
UBE1a Suppresses Herpes Simplex Virus-1 Replication


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Agents and Plasmids
2.2. Virus Infection and Cytopathic Effect (CPE) Assay
2.3. Western Blotting (WB), Immunofluorescence Analysis (IFA) and Antibodies
2.4. Cell Viability Assay
2.5. Quantification of HSV-1 DNA Synthesis
2.6. Establishment of Vero Cells Expressing Tetracycline/Doxycycline-Inducible shRNA
2.7. Transfection and Stable Cell Line Generation
2.8. Reverse Transcription Polymerase Chain Reaction (RT-PCR) and Real-Time-qPCR
2.9. In Situ Detergent Extraction, DNase, or RNase Treatment
2.10. Pulldown Assays
2.11. Statistics
3. Results
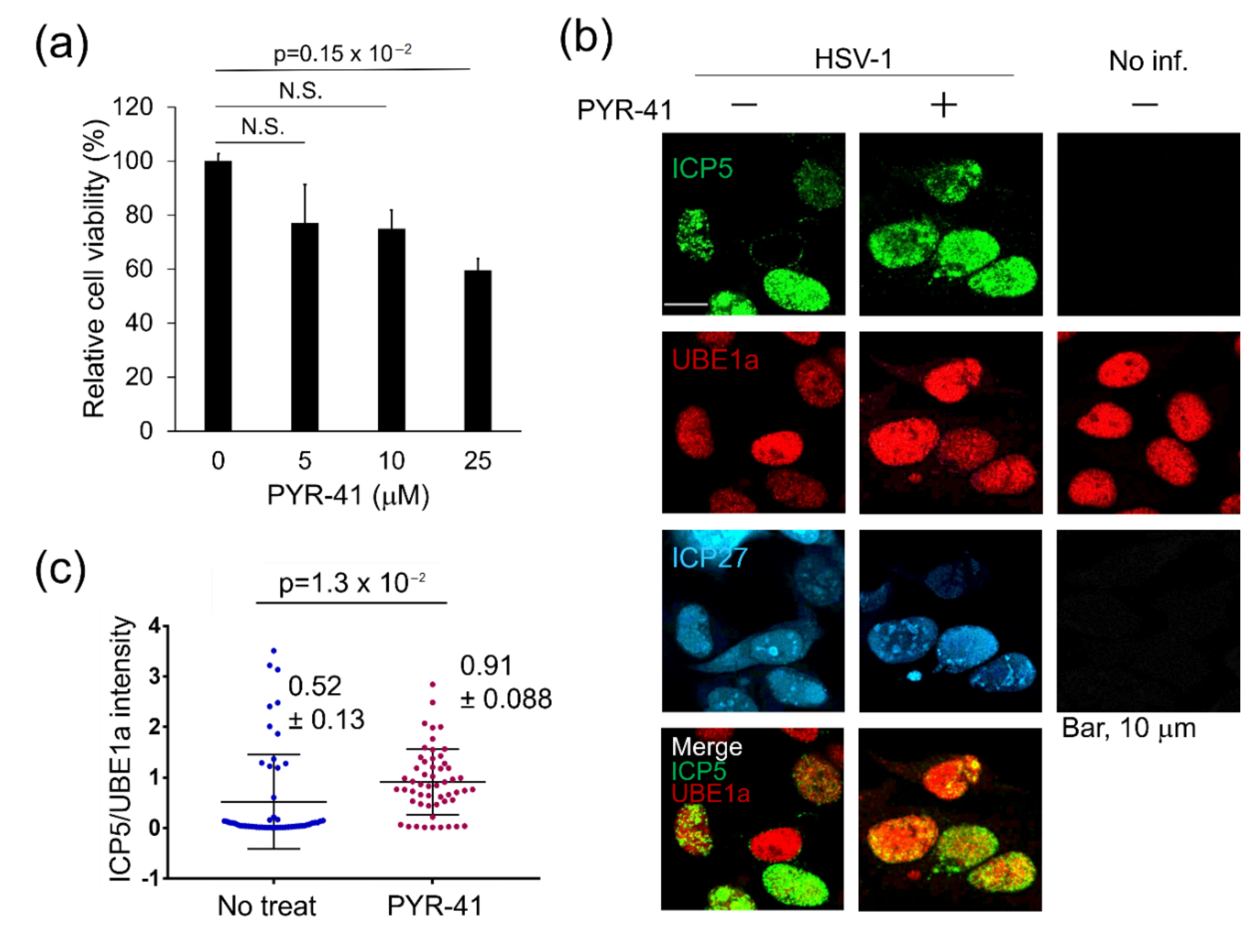
3.1. UBE1 Inhibitor PYR-41 Enhances HSV-1 Production
3.2. Expression Level of UBE1a Is Negatively Correlated with ICP5 Expression
3.3. UBE1a Inhibition Increases ICP5 Expression in HSV-1 Infected Cells
3.4. UBE1a Reduces and Retards ICP5 Protein Expression
3.5. UBE1a and ICP27 Partially Co-Localizes at the Hsc70 foci/VICE Domains
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Roizman, B.; Knipe, D.M. Whitley, R.J. Herpes Simplex Viruses. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Phiadelphia, PA, USA, 2007; Volume 2, pp. 2501–2601. [Google Scholar]
- Perng, G.-C.; Jones, C. Towards an Understanding of the Herpes Simplex Virus Type 1 Latency-Reactivation Cycle. Interdiscip. Perspect. Infect. Dis. 2010, 2010, 262415. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, C.; Weller, S.K. The herpes simplex virus type 1 UL6 protein is essential for cleavage and packaging but not for genomic inversion. Virology 1996, 226, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, M.P.; Chen, L.B.; Knipe, D.M. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 1984, 36, 857–868. [Google Scholar] [CrossRef]
- Burch, A.D.; Weller, S.K. Nuclear sequestration of cellular chaperone and proteasomal machinery during herpes simplex virus type 1 infection. J. Virol. 2004, 78, 7175–7185. [Google Scholar] [CrossRef] [PubMed]
- Burch, A.D.; Weller, S.K. Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone hsp90 for proper localization to the nucleus. J. Virol. 2005, 79, 10740–10749. [Google Scholar] [CrossRef]
- Wilkinson, D.E.; Weller, S.K. Herpes simplex virus type I disrupts the ATR-dependent DNA-damage response during lytic infection. J. Cell Sci. 2006, 119, 2695–2703. [Google Scholar] [CrossRef]
- Livingston, C.M.; Ifrim, M.F.; Cowan, A.E.; Weller, S.K. Virus-Induced Chaperone-Enriched (VICE) Domains Function as Nuclear Protein Quality Control Centers during HSV-1 Infection. PLoS Pathog. 2009, 5, e1000619. [Google Scholar] [CrossRef]
- Livingston, C.M.; DeLuca, N.A.; Wilkinson, D.E.; Weller, S.K. Oligomerization of ICP4 and Rearrangement of Heat Shock Proteins May Be Important for Herpes Simplex Virus Type 1 Prereplicative Site Formation. J. Virol. 2008, 82, 6324–6336. [Google Scholar] [CrossRef]
- Trempe, J.F. Reading the ubiquitin postal. code. Curr. Opin. Struct. Biol. 2011, 21, 792–801. [Google Scholar] [CrossRef]
- Weissman, A.M. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell Biol. 2001, 2, 169–178. [Google Scholar] [CrossRef]
- Kleiger, G.; Mayor, T. Perilous journey: A tour of the ubiquitin-proteasome system. Trends Cell Biol. 2014, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M. Mechanisms Underlying Ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Haas, A.L.; Warms, J.V.; Hershko, A.; Rose, I.A. Ubiquitin-activating enzyme. Mechanism and role in protein-ubiquitin conjugation. J. Biol. Chem. 1982, 257, 2543–2548. [Google Scholar] [PubMed]
- Hicke, L. Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol. 2001, 2, 195–201. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.P.; Jentsch, S.; Varshavsky, A. UBA 1: An essential yeast gene encoding ubiquitin-activating enzyme. EMBO J. 1991, 10, 227–236. [Google Scholar] [CrossRef]
- Stephen, A.G.; Trausch-Azar, J.S.; Ciechanover, A.; Schwartz, A.L. The ubiquitin-activating enzyme E1 is phosphorylated and localized to the nucleus in a cell cycle-dependent manner. J. Biol. Chem. 1996, 271, 15608–15614. [Google Scholar] [CrossRef]
- Groen, E.J.N.; Gillingwater, T.H. UBA1: At the Crossroads of Ubiquitin Homeostasis and Neurodegeneration. Trends Mol. Med. 2015, 21, 622–632. [Google Scholar] [CrossRef]
- Stephen, A.G.; Trausch-Azar, J.S.; Handley-Gearhart, P.M.; Ciechanover, A.; Schwartz, A.L. Identification of a region within the ubiquitin-activating enzyme required for nuclear targeting and phosphorylation. J. Biol. Chem. 1997, 272, 10895–10903. [Google Scholar] [CrossRef]
- Boutell, C.; Sadis, S.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 2002, 76, 841–850. [Google Scholar] [CrossRef]
- Boutell, C.; Everett, R.D. The Herpes Simplex Virus Type 1 (HSV-1) Regulatory Protein ICP0 Interacts with and Ubiquitinates p53. J. Biol. Chem. 2003, 278, 36596–36602. [Google Scholar] [CrossRef]
- Boutell, C.; Everett, R.D. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J. Gen. Virol. 2013, 94, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, C.E.; Wolberger, C. A spectrophotometric assay for conjugation of ubiquitin and ubiquitin-like proteins. Anal. Biochem. 2011, 418, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Watanabe, T.; Yagi, S.; Yamanaka, T.; Fujimuro, M. Kaposi’s sarcoma-associated herpesvirus ORF34 is essential for late gene expression and virus production. Sci. Rep. 2017, 7, 329. [Google Scholar] [CrossRef] [PubMed]
- Dejosez, M.; Levine, S.S.; Frampton, G.M.; Whyte, W.A.; Stratton, S.A.; Barton, M.C.; Gunaratne, P.H.; Young, R.A.; Zwaka, T.P. Ronin/Hcf-1 binds to a hyperconserved enhancer element and regulates genes involved in the growth of embryonic stem cells. Genes Dev. 2010, 24, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Shigemi, Z.; Hara, N.; Moriguchi, M.; Ikeda, M.; Watanabe, T.; Fujimuro, M. Arctigenin induces the apoptosis of primary effusion lymphoma cells under conditions of glucose deprivation. Int. J. Oncol. 2018, 52, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Shigemi, Z.; Furukawa, Y.; Hosokawa, K.; Minami, S.; Matsuhiro, J.; Nakata, S.; Watanabe, T.; Kagawa, H.; Nakagawa, K.; Takeda, H.; et al. Diallyl trisulfide induces apoptosis by suppressing NF-B signaling through destabilization of TRAF6 in primary effusion lymphoma. Int. J. Oncol. 2016, 48, 293–304. [Google Scholar] [CrossRef]
- Fujimuro, M.; Sawada, H.; Yokosawa, H. Production and characterization of monoclonal antibodies specific to multi-ubiquitin chains of polyubiquitinated proteins. FEBS Lett. 1994, 349, 172–180. [Google Scholar] [CrossRef]
- Chen, C.; Okayama, H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 1987, 7, 2745–2752. [Google Scholar] [CrossRef]
- Watanabe, T.; Nishimura, M.; Izumi, T.; Kuriyama, K.; Iwaisako, Y.; Hosokawa, K.; Takaori-Kondo, A.; Fujimuro, M. Kaposi’s Sarcoma-Associated Herpesvirus ORF66 Is Essential for Late Gene Expression and Virus Production via Interaction with ORF34. J. Virol. 2019, 94. [Google Scholar] [CrossRef]
- Fujimuro, M.; Sawada, H.; Yokosawa, H. Dynamics of ubiquitin conjugation during heat-shock response revealed by using a monoclonal antibody specific to multi-ubiquitin chains. Eur. J. Biochem. 1997, 249, 427–433. [Google Scholar] [CrossRef]
- Yang, Y.; Kitagaki, J.; Dai, R.-M.; Tsai, Y.C.; Lorick, K.L.; Ludwig, R.L.; Pierre, S.A.; Jensen, J.P.; Davydov, I.V.; Oberoi, P.; et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007, 67, 9472–9481. [Google Scholar] [CrossRef] [PubMed]
- Grenfell, S.J.; Trausch-Azar, J.S.; Handley-Gearhart, P.M.; Ciechanover, A.; Schwartz, A.L. Nuclear localization of the ubiquitin-activating enzyme, E1, is cell-cycle-dependent. Biochem. J. 1994, 300, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Fontaine-Rodriguez, E.C.; Knipe, D.M. Herpes Simplex Virus ICP27 Increases Translation of a Subset of Viral Late mRNAs. J. Virol. 2008, 82, 3538–3545. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.P.; Sandri-Goldin, R.M. Herpes simplex virus 1 regulatory protein ICP27 undergoes a head-to-tail intramolecular interaction. J. Virol. 2010, 84, 4124–4135. [Google Scholar] [CrossRef]
- Souki, S.K.; Gershon, P.D.; Sandri-Goldin, R.M. Arginine Methylation of the ICP27 RGG Box Regulates ICP27 Export and Is Required for Efficient Herpes Simplex Virus 1 Replication. J. Virol. 2009, 83, 5309–5320. [Google Scholar] [CrossRef]
- Souki, S.K.; Sandri-Goldin, R.M. Arginine Methylation of the ICP27 RGG Box Regulates the Functional Interactions of ICP27 with SRPK1 and Aly/REF during Herpes Simplex Virus 1 Infection. J. Virol. 2009, 83, 8970–8975. [Google Scholar] [CrossRef]
- Lieu, P.T.; Wagner, E.K. The Kinetics of VP5 mRNA Expression Is Not Critical for Viral Replication in Cultured Cells. J. Virol. 2000, 74, 2770–2776. [Google Scholar] [CrossRef][Green Version]
- Jin, F.; Li, S.; Zheng, K.; Zhuo, C.; Ma, K.; Chen, M.; Wang, Q.; Zhang, P.; Fan, J.; Ren, Z.; et al. Silencing herpes simplex virus type 1 capsid protein encoding genes by siRNA: A promising antiviral therapeutic approach. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Montellese, C.; van den Heuvel, J.; Ashiono, C.; Dörner, K.; Melnik, A.; Jonas, S.; Zemp, I.; Picotti, P.; Gillet, L.C.; Kutay, U. USP16 counteracts mono-ubiquitination of RPS27a and promotes maturation of the 40S ribosomal subunit. Elife 2020, 9. [Google Scholar] [CrossRef]
- Jung, Y.; Kim, H.D.; Yang, H.W.; Kim, H.J.; Jang, C.Y.; Kim, J. Modulating cellular balance of Rps3 mono-ubiquitination by both Hel2 E3 ligase and Ubp3 deubiquitinase regulates protein quality control. Exp. Mol. Med. 2017, 49. [Google Scholar] [CrossRef]
- Joazeiro, C.A.P. Mechanisms and functions of ribosome-associated protein quality control. Nat. Rev. Mol. Cell Biol. 2019, 20, 368–383. [Google Scholar] [CrossRef] [PubMed]
- Rojas, S.; Corbin-Lickfett, K.A.; Escudero-Paunetto, L.; Sandri-Goldin, R.M. ICP27 Phosphorylation Site Mutants Are Defective in Herpes Simplex Virus 1 Replication and Gene Expression. J. Virol. 2010, 84, 2200–2211. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Corbin-Lickfett, K.A.; Rojas, S.; Li, L.; Cocco, M.J.; Sandri-Goldin, R.M. ICP27 Phosphorylation Site Mutants Display Altered Functional Interactions with Cellular Export Factors Aly/REF and TAP/NXF1 but Are Able To Bind Herpes Simplex Virus 1 RNA. J. Virol. 2010, 84, 2212–2222. [Google Scholar] [CrossRef] [PubMed]
- Huffmaster, N.J.; Sollars, P.J.; Richards, A.L.; Pickard, G.E.; Smith, G.A. Dynamic ubiquitination drives herpesvirus neuroinvasion. Proc. Natl. Acad. Sci. USA 2015, 112, 12818–12823. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Gianni, T.; Salvioli, S.; Campadelli-Fiume, G. Herpes Simplex Virus Glycoproteins gH/gL and gB Bind Toll-Like Receptor 2, and Soluble gH/gL Is Sufficient To activate NF-κB. J. Virol. 2012, 86, 6555–6562. [Google Scholar] [CrossRef]
- Medici, M.A.; Sciortino, M.T.; Perri, D.; Amici, C.; Avitabile, E.; Ciotti, M.; Balestrieri, E.; De Smaele, E.; Franzoso, G.; Mastino, A. Protection by herpes simplex virus glycoprotein D against Fas-mediated apoptosis: Role of nuclear factor κB. J. Biol. Chem. 2003, 278, 36059–36067. [Google Scholar] [CrossRef]
- Skaug, B.; Jiang, X.; Chen, Z.J. The Role of Ubiquitin in NF-κB Regulatory Pathways. Annu. Rev. Biochem. 2009, 78, 769–796. [Google Scholar] [CrossRef]
- Su, C.; Zhan, G.; Zheng, C. Evasion of host antiviral innate immunity by HSV-1, an update. Virol. J. BioMed Cent. 2016, 13, 38. [Google Scholar] [CrossRef]
- Van Sant, C.; Hagglund, R.; Lopez, P.; Roizman, B. The infected cell protein 0 of herpes simplex virus 1 dynamically interacts with proteasomes, binds and activates the cdc34 E2 ubiquitin-conjugating enzyme, and possesses in vitro E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 2001, 98, 8815–8820. [Google Scholar] [CrossRef]
- Gu, H.; Roizman, B. The degradation of promyelocytic leukemia and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc. Natl. Acad. Sci. USA 2003, 100, 8963–8968. [Google Scholar] [CrossRef]
- Everett, R.D.; Meredith, M.; Orr, A.; Cross, A.; Kathoria, M.; Parkinson, J. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 1997, 16, 1519–1530. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikeda, M.; Ito, A.; Sekine, Y.; Fujimuro, M. UBE1a Suppresses Herpes Simplex Virus-1 Replication. Viruses 2020, 12, 1391. https://doi.org/10.3390/v12121391
Ikeda M, Ito A, Sekine Y, Fujimuro M. UBE1a Suppresses Herpes Simplex Virus-1 Replication. Viruses. 2020; 12(12):1391. https://doi.org/10.3390/v12121391
Chicago/Turabian StyleIkeda, Marina, Akihiro Ito, Yuichi Sekine, and Masahiro Fujimuro. 2020. "UBE1a Suppresses Herpes Simplex Virus-1 Replication" Viruses 12, no. 12: 1391. https://doi.org/10.3390/v12121391
APA StyleIkeda, M., Ito, A., Sekine, Y., & Fujimuro, M. (2020). UBE1a Suppresses Herpes Simplex Virus-1 Replication. Viruses, 12(12), 1391. https://doi.org/10.3390/v12121391