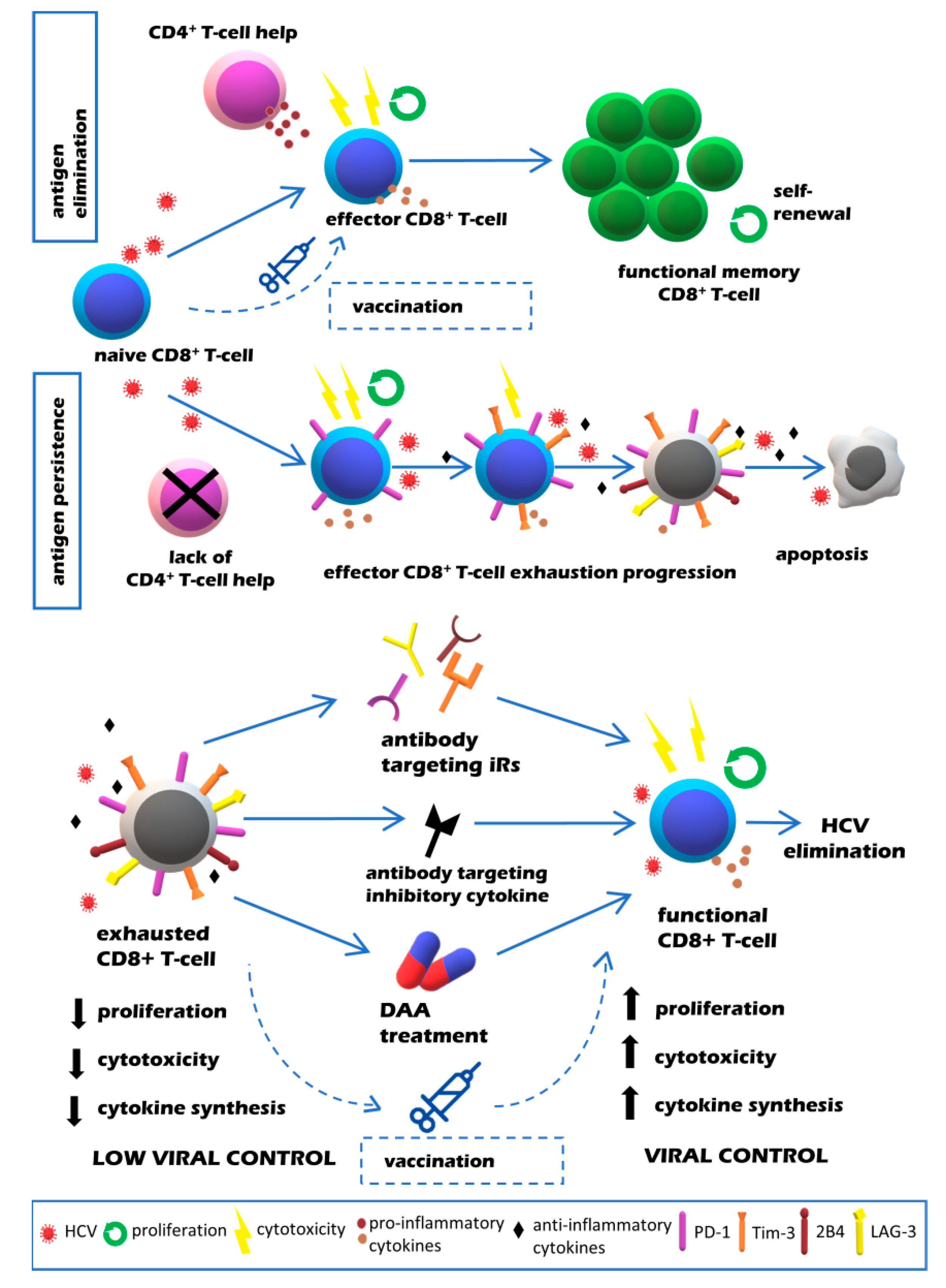
2. T Cell Exhaustion in HCV Infection
3. Reversibility of T Cell Exhaustion in HCV Infection
4. Impact of DAA Treatment of Chronic HCV Infection on T Cell Exhaustion
5. Effect of Vaccines on T Cell Exhaustion in Chronic HCV Infection
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- World Healh Organization. Available online: https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (accessed on 2 June 2020).
- Webster, D.P.; Klenerman, P.; Dusheiko, G.M. Hepatitis C. Lancet 2015, 385, 1124–1135. [Google Scholar] [CrossRef]
- Ansaldi, F.; Orsi, A.; Sticchi, L.; Bruzzone, B.; Icardi, G. Hepatitis C virus in the new era: Perspectives in epidemiology, prevention, diagnostics and predictors of response to therapy. World J. Gastroenterol. 2014, 20, 9633–9652. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.R.; Honegger, J.R. Hepatitis C virus in pregnancy. Am. J. Perinatol. 2013, 30, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Ueno, Y. Transmission of hepatitis C virus: Self-limiting hepatitis or chronic hepatitis? World J. Gastroenterol. 2013, 19, 6957–6961. [Google Scholar] [CrossRef] [PubMed]
- Wilder, J.M.; Muir, A.J. Strategies for treating chronic HCV infection in patients with cirrhosis: Latest evidence and clinical outcomes. Adv. Chronic Dis. 2015, 6, 314–327. [Google Scholar] [CrossRef]
- Goossens, N.; Hoshida, Y. Hepatitis C virus-induced hepatocellular carcinoma. Clin. Mol. Hepatol. 2015, 21, 105–114. [Google Scholar] [CrossRef]
- Conti, F.; Buonfiglioli, F.; Scuteri, A.; Crespi, C.; Bolondi, L.; Caraceni, P.; Foschi, F.G.; Lenzi, M.; Mazzella, G.; Verucchi, G.; et al. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J. Hepatol. 2016, 65, 727–733. [Google Scholar] [CrossRef]
- Martell, M.; Esteban, J.I.; Quer, J.; Genesca, J.; Weiner, A.; Esteban, R.; Guardia, J.; Gomez, J. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: Quasispecies nature of HCV genome distribution. J. Virol. 1992, 66, 3225–3229. [Google Scholar] [CrossRef]
- Burke, K.P.; Cox, A.L. Hepatitis C virus evasion of adaptive immune responses: A model for viral persistence. Immunol. Res. 2010, 47, 216–227. [Google Scholar] [CrossRef]
- Erickson, A.L.; Kimura, Y.; Igarashi, S.; Eichelberger, J.; Houghton, M.; Sidney, J.; McKinney, D.; Sette, A.; Hughes, A.L.; Walker, C.M. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity 2001, 15, 883–895. [Google Scholar] [CrossRef]
- Cox, A.L.; Mosbruger, T.; Mao, Q.; Liu, Z.; Wang, X.H.; Yang, H.C.; Sidney, J.; Sette, A.; Pardoll, D.; Thomas, D.L.; et al. Cellular immune selection with hepatitis C virus persistence in humans. J. Exp. Med. 2005, 201, 1741–1752. [Google Scholar] [CrossRef]
- Neumann-Haefelin, C.; Timm, J.; Spangenberg, H.C.; Wischniowski, N.; Nazarova, N.; Kersting, N.; Roggendorf, M.; Allen, T.M.; Blum, H.E.; Thimme, R. Virological and immunological determinants of intrahepatic virus-specific CD8+ T-cell failure in chronic hepatitis C virus infection. Hepatology 2008, 47, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B. Hepatitis C virus versus innate and adaptive immune responses: A tale of coevolution and coexistence. J. Clin. Investig. 2009, 119, 1745–1754. [Google Scholar] [CrossRef]
- Heim, M.H.; Thimme, R. Innate and adaptive immune responses in HCV infections. J. Hepatol. 2014, 61, S14–S25. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, K.; Flecken, T.; Schmidt, J.; Gostick, E.; Marget, M.; Neumann-Haefelin, C.; Blum, H.E.; Price, D.A.; Thimme, R. Tetramer enrichment reveals the presence of phenotypically diverse hepatitis C virus-specific CD8+ T cells in chronic infection. J. Virol. 2015, 89, 25–34. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Neumann-Haefelin, C.; Thimme, R. Adaptive immune responses in hepatitis C virus infection. Curr. Top. Microbiol. Immunol. 2013, 369, 243–262. [Google Scholar] [CrossRef] [PubMed]
- Penna, A.; Pilli, M.; Zerbini, A.; Orlandini, A.; Mezzadri, S.; Sacchelli, L.; Missale, G.; Ferrari, C. Dysfunction and functional restoration of HCV-specific CD8 responses in chronic hepatitis C virus infection. Hepatology 2007, 45, 588–601. [Google Scholar] [CrossRef]
- Dustin, L.B. Innate and Adaptive Immune Responses in Chronic HCV Infection. Curr. Drug Targets 2017, 18, 826–843. [Google Scholar] [CrossRef]
- Urbani, S.; Amadei, B.; Tola, D.; Massari, M.; Schivazappa, S.; Missale, G.; Ferrari, C. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J. Virol. 2006, 80, 11398–11403. [Google Scholar] [CrossRef]
- Thimme, R.; Oldach, D.; Chang, K.M.; Steiger, C.; Ray, S.C.; Chisari, F.V. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med. 2001, 194, 1395–1406. [Google Scholar] [CrossRef]
- Wolski, D.; Foote, P.K.; Chen, D.Y.; Lewis-Ximenez, L.L.; Fauvelle, C.; Aneja, J.; Walker, A.; Tonnerre, P.; Torres-Cornejo, A.; Kvistad, D.; et al. Early Transcriptional Divergence Marks Virus-Specific Primary Human CD8(+) T Cells in Chronic versus Acute Infection. Immunity 2017, 47, 648–663.e8. [Google Scholar] [CrossRef] [PubMed]
- Alfei, F.; Kanev, K.; Hofmann, M.; Wu, M.; Ghoneim, H.E.; Roelli, P.; Utzschneider, D.T.; von Hoesslin, M.; Cullen, J.G.; Fan, Y.; et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 2019, 571, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Boni, C.; Fisicaro, P.; Valdatta, C.; Amadei, B.; Di Vincenzo, P.; Giuberti, T.; Laccabue, D.; Zerbini, A.; Cavalli, A.; Missale, G.; et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J. Virol. 2007, 81, 4215–4225. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef]
- Rehermann, B.; Thimme, R. Insights From Antiviral Therapy Into Immune Responses to Hepatitis B and C Virus Infection. Gastroenterology 2019, 156, 369–383. [Google Scholar] [CrossRef]
- Li, H.; van der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; van Akkooi, A.C.J.; van den Braber, M.; Rozeman, E.A.; Haanen, J.; Blank, C.U.; et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2020, 181, 747. [Google Scholar] [CrossRef]
- Fuller, M.J.; Khanolkar, A.; Tebo, A.E.; Zajac, A.J. Maintenance, loss, and resurgence of T cell responses during acute, protracted, and chronic viral infections. J. Immunol. 2004, 172, 4204–4214. [Google Scholar] [CrossRef]
- Mackerness, K.J.; Cox, M.A.; Lilly, L.M.; Weaver, C.T.; Harrington, L.E.; Zajac, A.J. Pronounced virus-dependent activation drives exhaustion but sustains IFN-gamma transcript levels. J. Immunol. 2010, 185, 3643–3651. [Google Scholar] [CrossRef]
- Fuller, M.J.; Hildeman, D.A.; Sabbaj, S.; Gaddis, D.E.; Tebo, A.E.; Shang, L.; Goepfert, P.A.; Zajac, A.J. Cutting edge: Emergence of CD127high functionally competent memory T cells is compromised by high viral loads and inadequate T cell help. J. Immunol. 2005, 174, 5926–5930. [Google Scholar] [CrossRef]
- Lang, K.S.; Recher, M.; Navarini, A.A.; Harris, N.L.; Lohning, M.; Junt, T.; Probst, H.C.; Hengartner, H.; Zinkernagel, R.M. Inverse correlation between IL-7 receptor expression and CD8 T cell exhaustion during persistent antigen stimulation. Eur. J. Immunol. 2005, 35, 738–745. [Google Scholar] [CrossRef]
- Wherry, E.J.; Barber, D.L.; Kaech, S.M.; Blattman, J.N.; Ahmed, R. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc. Natl. Acad. Sci. USA 2004, 101, 16004–16009. [Google Scholar] [CrossRef] [PubMed]
- Speiser, D.E.; Utzschneider, D.T.; Oberle, S.G.; Munz, C.; Romero, P.; Zehn, D. T cell differentiation in chronic infection and cancer: Functional adaptation or exhaustion? Nat. Rev. Immunol. 2014, 14, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Doering, T.A.; Crawford, A.; Angelosanto, J.M.; Paley, M.A.; Ziegler, C.G.; Wherry, E.J. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 2012, 37, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Beltra, J.-C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020, 52, 825–841.e8. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef]
- Chihara, N.; Madi, A.; Kondo, T.; Zhang, H.; Acharya, N.; Singer, M.; Nyman, J.; Marjanovic, N.D.; Kowalczyk, M.S.; Wang, C.; et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 2018, 558, 454–459. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-cell exhaustion: Characteristics, causes and conversion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Fuertes Marraco, S.A.; Neubert, N.J.; Verdeil, G.; Speiser, D.E. Inhibitory Receptors Beyond T Cell Exhaustion. Front. Immunol. 2015, 6, 310. [Google Scholar] [CrossRef] [PubMed]
- Odorizzi, P.M.; Wherry, E.J. Inhibitory receptors on lymphocytes: Insights from infections. J. Immunol. 2012, 188, 2957–2965. [Google Scholar] [CrossRef]
- Hudson, W.H.; Gensheimer, J.; Hashimoto, M.; Wieland, A.; Valanparambil, R.M.; Li, P.; Lin, J.X.; Konieczny, B.T.; Im, S.J.; Freeman, G.J.; et al. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1(+) Stem-like CD8(+) T Cells during Chronic Infection. Immunity 2019, 51, 1043–1058.e4. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Freeman, G.J.; Wherry, E.J. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc. Natl. Acad. Sci. USA 2008, 105, 15016–15021. [Google Scholar] [CrossRef] [PubMed]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef]
- Wolski, D.; Lauer, G.M. Hepatitis C Virus as a Unique Human Model Disease to Define Differences in the Transcriptional Landscape of T Cells in Acute versus Chronic Infection. Viruses 2019, 11, 683. [Google Scholar] [CrossRef]
- Agnellini, P.; Wolint, P.; Rehr, M.; Cahenzli, J.; Karrer, U.; Oxenius, A. Impaired NFAT nuclear translocation results in split exhaustion of virus-specific CD8+ T cell functions during chronic viral infection. Proc. Natl. Acad. Sci. USA 2007, 104, 4565–4570. [Google Scholar] [CrossRef]
- Kao, C.; Oestreich, K.J.; Paley, M.A.; Crawford, A.; Angelosanto, J.M.; Ali, M.A.; Intlekofer, A.M.; Boss, J.M.; Reiner, S.L.; Weinmann, A.S.; et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat. Immunol. 2011, 12, 663–671. [Google Scholar] [CrossRef]
- Quigley, M.; Pereyra, F.; Nilsson, B.; Porichis, F.; Fonseca, C.; Eichbaum, Q.; Julg, B.; Jesneck, J.L.; Brosnahan, K.; Imam, S.; et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat. Med. 2010, 16, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Blackburn, S.D.; Intlekofer, A.M.; Kao, C.; Angelosanto, J.M.; Reiner, S.L.; Wherry, E.J. A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity 2009, 31, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, D.T.; Charmoy, M.; Chennupati, V.; Pousse, L.; Ferreira, D.P.; Calderon-Copete, S.; Danilo, M.; Alfei, F.; Hofmann, M.; Wieland, D.; et al. T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 2016, 45, 415–427. [Google Scholar] [CrossRef]
- Chen, Z.; Ji, Z.; Ngiow, S.F.; Manne, S.; Cai, Z.; Huang, A.C.; Johnson, J.; Staupe, R.P.; Bengsch, B.; Xu, C.; et al. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity 2019, 51, 840–855.e5. [Google Scholar] [CrossRef]
- Wu, T.; Ji, Y.; Moseman, E.A.; Xu, H.C.; Manglani, M.; Kirby, M.; Anderson, S.M.; Handon, R.; Kenyon, E.; Elkahloun, A.; et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 2016, 1, eaai8593. [Google Scholar] [CrossRef]
- Bentzen, A.K.; Hadrup, S.R. Evolution of MHC-based technologies used for detection of antigen-responsive T cells. Cancer Immunol. Immunother. CII 2017, 66, 657–666. [Google Scholar] [CrossRef]
- Holland, C.J.; Dolton, G.; Scurr, M.; Ladell, K.; Schauenburg, A.J.; Miners, K.; Madura, F.; Sewell, A.K.; Price, D.A.; Cole, D.K.; et al. Enhanced Detection of Antigen-Specific CD4+ T Cells Using Altered Peptide Flanking Residue Peptide-MHC Class II Multimers. J. Immunol. 2015, 195, 5827–5836. [Google Scholar] [CrossRef]
- Dong, Y.; Li, X.; Zhang, L.; Zhu, Q.; Chen, C.; Bao, J.; Chen, Y. CD4(+) T cell exhaustion revealed by high PD-1 and LAG-3 expression and the loss of helper T cell function in chronic hepatitis B. BMC Immunol. 2019, 20, 1–9. [Google Scholar] [CrossRef]
- Porichis, F.; Hart, M.G.; Zupkosky, J.; Barblu, L.; Kwon, D.S.; McMullen, A.; Brennan, T.; Ahmed, R.; Freeman, G.J.; Kavanagh, D.G.; et al. Differential impact of PD-1 and/or interleukin-10 blockade on HIV-1-specific CD4 T cell and antigen-presenting cell functions. J. Virol. 2014, 88, 2508–2518. [Google Scholar] [CrossRef]
- Osokine, I.; Snell, L.M.; Cunningham, C.R.; Yamada, D.H.; Wilson, E.B.; Elsaesser, H.J.; de la Torre, J.C.; Brooks, D. Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc. Natl. Acad. Sci. USA 2014, 111, 7409–7414. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.; Angelosanto, J.M.; Kao, C.; Doering, T.A.; Odorizzi, P.M.; Barnett, B.E.; Wherry, E.J. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity 2014, 40, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Major, M.E.; Dahari, H.; Mihalik, K.; Puig, M.; Rice, C.M.; Neumann, A.U.; Feinstone, S.M. Hepatitis C virus kinetics and host responses associated with disease and outcome of infection in chimpanzees. Hepatology 2004, 39, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Luxenburger, H.; Neumann-Haefelin, C.; Thimme, R.; Boettler, T. HCV-Specific T Cell Responses During and After Chronic HCV Infection. Viruses 2018, 10, 645. [Google Scholar] [CrossRef]
- Saeidi, A.; Zandi, K.; Cheok, Y.Y.; Saeidi, H.; Wong, W.F.; Lee, C.Y.Q.; Cheong, H.C.; Yong, Y.K.; Larsson, M.; Shankar, E.M. T-Cell Exhaustion in Chronic Infections: Reversing the State of Exhaustion and Reinvigorating Optimal Protective Immune Responses. Front. Immunol. 2018, 9, 2569. [Google Scholar] [CrossRef]
- Wieland, D.; Hofmann, M.; Thimme, R. Overcoming CD8+ T-Cell Exhaustion in Viral Hepatitis: Lessons from the Mouse Model and Clinical Perspectives. Dig. Dis. 2017, 35, 334–338. [Google Scholar] [CrossRef]
- Golden-Mason, L.; Palmer, B.; Klarquist, J.; Mengshol, J.A.; Castelblanco, N.; Rosen, H.R. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J. Virol. 2007, 81, 9249–9258. [Google Scholar] [CrossRef]
- Golden-Mason, L.; Palmer, B.E.; Kassam, N.; Townshend-Bulson, L.; Livingston, S.; McMahon, B.J.; Castelblanco, N.; Kuchroo, V.; Gretch, D.R.; Rosen, H.R. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J. Virol. 2009, 83, 9122–9130. [Google Scholar] [CrossRef]
- Urbani, S.; Amadei, B.; Tola, D.; Pedrazzi, G.; Sacchelli, L.; Cavallo, M.C.; Orlandini, A.; Missale, G.; Ferrari, C. Restoration of HCV-specific T cell functions by PD-1/PD-L1 blockade in HCV infection: Effect of viremia levels and antiviral treatment. J. Hepatol. 2008, 48, 548–558. [Google Scholar] [CrossRef]
- McMahan, R.H.; Golden-Mason, L.; Nishimura, M.I.; McMahon, B.J.; Kemper, M.; Allen, T.M.; Gretch, D.R.; Rosen, H.R. Tim-3 expression on PD-1+ HCV-specific human CTLs is associated with viral persistence, and its blockade restores hepatocyte-directed in vitro cytotoxicity. J. Clin. Investig. 2010, 120, 4546–4557. [Google Scholar] [CrossRef]
- Fuller, M.J.; Callendret, B.; Zhu, B.; Freeman, G.J.; Hasselschwert, D.L.; Satterfield, W.; Sharpe, A.H.; Dustin, L.B.; Rice, C.M.; Grakoui, A.; et al. Immunotherapy of chronic hepatitis C virus infection with antibodies against programmed cell death-1 (PD-1). Proc. Natl. Acad. Sci. USA 2013, 110, 15001–15006. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, D.; Lalezari, J.; Lawitz, E.; DiMicco, M.; Ghalib, R.; Reddy, K.R.; Chang, K.M.; Sulkowski, M.; Marro, S.O.; Anderson, J.; et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS ONE 2013, 8, e63818. [Google Scholar] [CrossRef] [PubMed]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Inarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef]
- Grover, S.; Rahma, O.E.; Hashemi, N.; Lim, R.M. Gastrointestinal and Hepatic Toxicities of Checkpoint Inhibitors: Algorithms for Management. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 13–19. [Google Scholar] [CrossRef]
- Richter, K.; Perriard, G.; Oxenius, A. Reversal of chronic to resolved infection by IL-10 blockade is LCMV strain dependent. Eur. J. Immunol. 2013, 43, 649–654. [Google Scholar] [CrossRef]
- Rigopoulou, E.I.; Abbott, W.G.; Haigh, P.; Naoumov, N.V. Blocking of interleukin-10 receptor--a novel approach to stimulate T-helper cell type 1 responses to hepatitis C virus. Clin. Immunol. 2005, 117, 57–64. [Google Scholar] [CrossRef]
- Vermehren, J.; Park, J.S.; Jacobson, I.M.; Zeuzem, S. Challenges and perspectives of direct antivirals for the treatment of hepatitis C virus infection. J. Hepatol. 2018, 69, 1178–1187. [Google Scholar] [CrossRef]
- Wilkins, T.; Akhtar, M.; Gititu, E.; Jalluri, C.; Ramirez, J. Diagnosis and Management of Hepatitis C. Am. Fam. Physician 2015, 91, 835–842. [Google Scholar]
- Geddawy, A.; Ibrahim, Y.F.; Elbahie, N.M.; Ibrahim, M.A. Direct Acting Anti-hepatitis C Virus Drugs: Clinical Pharmacology and Future Direction. J. Transl. Int. Med. 2017, 5, 8–17. [Google Scholar] [CrossRef]
- Pawlotsky, J.M. New hepatitis C therapies: The toolbox, strategies, and challenges. Gastroenterology 2014, 146, 1176–1192. [Google Scholar] [CrossRef] [PubMed]
- Ghany, M.G.; Marks, K.M.; Morgan, T.R.; Wyles, D.L.; Aronsohn, A.I.; Bhattacharya, D.; Broder, T.; Falade-Nwulia, O.O.; Feld, J.J.; Gordon, S.C.; et al. Hepatitis C Guidance 2019 Update: AASLD-IDSA Recommendations for Testing, Managing, and Treating Hepatitis C Virus Infection. Hepatology 2019, 71, 686–721. [Google Scholar] [CrossRef] [PubMed]
- Kamal, S.M.; Fehr, J.; Roesler, B.; Peters, T.; Rasenack, J.W. Peginterferon alone or with ribavirin enhances HCV-specific CD4 T-helper 1 responses in patients with chronic hepatitis C. Gastroenterology 2002, 123, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Cramp, M.E.; Rossol, S.; Chokshi, S.; Carucci, P.; Williams, R.; Naoumov, N.V. Hepatitis C virus-specific T-cell reactivity during interferon and ribavirin treatment in chronic hepatitis C. Gastroenterology 2000, 118, 346–355. [Google Scholar] [CrossRef]
- Barnes, E.; Harcourt, G.; Brown, D.; Lucas, M.; Phillips, R.; Dusheiko, G.; Klenerman, P. The dynamics of T-lymphocyte responses during combination therapy for chronic hepatitis C virus infection. Hepatology 2002, 36, 743–754. [Google Scholar] [CrossRef]
- Kaplan, D.E.; Sugimoto, K.; Ikeda, F.; Stadanlick, J.; Valiga, M.; Shetty, K.; Reddy, K.R.; Chang, K.M. T-cell response relative to genotype and ethnicity during antiviral therapy for chronic hepatitis C. Hepatology 2005, 41, 1365–1375. [Google Scholar] [CrossRef]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale, M., Jr.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef]
- Clausznitzer, D.; Harnisch, J.; Kaderali, L. Multi-scale model for hepatitis C viral load kinetics under treatment with direct acting antivirals. Virus Res. 2016, 218, 96–101. [Google Scholar] [CrossRef]
- Perpinan, E.; Caro-Perez, N.; Garcia-Gonzalez, N.; Gregori, J.; Gonzalez, P.; Bartres, C.; Soria, M.E.; Perales, C.; Lens, S.; Marino, Z.; et al. Hepatitis C virus early kinetics and resistance-associated substitution dynamics during antiviral therapy with direct-acting antivirals. J. Viral Hepat. 2018, 25, 1515–1525. [Google Scholar] [CrossRef]
- Barnes, E.; Gelderblom, H.C.; Humphreys, I.; Semmo, N.; Reesink, H.W.; Beld, M.G.; van Lier, R.A.; Klenerman, P. Cellular immune responses during high-dose interferon-alpha induction therapy for hepatitis C virus infection. J. Infect. Dis. 2009, 199, 819–828. [Google Scholar] [CrossRef]
- Missale, G.; Pilli, M.; Zerbini, A.; Penna, A.; Ravanetti, L.; Barili, V.; Orlandini, A.; Molinari, A.; Fasano, M.; Santantonio, T.; et al. Lack of full CD8 functional restoration after antiviral treatment for acute and chronic hepatitis C virus infection. Gut 2012, 61, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hakeem, M.S.; Bedard, N.; Badr, G.; Ostrowski, M.; Sekaly, R.P.; Bruneau, J.; Willems, B.; Heathcote, E.J.; Shoukry, N.H. Comparison of immune restoration in early versus late alpha interferon therapy against hepatitis C virus. J. Virol. 2010, 84, 10429–10435. [Google Scholar] [CrossRef] [PubMed]
- Larrubia, J.R.; Moreno-Cubero, E.; Miquel, J.; Sanz-de-Villalobos, E. Hepatitis C virus-specific cytotoxic T cell response restoration after treatment-induced hepatitis C virus control. World J. Gastroenterol. 2015, 21, 3480–3491. [Google Scholar] [CrossRef] [PubMed]
- Sidharthan, S.; Kohli, A.; Sims, Z.; Nelson, A.; Osinusi, A.; Masur, H.; Kottilil, S. Utility of hepatitis C viral load monitoring on direct-acting antiviral therapy. Clin. Infect. Dis. 2015, 60, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Maasoumy, B.; Buggisch, P.; Mauss, S.; Boeker, K.H.W.; Muller, T.; Gunther, R.; Zimmermann, T.; Manns, M.P.; Sarrazin, C.; Huppe, D.; et al. Clinical significance of detectable and quantifiable HCV RNA at the end of treatment with ledipasvir/sofosbuvir in GT1 patients. Liver Int. 2018, 38, 1906–1910. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Bhatta, M.; Ward, H.; Romani, S.; Lee, R.; Rosenthal, E.; Osinusi, A.; Kohli, A.; Masur, H.; Kottilil, S.; et al. Multitarget Direct-Acting Antiviral Therapy Is Associated With Superior Immunologic Recovery in Patients Coinfected With Human Immunodeficiency Virus and Hepatitis C Virus. Hepatol. Commun. 2018, 2, 1451–1466. [Google Scholar] [CrossRef]
- Burchill, M.A.; Golden-Mason, L.; Wind-Rotolo, M.; Rosen, H.R. Memory re-differentiation and reduced lymphocyte activation in chronic HCV-infected patients receiving direct-acting antivirals. J. Viral Hepat. 2015, 22, 983–991. [Google Scholar] [CrossRef]
- Najafi Fard, S.; Schietroma, I.; Corano Scheri, G.; Giustini, N.; Serafino, S.; Cavallari, E.N.; Pinacchio, C.; De Girolamo, G.; Ceccarelli, G.; Scagnolari, C.; et al. Direct-acting antiviral therapy enhances total CD4+ and CD8+ T-cells responses, but does not alter T-cells activation among HCV mono-infected, and HCV/HIV-1 co-infected patients. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 319–329. [Google Scholar] [CrossRef]
- Meissner, E.G.; Kohli, A.; Higgins, J.; Lee, Y.J.; Prokunina, O.; Wu, D.; Orr, C.; Masur, H.; Kottilil, S. Rapid changes in peripheral lymphocyte concentrations during interferon-free treatment of chronic hepatitis C virus infection. Hepatol. Commun. 2017, 1, 586–594. [Google Scholar] [CrossRef]
- Lattanzi, B.; Baroncelli, S.; De Santis, A.; Galluzzo, C.M.; Mennini, G.; Michelini, Z.; Lupo, M.; Ginanni Corradini, S.; Rossi, M.; Palmisano, L.; et al. Microbial translocation and T cell activation are modified by direct-acting antiviral therapy in HCV-infected patients. Aliment. Pharmacol. Ther. 2018, 48, 1146–1155. [Google Scholar] [CrossRef]
- Emmanuel, B.; El-Kamary, S.S.; Magder, L.S.; Stafford, K.A.; Charurat, M.E.; Poonia, B.; Chairez, C.; McLaughlin, M.; Hadigan, C.; Masur, H.; et al. Immunological recovery in T-cell activation after sustained virologic response among HIV positive and HIV negative chronic Hepatitis C patients. Hepatol. Int. 2019, 13, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Vranjkovic, A.; Deonarine, F.; Kaka, S.; Angel, J.B.; Cooper, C.L.; Crawley, A.M. Direct-Acting Antiviral Treatment of HCV Infection Does Not Resolve the Dysfunction of Circulating CD8(+) T-Cells in Advanced Liver Disease. Front. Immunol. 2019, 10, 1926. [Google Scholar] [CrossRef] [PubMed]
- Romani, S.; Stafford, K.; Nelson, A.; Bagchi, S.; Kottilil, S.; Poonia, B. Peripheral PD-1(+) T Cells Co-expressing Inhibitory Receptors Predict SVR With Ultra Short Duration DAA Therapy in HCV Infection. Front. Immunol. 2019, 10, 1470. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.; Hennecke, N.; Lohmann, V.; Kayser, A.; Neumann-Haefelin, C.; Kukolj, G.; Bocher, W.O.; Thimme, R. Restoration of HCV-specific CD8+ T cell function by interferon-free therapy. J. Hepatol. 2014, 61, 538–543. [Google Scholar] [CrossRef]
- Wieland, D.; Kemming, J.; Schuch, A.; Emmerich, F.; Knolle, P.; Neumann-Haefelin, C.; Held, W.; Zehn, D.; Hofmann, M.; Thimme, R. TCF1(+) hepatitis C virus-specific CD8(+) T cells are maintained after cessation of chronic antigen stimulation. Nat. Commun. 2017, 8, 15050. [Google Scholar] [CrossRef]
- Han, J.W.; Sung, P.S.; Kim, K.H.; Hong, S.H.; Shin, E.C.; Jun Song, M.; Park, S.H. Dynamic Changes in Ex Vivo T-Cell Function After Viral Clearance in Chronic HCV Infection. J. Infect. Dis. 2019, 220, 1290–1301. [Google Scholar] [CrossRef]
- Aregay, A.; Owusu Sekyere, S.; Deterding, K.; Port, K.; Dietz, J.; Berkowski, C.; Sarrazin, C.; Manns, M.P.; Cornberg, M.; Wedemeyer, H. Elimination of hepatitis C virus has limited impact on the functional and mitochondrial impairment of HCV-specific CD8+ T cell responses. J. Hepatol. 2019, 71, 889–899. [Google Scholar] [CrossRef]
- Hartnell, F.; Esposito, I.; Swadling, L.; Brown, A.; Phetsouphanh, C.; de Lara, C.; Gentile, C.; Turner, B.; Kopycinski, J.; Dorrell, L.; et al. Characterising HCV specific CD4+ T-cells following viral-vectored vaccination, directly acting anti-virals and spontaneous viral cure. Hepatology 2020. [Google Scholar] [CrossRef]
- Smits, M.; Zoldan, K.; Ishaque, N.; Gu, Z.; Jechow, K.; Wieland, D.; Conrad, C.; Eils, R.; Fauvelle, C.; Baumert, T.F.; et al. Follicular T helper cells shape the HCV-specific CD4+ T cell repertoire after virus elimination. J. Clin. Investig. 2020, 130, 998–1009. [Google Scholar] [CrossRef]
- Reig, M.; Boix, L.; Bruix, J. The impact of direct antiviral agents on the development and recurrence of hepatocellular carcinoma. Liver Int. 2017, 37, 136–139. [Google Scholar] [CrossRef]
- Nyberg, A.H. The Association of Extrahepatic Cancers With Chronic Hepatitis C Virus Infection. Gastroenterol. Hepatol. 2016, 12, 185–187. [Google Scholar]
- Houghton, M. Prospects for prophylactic and therapeutic vaccines against the hepatitis C viruses. Immunol. Rev. 2011, 239, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Ingiliz, P.; Martin, T.C.; Rodger, A.; Stellbrink, H.J.; Mauss, S.; Boesecke, C.; Mandorfer, M.; Bottero, J.; Baumgarten, A.; Bhagani, S.; et al. HCV reinfection incidence and spontaneous clearance rates in HIV-positive men who have sex with men in Western Europe. J. Hepatol. 2017, 66, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Callendret, B.; Eccleston, H.B.; Hall, S.; Satterfield, W.; Capone, S.; Folgori, A.; Cortese, R.; Nicosia, A.; Walker, C.M. T-cell immunity and hepatitis C virus reinfection after cure of chronic hepatitis C with an interferon-free antiviral regimen in a chimpanzee. Hepatology 2014, 60, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Parga, T.; Sehrawat, S.; Rouse, B.T. Role of regulatory T cells during virus infection. Immunol. Rev. 2013, 255, 182–196. [Google Scholar] [CrossRef]
- Langhans, B.; Nischalke, H.D.; Kramer, B.; Hausen, A.; Dold, L.; van Heteren, P.; Huneburg, R.; Nattermann, J.; Strassburg, C.P.; Spengler, U. Increased peripheral CD4(+) regulatory T cells persist after successful direct-acting antiviral treatment of chronic hepatitis C. J. Hepatol. 2017, 66, 888–896. [Google Scholar] [CrossRef]
- Wu, S.F.; Tseng, C.W.; Ho, Y.C.; Chen, Y.C.; Ko, P.H.; He, Y.T.; Tseng, K.C. Regulatory T Cell Function Modulated After Successful Direct-Acting Antiviral Treatment for Chronic Hepatitis C Patients. Dig. Dis. Sci. 2020, 65, 1385–1395. [Google Scholar] [CrossRef]
- Hill, A.M.; Nath, S.; Simmons, B. The road to elimination of hepatitis C: Analysis of cures versus new infections in 91 countries. J. Virus Erad. 2017, 3, 117–123. [Google Scholar] [CrossRef]
- Grady, B.P.; Schinkel, J.; Thomas, X.V.; Dalgard, O. Hepatitis C virus reinfection following treatment among people who use drugs. Clin. Infect. Dis. 2013, 57, S105–S110. [Google Scholar] [CrossRef]
- Swadling, L.; Halliday, J.; Kelly, C.; Brown, A.; Capone, S.; Ansari, M.A.; Bonsall, D.; Richardson, R.; Hartnell, F.; Collier, J.; et al. Highly-Immunogenic Virally-Vectored T-cell Vaccines Cannot Overcome Subversion of the T-cell Response by HCV during Chronic Infection. Vaccines 2016, 4, 27. [Google Scholar] [CrossRef]
- Kelly, C.; Swadling, L.; Capone, S.; Brown, A.; Richardson, R.; Halliday, J.; von Delft, A.; Oo, Y.; Mutimer, D.; Kurioka, A.; et al. Chronic hepatitis C viral infection subverts vaccine-induced T-cell immunity in humans. Hepatology 2016, 63, 1455–1470. [Google Scholar] [CrossRef] [PubMed]
- Callendret, B.; Eccleston, H.B.; Satterfield, W.; Capone, S.; Folgori, A.; Cortese, R.; Nicosia, A.; Walker, C.M. Persistent hepatitis C viral replication despite priming of functional CD8+ T cells by combined therapy with a vaccine and a direct-acting antiviral. Hepatology 2016, 63, 1442–1454. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Reference | Immune Checkpoint Blocked | Stage of HCV Infection | Number of Subjects | Character of the Study | Results |
|---|---|---|---|---|---|
| Golden Mason et al. [68] | PD-ligand 1 (PD-L1) PD-ligand 2 (PD-L2) | chronic | 7 | In vitro | ↑ proliferation of HCV-specific CD8+ T cells ↑ IFN-ϒ and IL-2 secretion by HCV-specific CD8+T cells |
| Golden Mason et al. [69] | Tim-3 | chronic | 4 | In vitro | ↑ proliferation of HCV-specific CD8+ T cells ↑ IFN-ϒ secretion by HCV-specific CD8+ T cells ↓ IL-10 secretion by HCV-specific CD8+ T cells |
| Penna et al. [18] | PD-L1 | chronic | 8 | In vitro | ↑ expansion of HCV-specific CD8+ T cells ↑ frequency of both IFN-γ– and IL-2–secreting HCV-specific CD8+ T cells |
| Urbani et al. [70] | PD-L1 | acute | 8 | In vitro | ↑ expansion and IFN-γ and IL-2 production but not the cytolytic activity of HCV-specific CD8+ T cells. |
| McMahan et al. [71] | Tim-3, PD-L1, PD-L2 | acute/chronic | 6/4 | In vitro | ↑ proliferation of HCV-specific CD8+ T cells achieved by either PD-1 or Tim-3 blockade ↑ cytotoxicity of HCV-specific CD8+ T cells (increased expression of CD107a, killing of hepatocytes cell line expressing cognate HCV epitopes) achieved exclusively by Tim-3 blockade |
| Fuller et al. [72] | PD-1 | chronic | 3 chimpanzees | In vivo | ↓ HCV viral load in one of three treated animals ↑ frequencies and IFN-ϒ production of intrahepatic HCV-specific CD4+ and CD8+ T cells in the same animal |
| Gardiner et al. [73] | PD-1 | chronic | 54 | In vivo | ↓ viral load in five patients (two patients achieved undetectable HCV RNA) |
| Sangro et al. [74] | CTLA-4 | chronic | 20 | In vivo | ↓ viral load sustained in most patients for 3 months follow-up; transient complete viral response in 15% of patients during follow-up ↑ HCV-specific T cell response (IFN-ϒ production) |
| Reference | HCV Genotype | Number of Subjects | Effect of Treatment/Effect of Successful Treatment | Follow-up | Results |
|---|---|---|---|---|---|
| Shrivastava et al. [97] | 1 | 22 HIV/HCV co-infected | Effect of successful treatment | 12 weeks after the end of treatment (EOT) (sustained virologic response (SVR) 12) | ↓ PD1 and TIGIT expression on CD4+ and CD8+ T cells ↓ Eomeshi T-betlo CD4+ and CD8+ T cells ↑ T-bethi Eomeslo CD4+ and CD8+ T cells ↓ BLIMP-1 expression on CD4+ T cells ↓ CD38 expression on both CD4+ and CD8+ T cells ↑ Tem (effector memory) population ↓ naïve T cell subset |
| Burchill et al. [98] | 1a/1b | 19 | Effect of successful treatment | 24 weeks post-EOT (SVR24) | ↑ frequency of CD4+ T cells; ↓ expression of TIGIT on CD4+ and CD8+ T cells; ↑ percentage of Tem in both CD4+ and CD8+ T cells compartments |
| Najafi Fard et al. [99] | 1–4 | HCV mono-infection n = 18; HCV/HIV-1 co-infection (n = 17) | Effect of successful treatment | 12 weeks post-EOT (SVR12) | ↑ peripheral CD4+ and CD8+ T cells producing IFN-γ, IL-17, and IL-22 no significant impact on the status of CD4+ and CD8+ T cells activation |
| Meissner et al. [100] | 1 | 95 | Effect of treatment | up to 20 weeks after treatment initiation | ↑ peripheral CD4+ and CD8+ T cells early after treatment initiation ↓ HLA-DR+CD38+ T-cells during observation ↑ expression CXCR3 on T cells early after treatment initiation |
| Lattanzi et al. [101] | 1–4 | 45 | Effect of treatment | at first month of treatment (T1), at EOT (T2) and 12 weeks post-EOT (T3, SVR12) | stable percentage of CD4+ and CD8+ T cells at T1 when compared to baseline ↓ HLA-DR+ CD38+ CD4+ and CD8+ T cells at T3 with respect to baseline |
| Emmanuel et al. [102] | NA | HCV mono-infection (n = 161); HIV/HCV co-infection (n = 59) | Effect of successful treatment | 1 or 2 years post-SVR | ↓ HLA-DR+CD38+ CD4+ and CD8+ T cells in both HCV infection and HIV/HCV co-infection |
| Vranjkovic et al. [103] | NA | 18 | Effect of successful treatment | 24 weeks post-SVR12 | phenotypic distribution of peripheral CD8+ T cell subsets in patients with advanced liver fibrosis (F4) different from those with minimal fibrosis (F0-1) which remained unchanged after viral elimination sustained hyperfunctional activity (perforin production and cytotoxicity) of CD8+ T cell subsets in patients with liver fibrosis (F4) up to a year post-treatment initiation sustained elevated concentrations of systemic inflammatory cytokines and decreased levels of TGF-β in plasma of patients with liver fibrosis (F4) |
| Reference | HCV Genotype | Number of Subjects | Effect of Treatment/Effect of Successful/Unsuccessful Treatment | Follow-up | Results |
|---|---|---|---|---|---|
| Romani et al. [104] | 1a/1b | 26 | Effect of successful/unsuccessful treatment | at the end of treatment (EOT), at week 4 and 12 weeks post-EOT (sustained virologic response (SVR) 12) | higher levels of PD-1+ HCV-specific T cells at baseline and at EOT in patients who achieved SVR ↓ PD-1+ HCV-specific T cell subset at SVR12 in responders |
| Burchill et al. [98] | 1a/1b | 7 | Effect of successful treatment | 24 weeks post -EOT (SVR24) | no significant change in the frequency of HCV-specific CD8+ T cells ↓ PD-1 expression |
| Martin et al. [105] | 1 | 51 | Effect of successful/unsuccessful treatment | treatment week 4, 12 and 24 weeks post-treatment (SVR24) | ↑ HCV-specific CD8+ T cells frequency after in vitro expansion in patients with SVR from baseline to 24 weeks after completion of treatment no change in HCV-specific CD8+ T cells frequency in patients with treatment failure |
| Shrivastava et al. [97] | 1 | 22 HIV-1/HCV co-infected | Effect of successful treatment | 12 weeks post-EOT (SVR12) | ↑ HCV-specific CD8+ T cells ↑ IL-2 and IFN-γ production ↑ polyfunctionality (co-expression of IFN-γ and TNF-α) ↑ cytolytic capacity (CD107A expression and perforin and granzyme B secretion) |
| Wieland et al. [106] | 1a/1b | 21 | Effect of successful treatment | at EOT and 12 weeks post-EOT (SVR12) | ↓ terminally exhausted HCV-specific CD8+ T cells (TCF-1-CD127-PD1hi) after antigen elimination persistence of memory-like HCV-specific CD8+ T cells (TCF-1+CD127+PD-1+) with ability of self-renewal and proliferation |
| Han et al. [107] | 1b/2a | 41 | Effect of successful/unsuccessful treatment | treatment week 4, 12, 24 (EOT) and 12 weeks post-treatment (SVR12) or week 4, 12 (EOT), and 12 weeks post-treatment (SVR12) | ↑ HCV-specific CD8+ T cell response (IFN-ϒ production, cytotoxicity) at week 4, which diminished at later weeks ↓ PD-1+EomeshiT-betlow HCV-specific CD8+ T cells at week 4 ↓ HCV-specific CD8+ T cell frequency at SVR12, including antigen-experienced KLRG1+CCR7− HCV-specific T cells no change in TCF-1+CD127+PD-1+ HCV-specific CD8+ T cells responsible for recall proliferation over observation time defective restoration of HCV-specific T cell responses in SVR- group |
| Aregay et al. [108] | 1a/1b | 40 | Effect of successful treatment | at EOT and 24 weeks post-EOT (SVR24) | unaltered expression of PD-1, Tim-3, LAG-3 and CD5 on HCV-specific CD8+ T cells sustained impaired IFN-ϒ, MIP-1β production, mitochondrial dysfunction and metabolic deregulation ↓ HLA-DR+CD38+ HCV-specific CD8+ T cells maintenance of memory-like TCF-1+CD127+PD-1+ HCV-specific CD8+ T cells |
| Hartnell et al. [109] | NA | 21 | Effect of successful treatment | average 6 weeks post-treatment (range 0–26 weeks) | unchanged proliferative capacity and cytokine production (TNF-α, IFN-ϒ MIP-1β) of exhausted HCV-specific CD4+ T cells |
| Smits et al. [110] | 1, 2, 3 | 40 | Effect of successful treatment | week 2, either 8, 12, 16 or 24 week of treatment (EOT) and 24 weeks post-treatment (SVR24) | ↑ HCV-specific CD4+ T cells within the initial two weeks of treatment unchanged percentages of HCV-specific CD4+ T cells expressing PD-1, BTLA and TIGIT ↑ follicular T helper cells (Tfh) ↓ germinal center activity and HCV-specific neutralizing antibodies |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osuch, S.; Metzner, K.J.; Caraballo Cortés, K. Reversal of T Cell Exhaustion in Chronic HCV Infection. Viruses 2020, 12, 799. https://doi.org/10.3390/v12080799
Osuch S, Metzner KJ, Caraballo Cortés K. Reversal of T Cell Exhaustion in Chronic HCV Infection. Viruses. 2020; 12(8):799. https://doi.org/10.3390/v12080799
Chicago/Turabian StyleOsuch, Sylwia, Karin J. Metzner, and Kamila Caraballo Cortés. 2020. "Reversal of T Cell Exhaustion in Chronic HCV Infection" Viruses 12, no. 8: 799. https://doi.org/10.3390/v12080799
APA StyleOsuch, S., Metzner, K. J., & Caraballo Cortés, K. (2020). Reversal of T Cell Exhaustion in Chronic HCV Infection. Viruses, 12(8), 799. https://doi.org/10.3390/v12080799