
Genetic Diversity of Rift Valley Fever Strains Circulating in Namibia in 2010 and 2011


 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Strains
2.2. Sequencing
2.3. Phylogenetic Analysis
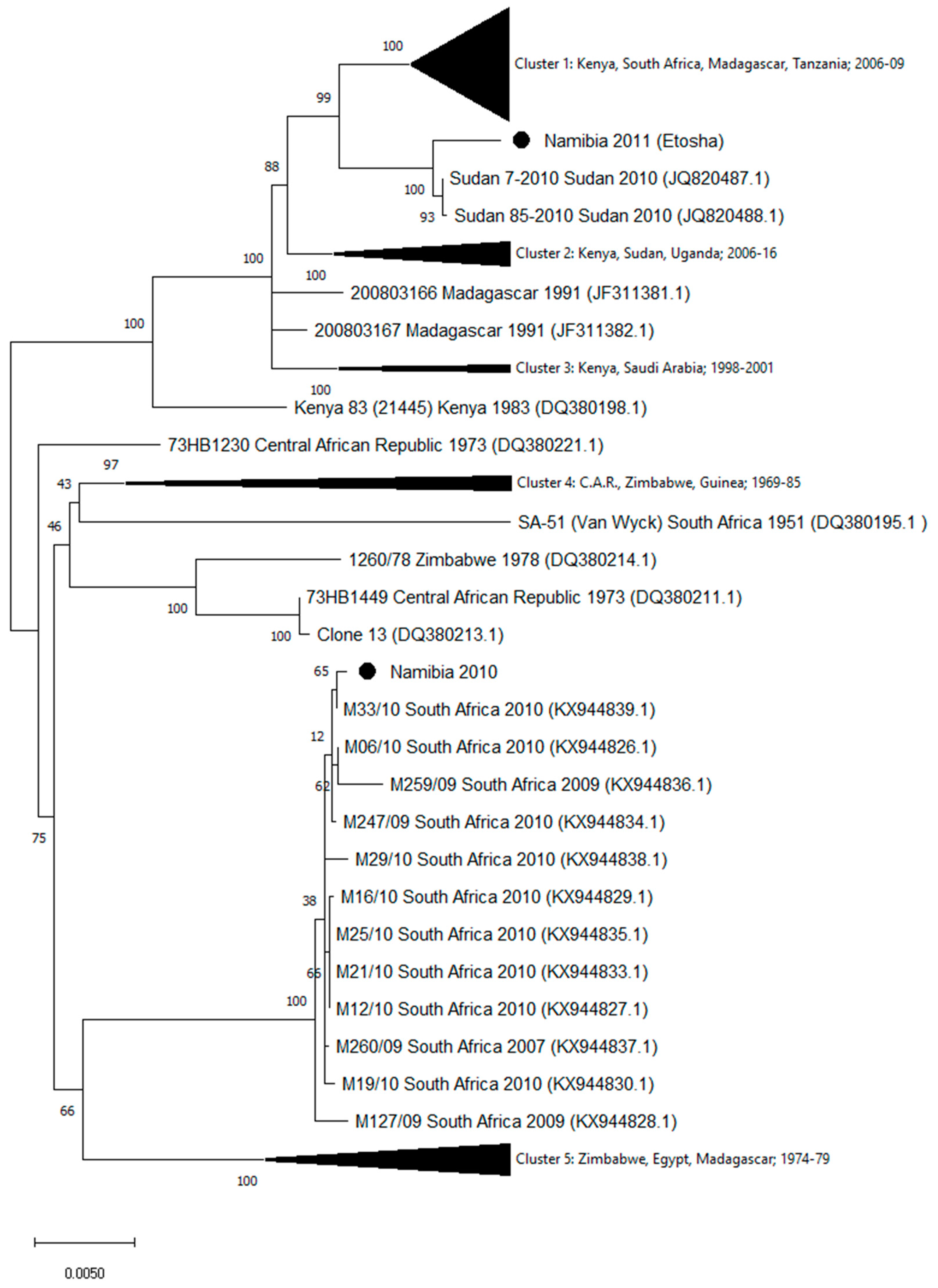
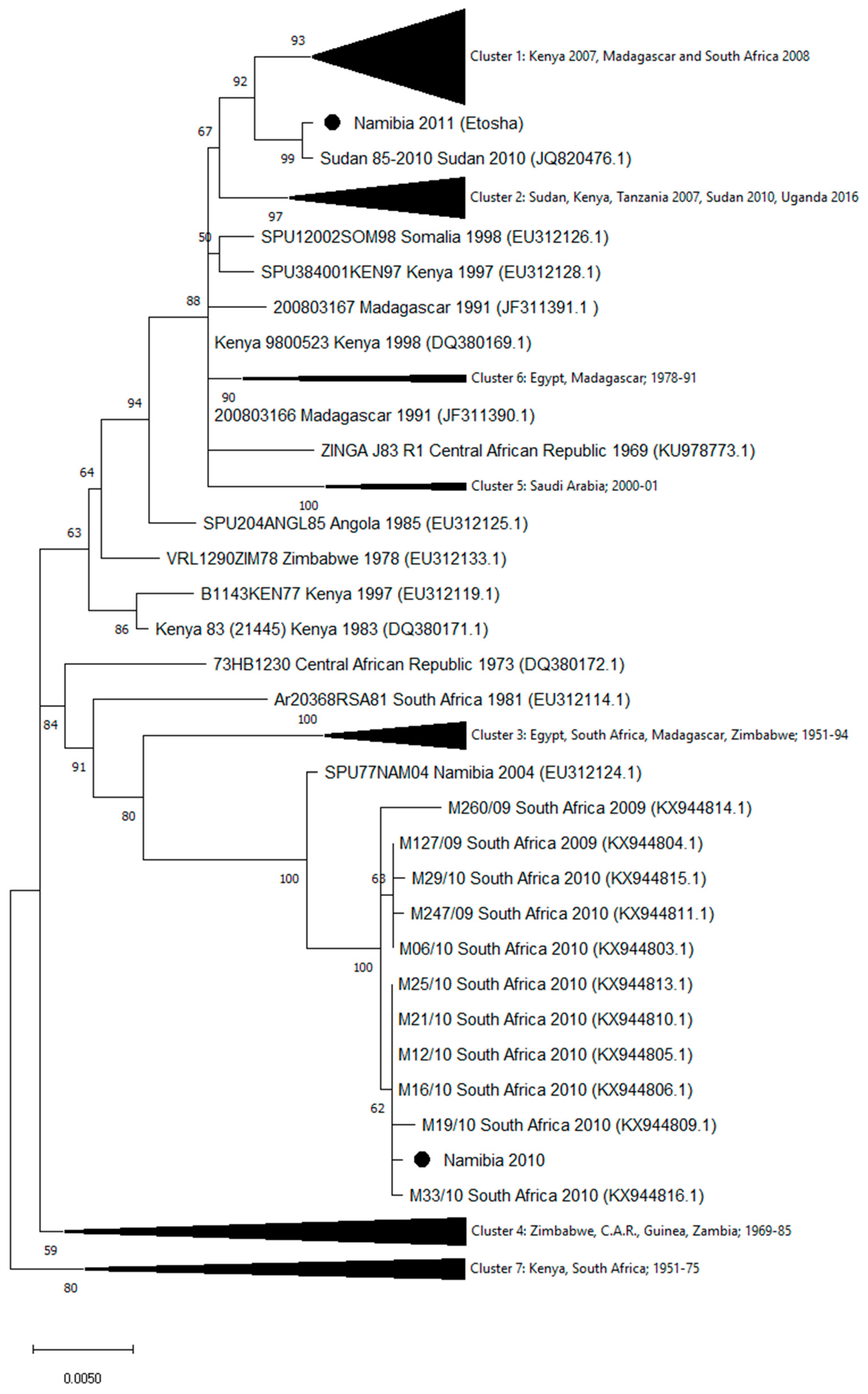
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hartman, A. Rift Valley Fever. Clin. Lab. Med. 2017, 37, 285–301. [Google Scholar] [CrossRef] [PubMed]
- Meegan, J.M.; Khalil, G.M.; Hoogstraal, H.; Adham, F.K. Experimental Transmission and Field Isolation Studies Implicating Culex Pipiens as a Vector of Rift Valley Fever Virus in Egypt. Am. J. Trop. Med. Hyg. 1980, 29, 1405–1410. [Google Scholar] [CrossRef]
- Pepin, M.; Bouloy, M.; Bird, B.H.; Kemp, A.; Paweska, J. Rift Valley fever virus (Bunyaviridae: Phlebovirus): An update on pathogenesis, molecular epidemiology, vectors, diagnostics and prevention. Vet. Res. 2010, 41, 61. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.; Dobler, G. Emergence of zoonotic arboviruses by animal trade and migration. Parasites Vectors 2010, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, V.; Pépin, M.; Plée, L.; Lancelot, R. Rift Valley fever—A threat for Europe? Eurosurveillance 2010, 15, 19506. [Google Scholar] [PubMed]
- Daubney, R.; Hudson, J.R.; Garnham, P.C. Enzootic hepatitis or rift valley fever. An undescribed virus disease of sheep cattle and man from east africa. J. Pathol. Bacteriol. 1931, 34, 545–579. [Google Scholar] [CrossRef]
- Daubney, R.; Hudson, J.R. Rift valley fever. Lancet 1932, 219, 611–612. [Google Scholar] [CrossRef]
- Nanyingi, M.O.; Munyua, P.; Kiama, S.G.; Muchemi, G.M.; Thumbi, S.M.; Bitek, A.O.; Bett, B.; Muriithi, R.M.; Njenga, M.K. A systematic review of Rift Valley Fever epidemiology 1931–2014. Infect. Ecol. Epidemiol. 2015, 5, 28024. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention, National Center for Emerging and Zoonotic Infectious Diseases (NCEZID), Division of High-Consequence Pathogens and Pathology (DHCPP), Viral Special Pathogens Branch (VSPB). RVF Distribution Map. Available online: https://www.cdc.gov/vhf/rvf/outbreaks/distribution-map.html (accessed on 9 November 2020).
- Bird, B.H.; Khristova, M.L.; Rollin, P.E.; Ksiazek, T.G.; Nichol, S.T. Complete Genome Analysis of 33 Ecologically and Biologically Diverse Rift Valley Fever Virus Strains Reveals Widespread Virus Movement and Low Genetic Diversity due to Recent Common Ancestry. J. Virol. 2006, 81, 2805–2816. [Google Scholar] [CrossRef]
- Pienaar, N.J.; Thompson, P.N. Temporal and spatial history of Rift Valley fever in South Africa: 1950 to 2011. Onderstepoort J. Vet. Res. 2013, 80, 13. [Google Scholar] [CrossRef]
- Paweska, J.; Weyner, J.; van Vuren, P.J.; Kemp, A.; le Roux, C.; Leman, P.; Grobbelaar, A.; Archer, B.; Thomas, J.; Last, R.; et al. Update on the 2010 Rift Valley fever outbreak in South Africa. Arbo-Zoonet News 2011, 5, 7–13. Available online: http://www.arbo-zoo.net/_data/arbozoonet-news_No5.pdf (accessed on 31 May 2011).
- Monaco, F.; Pinoni, C.; Cosseddu, G.M.; Khaiseb, S.; Calistri, P.; Molini, U.; Bishi, A.; Conte, A.; Scacchia, M.; Lelli, R. Rift Valley Fever in Namibia, 2010. Emerg. Infect. Dis. 2013, 19, 2025–2027. [Google Scholar] [CrossRef] [PubMed]
- Grobbelaar, A.A.; Weyer, J.; Leman, P.A.; Kemp, A.; Paweska, J.T.; Swanepoel, R. Molecular Epidemiology of Rift Valley Fever Virus. Emerg. Infect. Dis. 2011, 17, 2270–2276. [Google Scholar] [CrossRef] [PubMed]
- Office International des Epizooties. World Animal Health Information Database, 2012: Event Summary, Rift Valley Fever, Namibia. Available online: http://www.oie.int/wahis_2/public/wahid.php/Reviewreport/Review/viewsummary?reportid=10680 (accessed on 14 April 2020).
- Dondona, A.C.; Aschenborn, O.; Pinoni, C.; Di Gialleonardo, L.; Maseke, A.; Bortone, G.; Polci, A.; Scacchia, M.; Molini, U.; Monaco, F. Rift Valley Fever Virus among Wild Ruminants, Etosha National Park, Namibia, 2011. Emerg. Infect. Dis. 2016, 22, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Sall, A.A.; Zanotto, P.M.D.A.; Sene, O.K.; Zeller, H.G.; Digoutte, J.P.; Thiongane, Y.; Bouloy, M. Genetic Reassortment of Rift Valley Fever Virus in Nature. J. Virol. 1999, 73, 8196–8200. [Google Scholar] [CrossRef]
- Liu, L.; Celma, C.C.; Roy, P. Rift Valley fever virus structural proteins: Expression, characterization and assembly of recombinant proteins. Virol. J. 2008, 5, 82. [Google Scholar] [CrossRef]
- Bouloy, M.; Weber, F. Molecular Biology of Rift Valley Fever Virus. Open Virol. J. 2010, 4, 8–14. [Google Scholar] [CrossRef]
- Coffey, L.L.; Vasilakis, N.; Brault, A.C.; Powers, A.M.; Tripet, F.; Weaver, S. Arbovirus evolution in vivo is constrained by host alternation. Proc. Natl. Acad. Sci. USA 2008, 105, 6970–6975. [Google Scholar] [CrossRef]
- Moutailler, S.; Roche, B.; Thiberge, J.-M.; Caro, V.; Rougeon, F.; Failloux, A.-B. Host Alternation Is Necessary to Maintain the Genome Stability of Rift Valley Fever Virus. PLoS Negl. Trop. Dis. 2011, 5, e1156. [Google Scholar] [CrossRef]
- Freire, C.C.M.; Iamarino, A.; Soumaré, P.O.L.; Faye, O.; Sall, A.A.; Zanotto, P.M.D.A. Reassortment and distinct evolutionary dynamics of Rift Valley Fever virus genomic segments. Sci. Rep. 2015, 5, 11353. [Google Scholar] [CrossRef]
- Allander, T.; Tammi, M.T.; Eriksson, M.; Bjerkner, A.; Tiveljung-Lindell, A.; Andersson, B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc. Natl. Acad. Sci. USA 2005, 102, 12891–12896. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Aradaib, I.E.; Erickson, B.R.; Elageb, R.M.; Khristova, M.L.; Carroll, S.A.; Elkhidir, I.M.; Karsany, M.E.; Karrar, A.E.; Elbashir, M.I.; Nichol, S.T. Rift Valley Fever, Sudan, 2007 and 2010. Emerg. Infect. Dis. 2013, 19, 246–253. [Google Scholar] [CrossRef]
- Bird, B.H.; Githinji, J.W.K.; Macharia, J.M.; Kasiiti, J.L.; Muriithi, R.M.; Gacheru, S.G.; Musaa, J.O.; Towner, J.S.; Reeder, S.A.; Oliver, J.B.; et al. Multiple Virus Lineages Sharing Recent Common Ancestry Were Associated with a Large Rift Valley Fever Outbreak among Livestock in Kenya during 2006–2007. J. Virol. 2008, 82, 11152–11166. [Google Scholar] [CrossRef] [PubMed]
- Soumaré, P.O.L.; Freire, C.C.M.; Faye, O.; Diallo, M.; De Oliveira, J.V.C.; Zanotto, P.M.A.; Sall, A.A. Phylogeography of Rift Valley Fever Virus in Africa Reveals Multiple Introductions in Senegal and Mauritania. PLoS ONE 2012, 7, e35216. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cosseddu, G.M.; Magwedere, K.; Molini, U.; Pinoni, C.; Khaiseb, S.; Scacchia, M.; Marcacci, M.; Capobianco Dondona, A.; Valleriani, F.; Polci, A.; et al. Genetic Diversity of Rift Valley Fever Strains Circulating in Namibia in 2010 and 2011. Viruses 2020, 12, 1453. https://doi.org/10.3390/v12121453
Cosseddu GM, Magwedere K, Molini U, Pinoni C, Khaiseb S, Scacchia M, Marcacci M, Capobianco Dondona A, Valleriani F, Polci A, et al. Genetic Diversity of Rift Valley Fever Strains Circulating in Namibia in 2010 and 2011. Viruses. 2020; 12(12):1453. https://doi.org/10.3390/v12121453
Chicago/Turabian StyleCosseddu, Gian Mario, Kudakwashe Magwedere, Umberto Molini, Chiara Pinoni, Sigfried Khaiseb, Massimo Scacchia, Maurilia Marcacci, Andrea Capobianco Dondona, Fabrizia Valleriani, Andrea Polci, and et al. 2020. "Genetic Diversity of Rift Valley Fever Strains Circulating in Namibia in 2010 and 2011" Viruses 12, no. 12: 1453. https://doi.org/10.3390/v12121453
APA StyleCosseddu, G. M., Magwedere, K., Molini, U., Pinoni, C., Khaiseb, S., Scacchia, M., Marcacci, M., Capobianco Dondona, A., Valleriani, F., Polci, A., & Monaco, F. (2020). Genetic Diversity of Rift Valley Fever Strains Circulating in Namibia in 2010 and 2011. Viruses, 12(12), 1453. https://doi.org/10.3390/v12121453