
The RNA Architecture of the SARS-CoV-2 3′-Untranslated Region





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
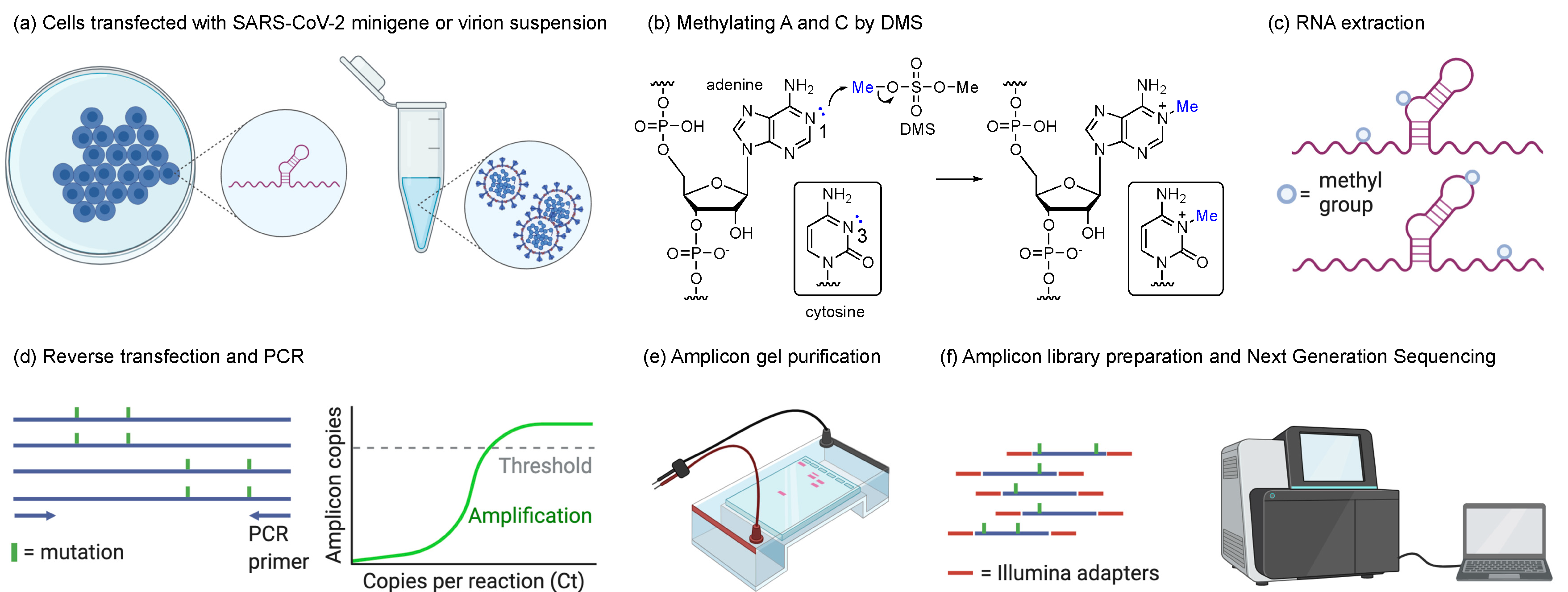
2.2. Transfection and DMS Treatment
2.3. SARS-CoV-2 Propagation and DMS Treatment
2.4. Amplicon Library Preparation and Next-Generation Sequencing
2.5. DMS-MaPseq and DREEM Analysis Pipelines
3. Results
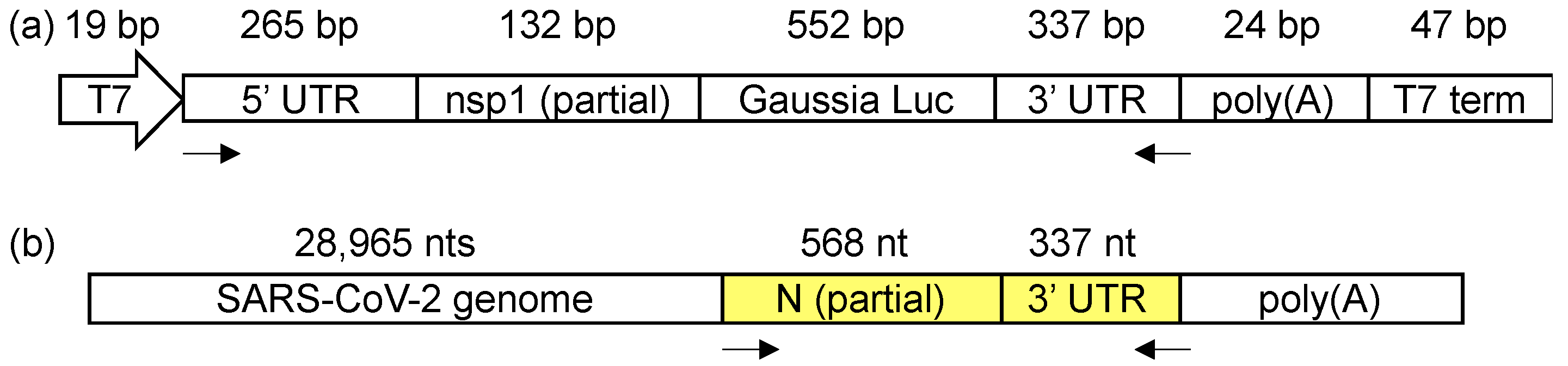
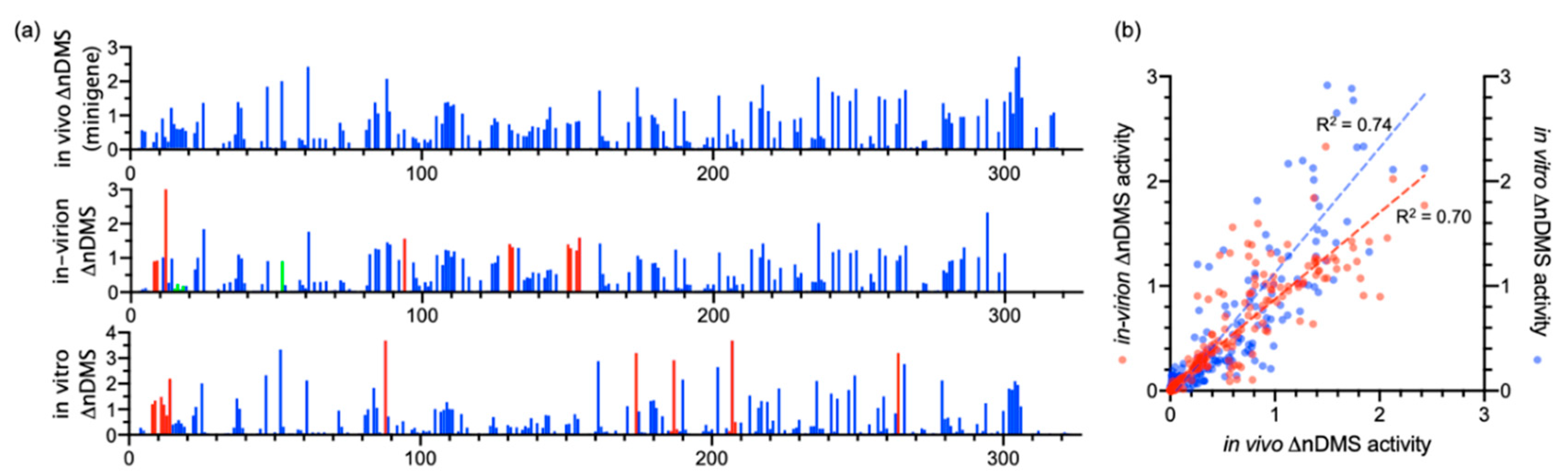
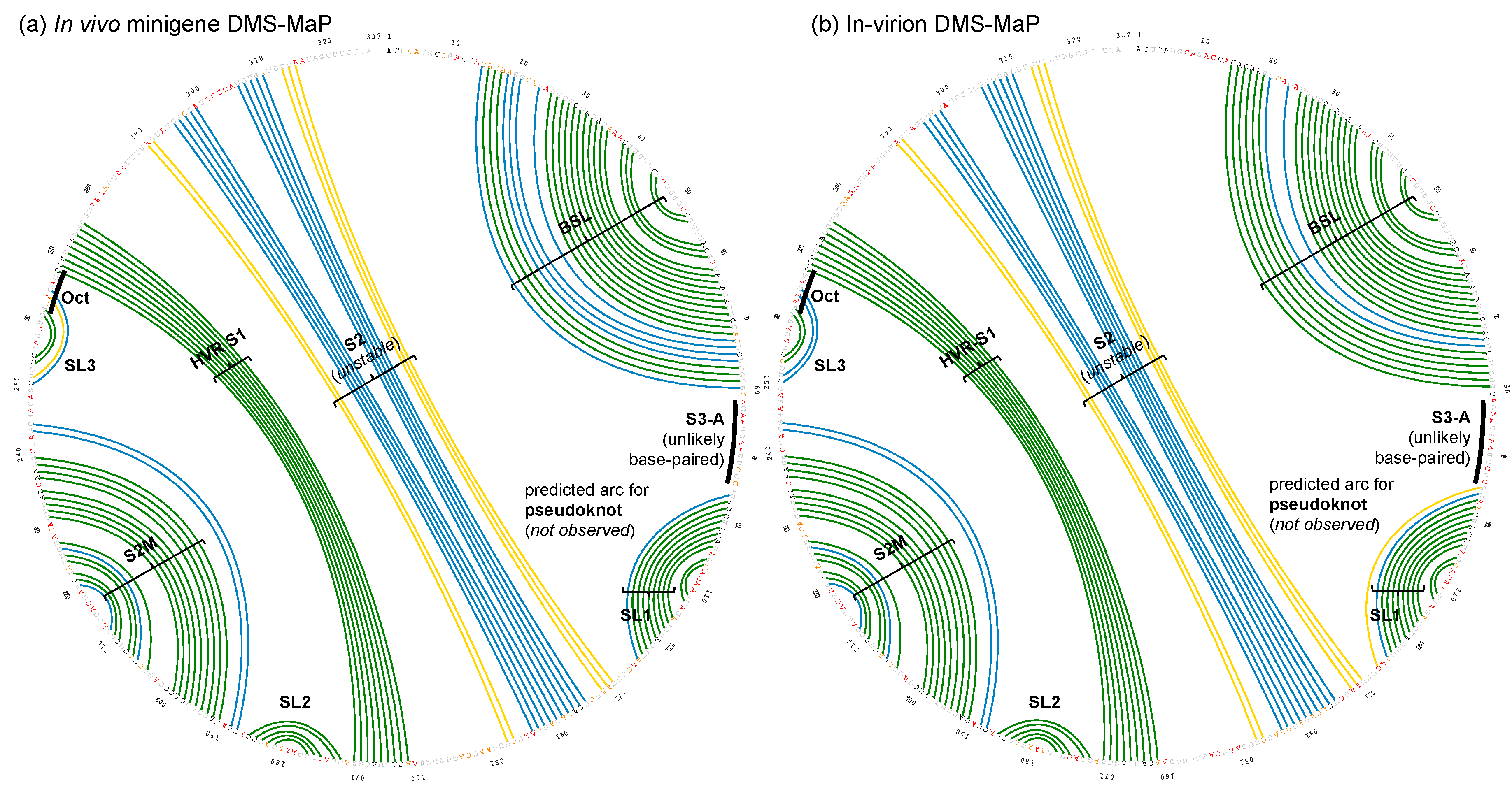
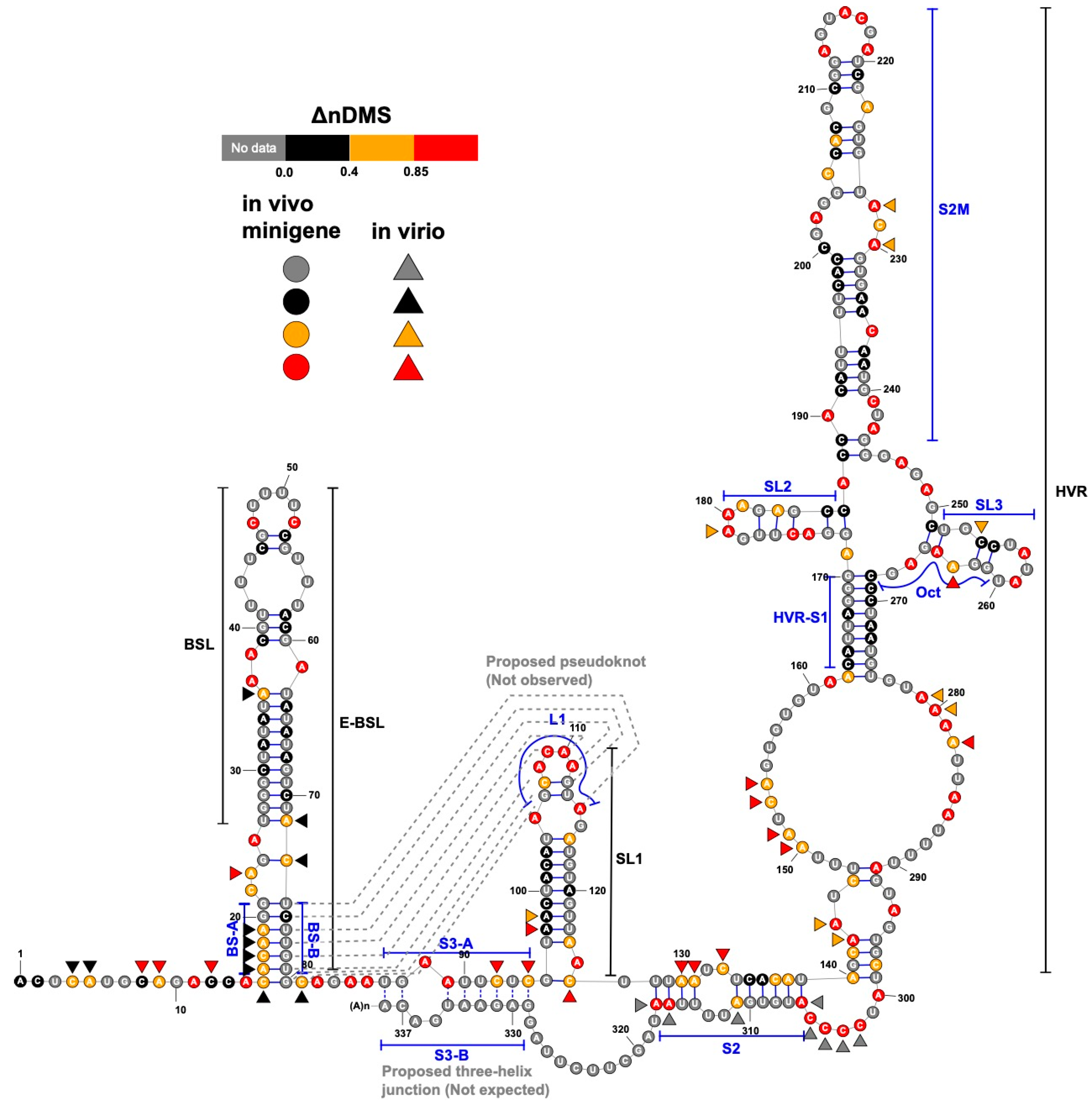
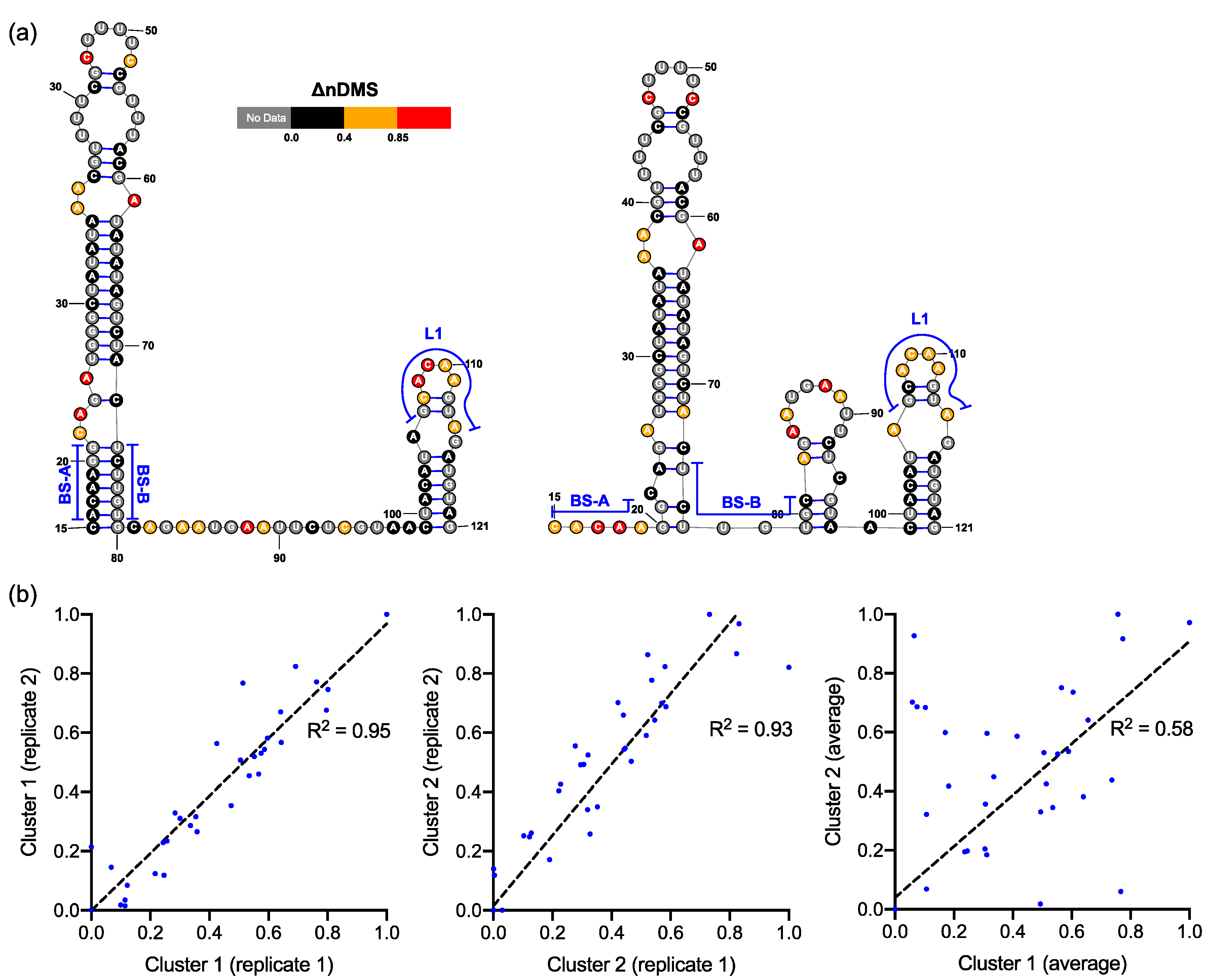
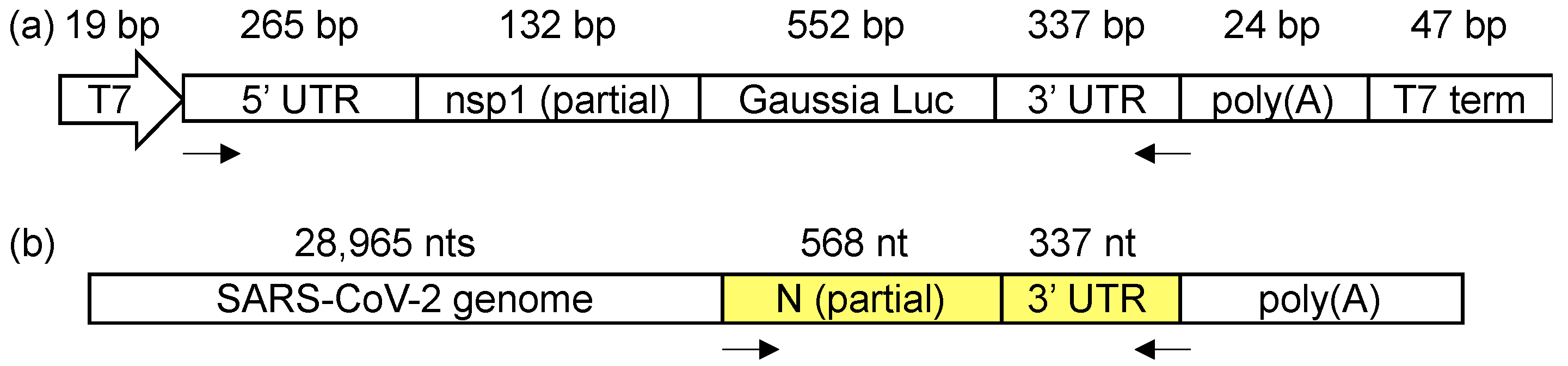
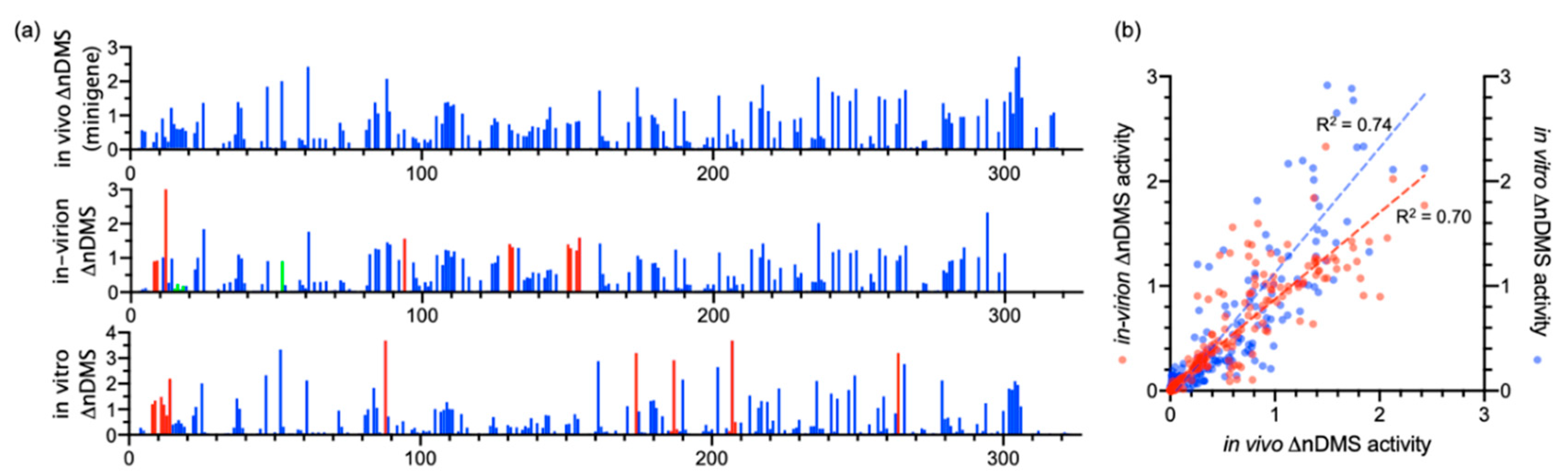
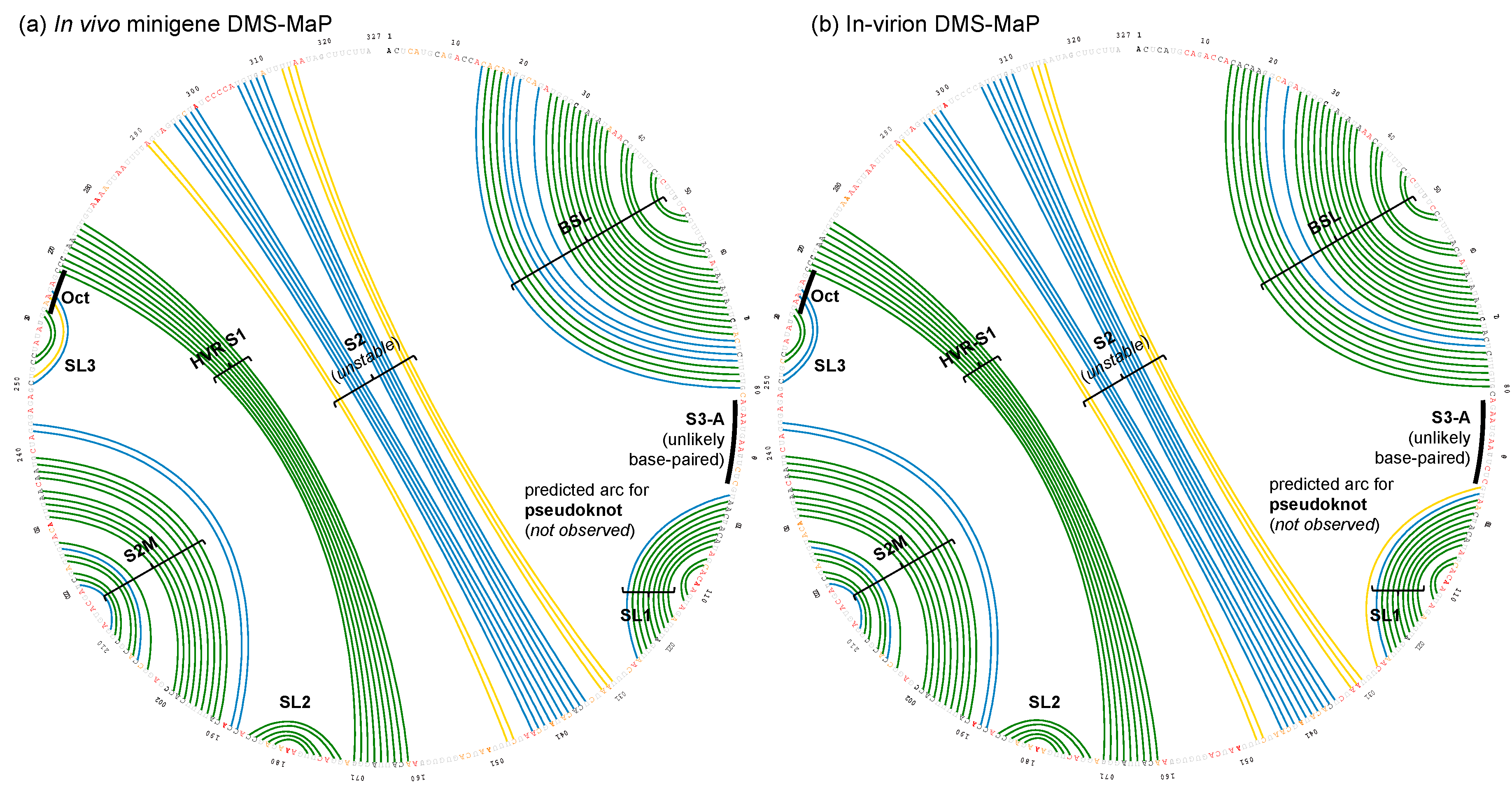
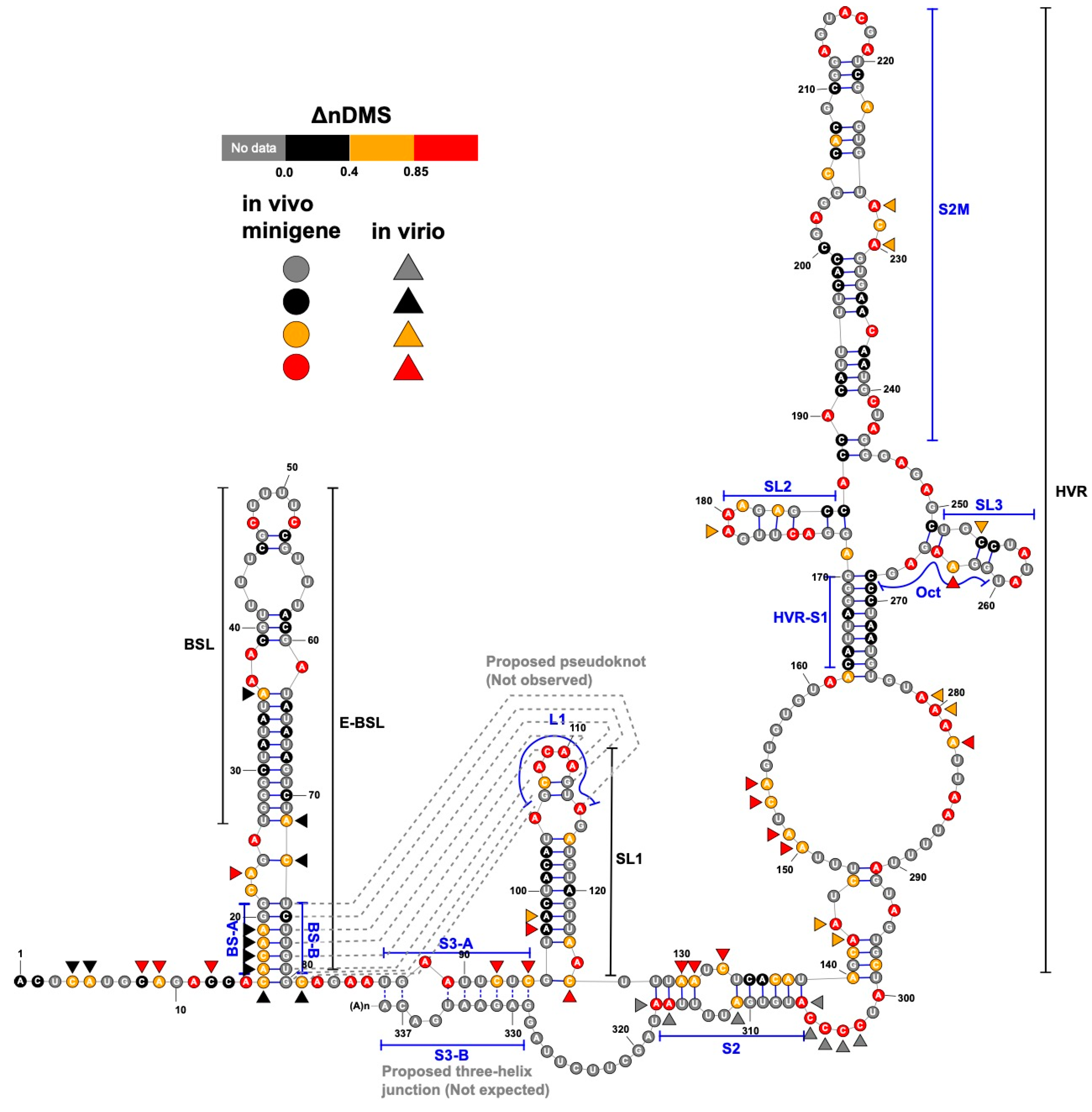
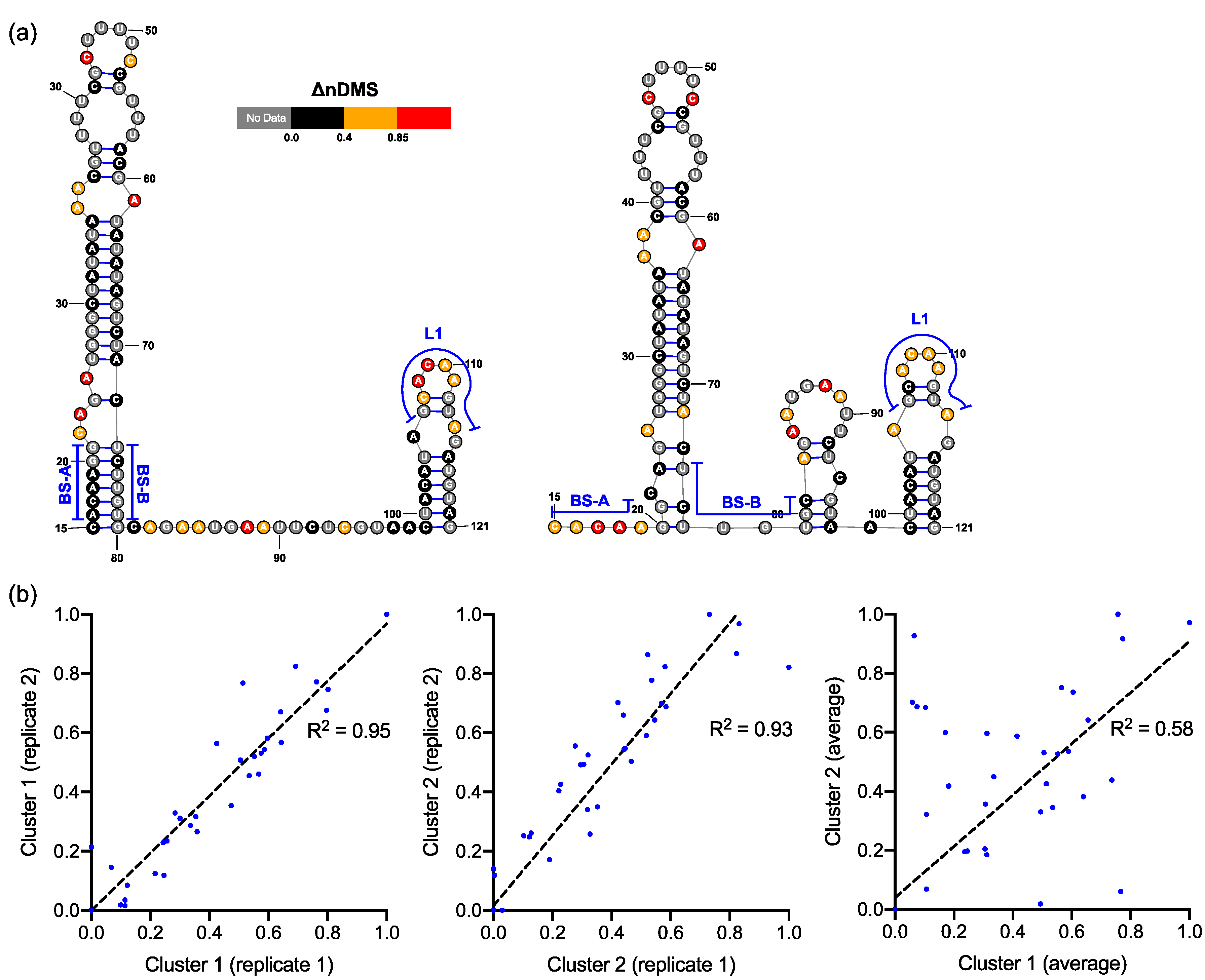
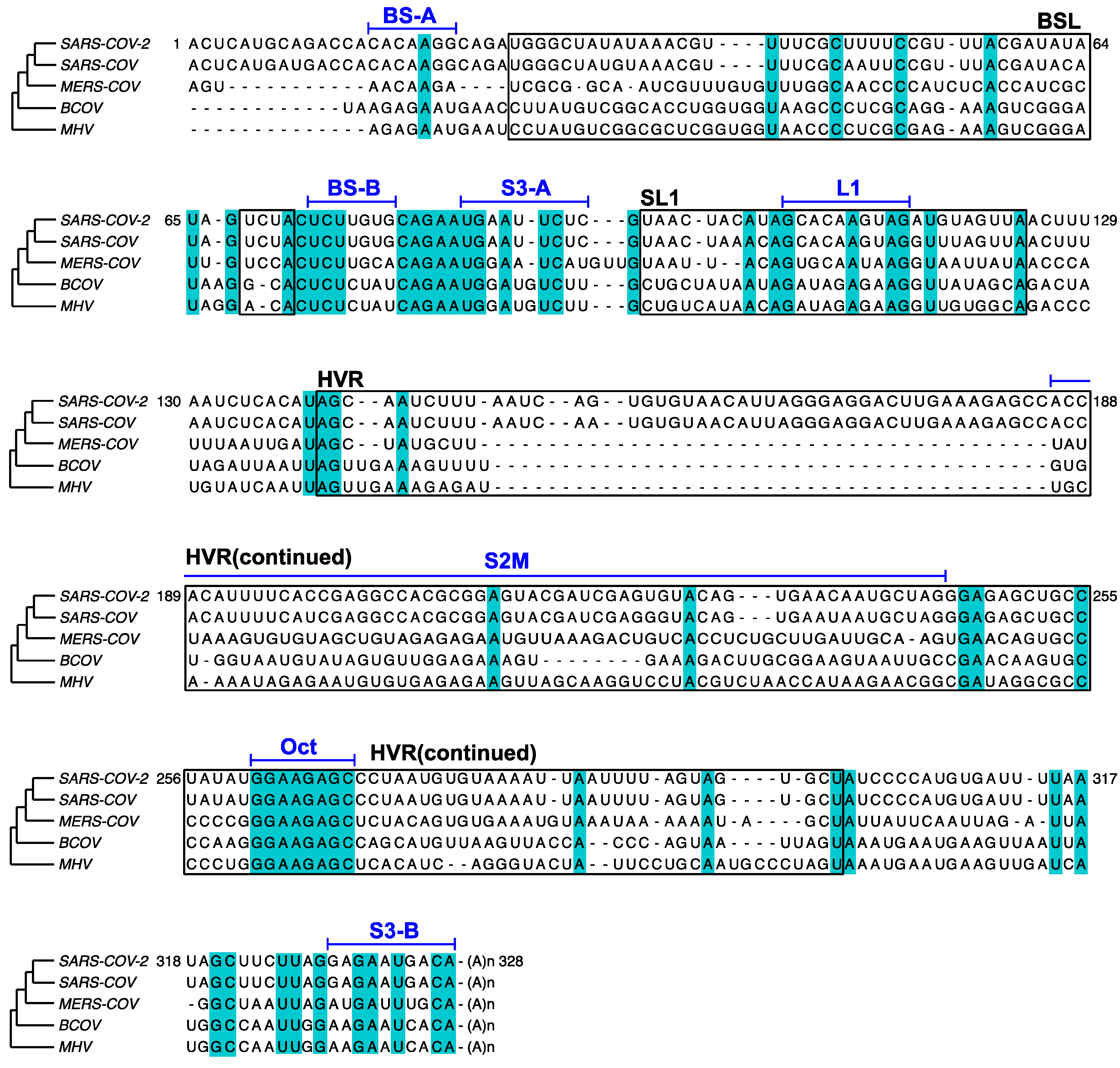
3.1. DMS-MaPseq Uncovered almost Identical SARS-CoV-2 3′ UTR RNA Structures in Minigene-Transfected Cells and Infectious Virions
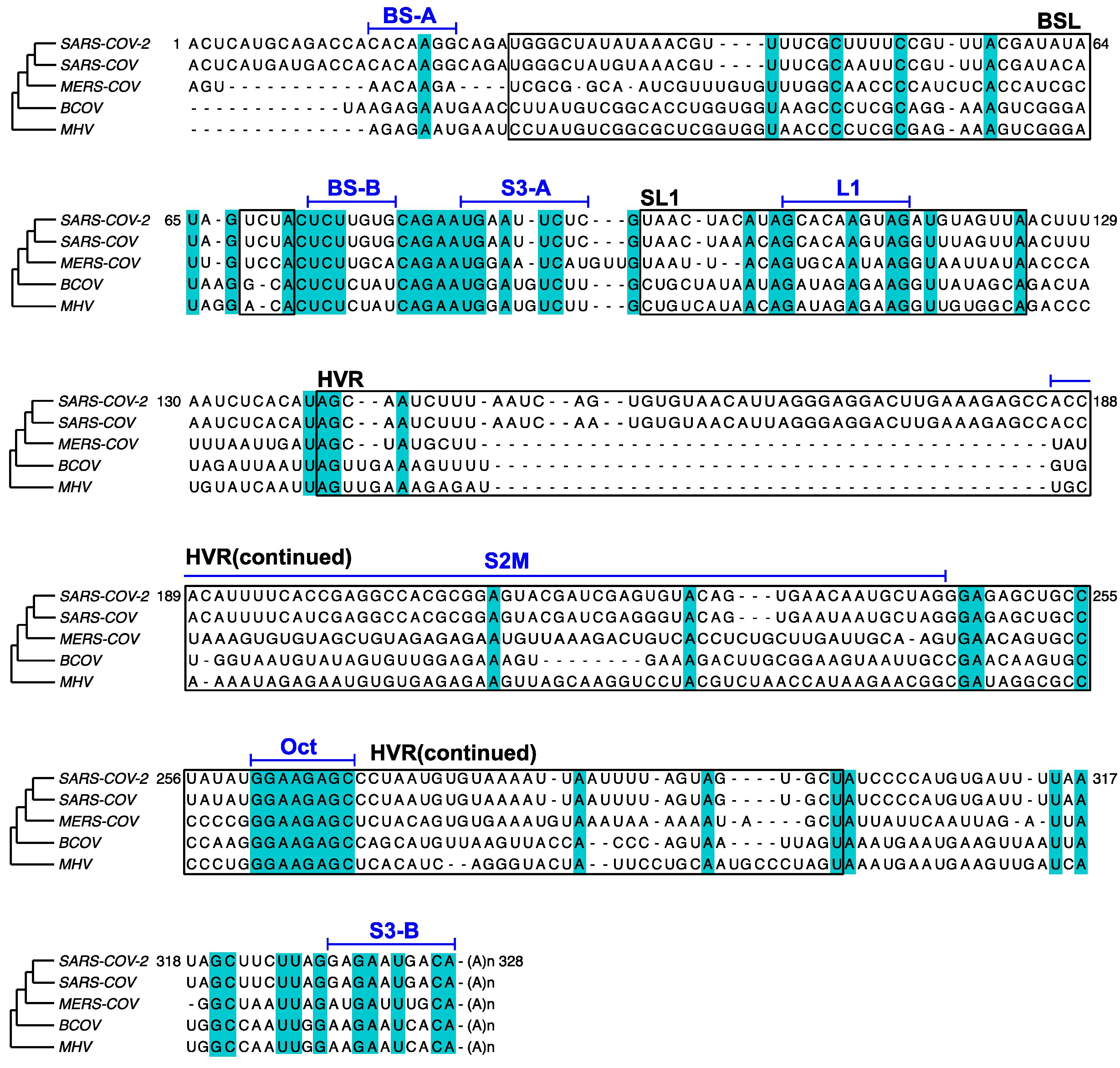
3.2. DREEM Analysis Uncovered an Unexpected Lack of the Pseudoknot and Three-Helix Junction in the 3′ UTR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef] [PubMed]
- van Hemert, M.J.; van den Worm, S.H.E.; Knoops, K.; Mommaas, A.M.; Gorbalenya, A.E.; Snijder, E.J. SARS-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathog. 2008, 4, e1000054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, T.S.; Liu, D.X. Human Coronavirus: Host-Pathogen Interaction. Annu. Rev. Microbiol. 2019, 73, 529–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Leibowitz, J.L. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 2015, 206, 120–133. [Google Scholar] [CrossRef]
- Cagliani, R.; Forni, D.; Clerici, M.; Sironi, M. Coding potential and sequence conservation of SARS-CoV-2 and related animal viruses. Infect. Genet. Evol. 2020, 83, 104353. [Google Scholar] [CrossRef]
- Goebel, S.J.; Hsue, B.; Dombrowski, T.F.; Masters, P.S. Characterization of the RNA components of a putative molecular switch in the 3′ untranslated region of the murine coronavirus genome. J. Virol. 2004, 78, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Züst, R.; Miller, T.B.; Goebel, S.J.; Thiel, V.; Masters, P.S. Genetic interactions between an essential 3′ cis-acting RNA pseudoknot, replicase gene products, and the extreme 3′ end of the mouse coronavirus genome. J. Virol. 2008, 82, 1214–1228. [Google Scholar] [CrossRef] [Green Version]
- Andrews, R.J.; Peterson, J.M.; Haniff, H.S.; Chen, J.; Williams, C.; Grefe, M.; Disney, M.D.; Moss, W.N. An in silico map of the SARS-CoV-2 RNA Structurome. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Rangan, R.; Zheludev, I.N.; Das, R. RNA genome conservation and secondary structure in SARS-CoV-2 and SARS-related viruses: A first look. RNA 2020. [Google Scholar] [CrossRef]
- Lan, T.C.T.; Allan, M.F.; Malsick, L.E.; Khandwala, S.; Nyeo, S.S.Y.; Bathe, M.; Griffiths, A.; Rouskin, S. Structure of the full SARS-CoV-2 RNA genome in infected cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Manfredonia, I.; Nithin, C.; Ponce-Salvatierra, A.; Ghosh, P.; Wirecki, T.K.; Marinus, T.; Ogando, N.S.; Snijder, E.J.; van Hemert, M.J.; Bujnicki, J.M.; et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Huston, N.C.; Wan, H.; de Tavares, R.C.A.; Wilen, C.; Pyle, A.M. Comprehensive in-vivo secondary structure of the SARS-CoV-2 genome reveals novel regulatory motifs and mechanisms. BioRxiv 2020. [Google Scholar] [CrossRef]
- Sun, L.; Li, P.; Ju, X.; Rao, J.; Huang, W.; Zhang, S.; Xiong, T.; Xu, K.; Zhou, X.; Ren, L.; et al. In vivo structural characterization of the whole SARS-CoV-2 RNA genome identifies host cell target proteins vulnerable to re-purposed drugs. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sanders, W.; Fritch, E.J.; Madden, E.A.; Graham, R.L.; Vincent, H.A.; Heise, M.T.; Baric, R.S.; Moorman, N.J. Comparative analysis of coronavirus genomic RNA structure reveals conservation in SARS-like coronaviruses. bioRxiv 2020. [Google Scholar] [CrossRef]
- Miao, Z.; Tidu, A.; Eriani, G.; Martin, F. Secondary structure of the SARS-CoV-2 5′-UTR. RNA Biol. 2020, 1–10. [Google Scholar] [CrossRef]
- Wacker, A.; Weigand, J.E.; Akabayov, S.R.; Altincekic, N.; Bains, J.K.; Banijamali, E.; Binas, O.; Castillo-Martinez, J.; Cetiner, E.; Ceylan, B.; et al. Secondary structure determination of conserved SARS-CoV-2 RNA elements by NMR spectroscopy. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Rouskin, S.; Zubradt, M.; Washietl, S.; Kellis, M.; Weissman, J.S. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 2014, 505, 701–705. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, K.A.; Merino, E.J.; Weeks, K.M. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): Quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006, 1, 1610–1616. [Google Scholar] [CrossRef]
- Xie, X.; Muruato, A.; Lokugamage, K.G.; Narayanan, K.; Zhang, X.; Zou, J.; Liu, J.; Schindewolf, C.; Bopp, N.E.; Aguilar, P.V.; et al. An Infectious cDNA Clone of SARS-CoV-2. Cell Host Microbe 2020, 27, 841–848.e3. [Google Scholar] [CrossRef]
- Hao, S.; Ning, K.; Kuz, C.A.; Vorhies, K.; Yan, Z.; Qiu, J. Long-Term Modeling of SARS-CoV-2 Infection of In vitro Cultured Polarized Human Airway Epithelium. MBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Zubradt, M.; Gupta, P.; Persad, S.; Lambowitz, A.M.; Weissman, J.S.; Rouskin, S. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods 2017, 14, 75–82. [Google Scholar] [CrossRef] [PubMed]
- FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 6 April 2010).
- Trim Galore. Available online: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 14 March 2012).
- Busan, S.; Weeks, K.M. Accurate detection of chemical modifications in RNA by mutational profiling (MaP) with ShapeMapper 2. RNA 2018, 24, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smola, M.J.; Rice, G.M.; Busan, S.; Siegfried, N.A.; Weeks, K.M. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat. Protoc. 2015, 10, 1643–1669. [Google Scholar] [CrossRef]
- Busan, S.; Weeks, K.M. Visualization of RNA structure models within the Integrative Genomics Viewer. RNA 2017, 23, 1012–1018. [Google Scholar] [CrossRef] [Green Version]
- Hajdin, C.E.; Bellaousov, S.; Huggins, W.; Leonard, C.W.; Mathews, D.H.; Weeks, K.M. Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc. Natl. Acad. Sci. USA 2013, 110, 5498–5503. [Google Scholar] [CrossRef] [Green Version]
- Tomezsko, P.J.; Corbin, V.D.A.; Gupta, P.; Swaminathan, H.; Glasgow, M.; Persad, S.; Edwards, M.D.; Mcintosh, L.; Papenfuss, A.T.; Emery, A.; et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature 2020, 582, 438–442. [Google Scholar] [CrossRef]
- Wendt, L.; Bostedt, L.; Hoenen, T.; Groseth, A. High-throughput screening for negative-stranded hemorrhagic fever viruses using reverse genetics. Antivir. Res. 2019, 170, 104569. [Google Scholar] [CrossRef]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [Green Version]
- Smola, M.J.; Calabrese, J.M.; Weeks, K.M. Detection of RNA-Protein Interactions in Living Cells with SHAPE. Biochemistry 2015, 54, 6867–6875. [Google Scholar] [CrossRef]
- Homan, P.J.; Favorov, O.V.; Lavender, C.A.; Kursun, O.; Ge, X.; Busan, S.; Dokholyan, N.V.; Weeks, K.M. Single-molecule correlated chemical probing of RNA. Proc. Natl. Acad. Sci. USA 2014, 111, 13858–13863. [Google Scholar] [CrossRef] [Green Version]
- Mustoe, A.M.; Lama, N.N.; Irving, P.S.; Olson, S.W.; Weeks, K.M. RNA base-pairing complexity in living cells visualized by correlated chemical probing. Proc. Natl. Acad. Sci. USA 2019, 116, 24574–24582. [Google Scholar] [CrossRef]
- Ziv, O.; Price, J.; Shalamova, L.; Kamenova, T.; Goodfellow, I.; Weber, F.; Miska, E.A. The Short- and Long-Range RNA-RNA Interactome of SARS-CoV-2. Mol. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Stammler, S.N.; Cao, S.; Chen, S.-J.; Giedroc, D.P. A conserved RNA pseudoknot in a putative molecular switch domain of the 3′-untranslated region of coronaviruses is only marginally stable. RNA 2011, 17, 1747–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, G.D.; Chang, R.Y.; Brian, D.A. A phylogenetically conserved hairpin-type 3′ untranslated region pseudoknot functions in coronavirus RNA replication. J. Virol. 1999, 73, 8349–8355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathews, D.H.; Disney, M.D.; Childs, J.L.; Schroeder, S.J.; Zuker, M.; Turner, D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7287–7292. [Google Scholar] [CrossRef] [Green Version]
- Aird, D.; Ross, M.G.; Chen, W.-S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef] [Green Version]
- Goebel, S.J.; Miller, T.B.; Bennett, C.J.; Bernard, K.A.; Masters, P.S. A hypervariable region within the 3′ cis-acting element of the murine coronavirus genome is nonessential for RNA synthesis but affects pathogenesis. J. Virol. 2007, 81, 1274–1287. [Google Scholar] [CrossRef] [Green Version]
- Robertson, M.P.; Igel, H.; Baertsch, R.; Haussler, D.; Ares, M.; Scott, W.G. The structure of a rigorously conserved RNA element within the SARS virus genome. PLoS Biol. 2005, 3, e5. [Google Scholar] [CrossRef]
- Liu, P.; Yang, D.; Carter, K.; Masud, F.; Leibowitz, J.L. Functional analysis of the stem loop S3 and S4 structures in the coronavirus 3′UTR. Virology 2013, 443, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Tange, O. GNU Parallel 20200722. Zenodo 2020. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Qiu, J.; Aryal, S.; Hackett, J.L.; Wang, J. The RNA Architecture of the SARS-CoV-2 3′-Untranslated Region. Viruses 2020, 12, 1473. https://doi.org/10.3390/v12121473
Zhao J, Qiu J, Aryal S, Hackett JL, Wang J. The RNA Architecture of the SARS-CoV-2 3′-Untranslated Region. Viruses. 2020; 12(12):1473. https://doi.org/10.3390/v12121473
Chicago/Turabian StyleZhao, Junxing, Jianming Qiu, Sadikshya Aryal, Jennifer L. Hackett, and Jingxin Wang. 2020. "The RNA Architecture of the SARS-CoV-2 3′-Untranslated Region" Viruses 12, no. 12: 1473. https://doi.org/10.3390/v12121473
APA StyleZhao, J., Qiu, J., Aryal, S., Hackett, J. L., & Wang, J. (2020). The RNA Architecture of the SARS-CoV-2 3′-Untranslated Region. Viruses, 12(12), 1473. https://doi.org/10.3390/v12121473