Identification of RUVBL1 and RUVBL2 as Novel Cellular Interactors of the Ebola Virus Nucleoprotein

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
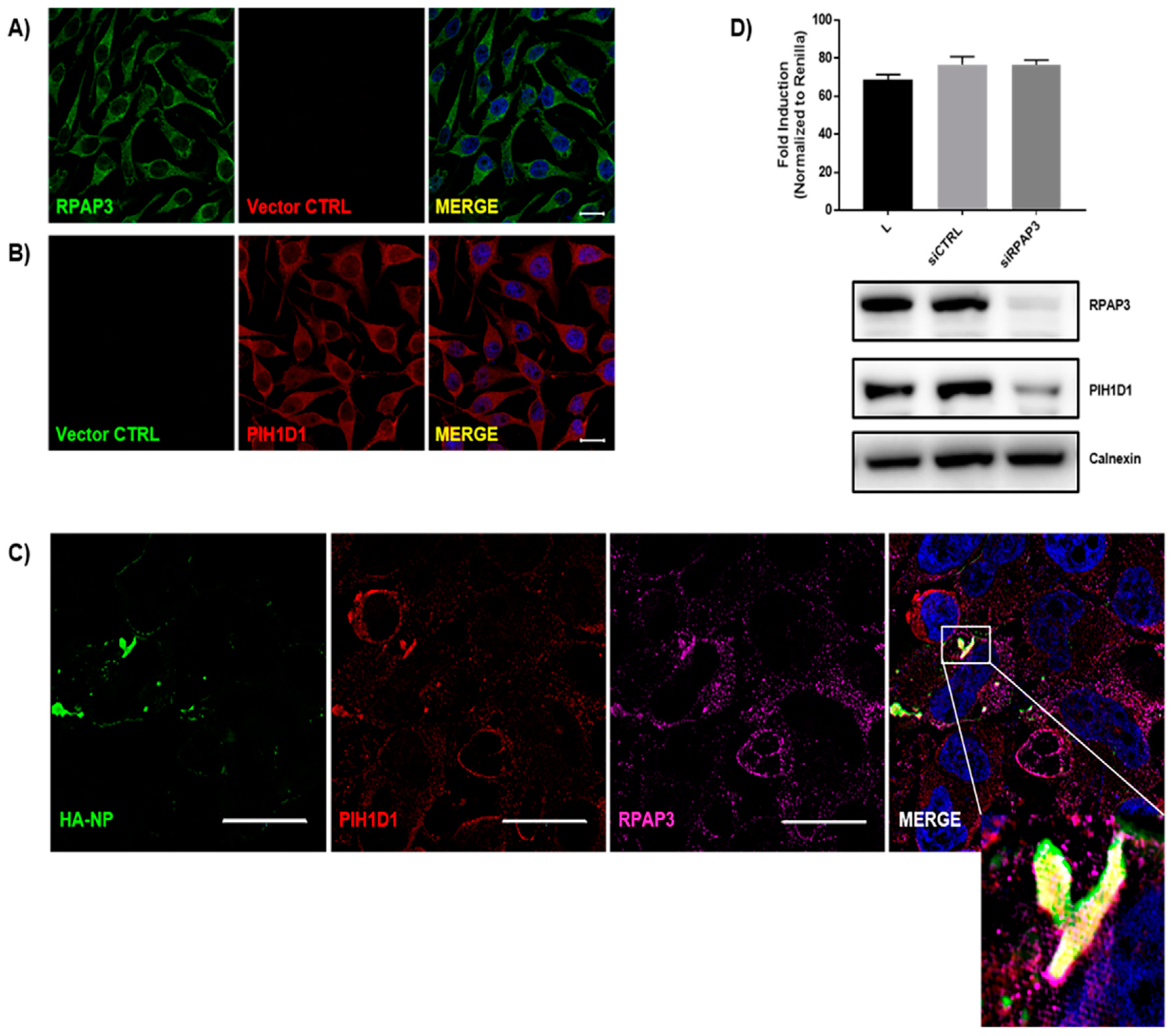{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Expression Vectors and Reagents
2.3. Gel-Liquid Chromatography/Tandem Mass Spectrometry Analysis
2.4. Protein Co-Immunoprecipitation
2.5. Immunoblotting
2.6. Confocal Microscopy
2.7. siRNA Transfection and Minigenome Assay
3. Results
3.1. Identification of NP Candidate Cellular Interactors by Affinity Purification-Mass Spectrometry (AP-MS)
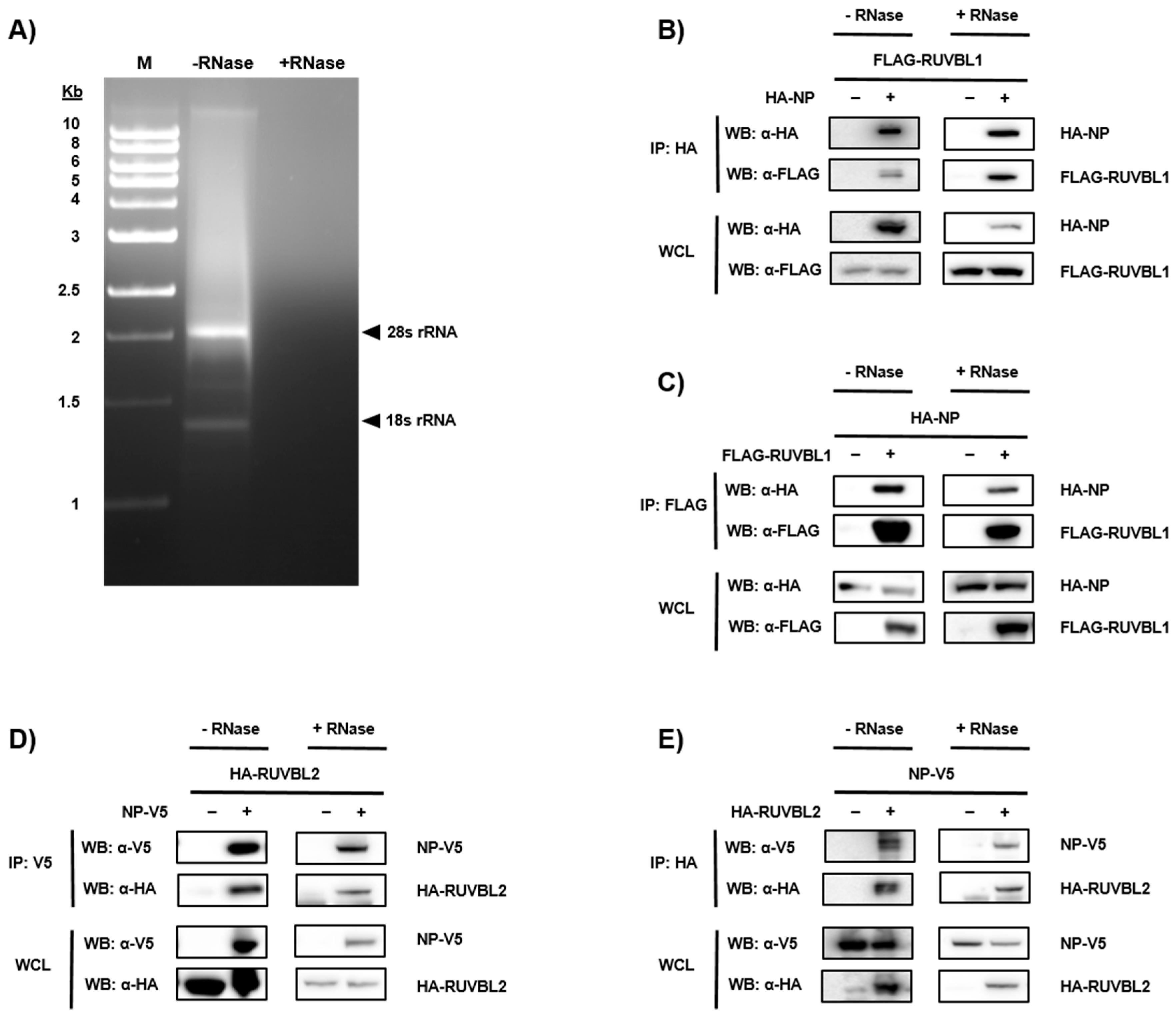
3.2. Validation of NP-Candidate Interactions by Co-Immunoprecipitation (Co-IP)
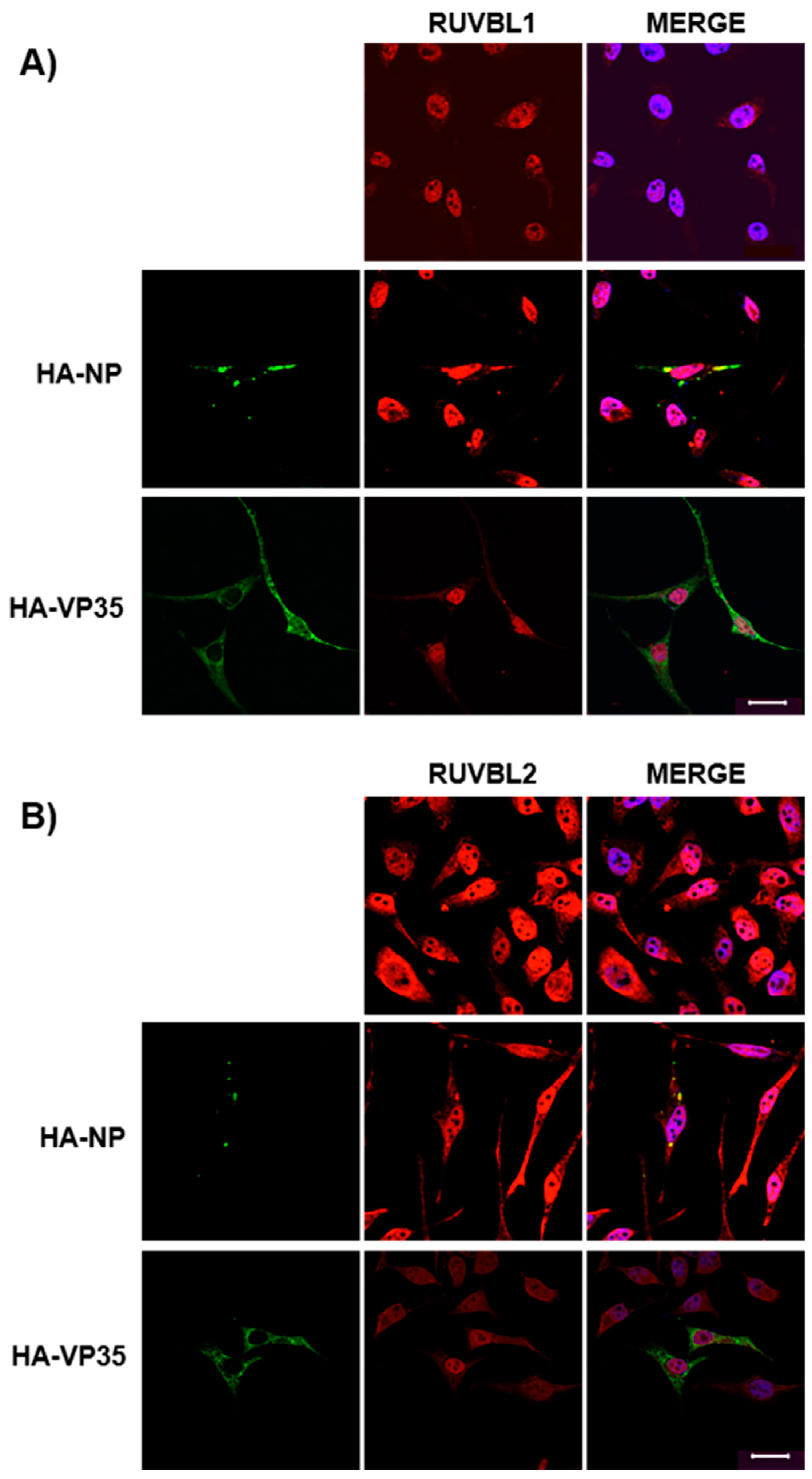
3.3. Assessing Localization of RUVBL1 and RUVBL2 in the Presence of NP
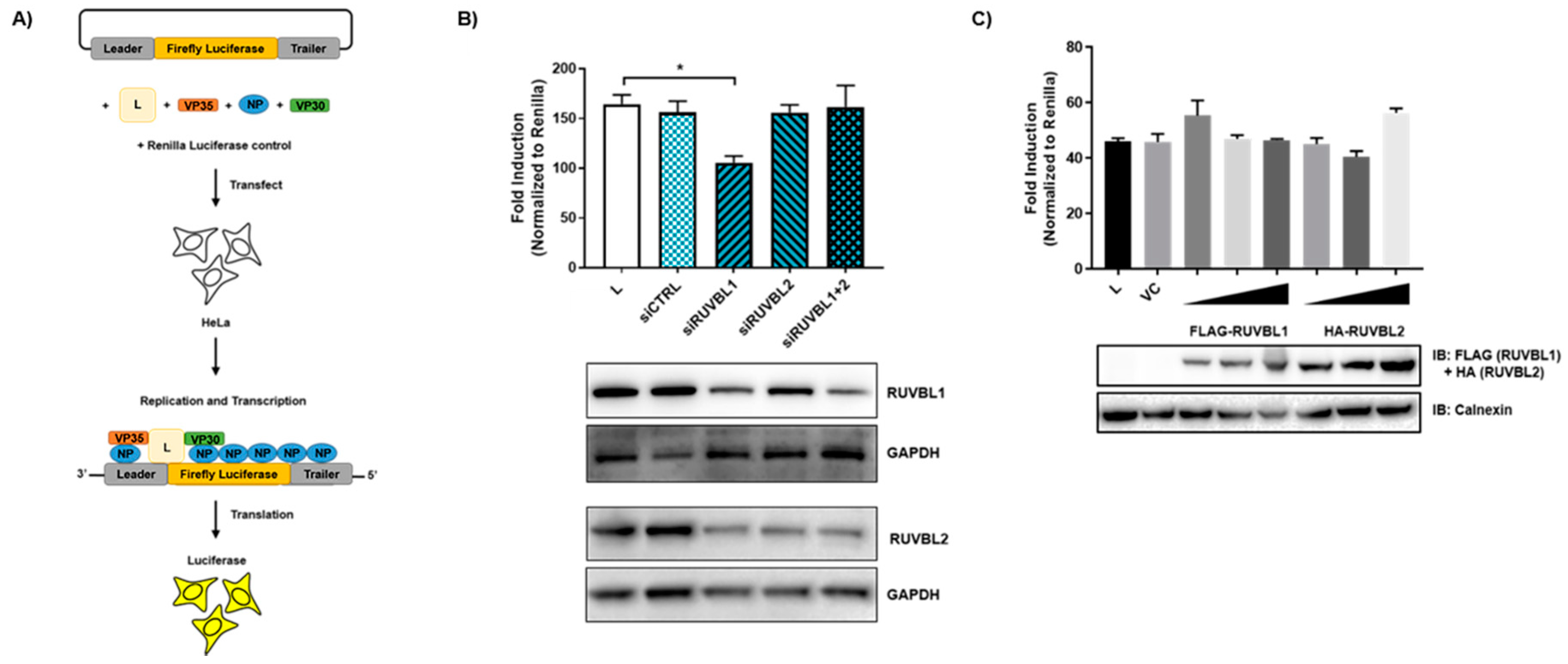
3.4. Evaluating the Effects of RUVBL1 and RUVBL2 on EBOV Minigenome Activity
3.5. Identification of the R2TP Complex Recruitment to NP Inclusions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Feldmann, H.; Sanchez, A.; Geisbert, T. Filoviridae: Marburg and ebola viruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 923–956. [Google Scholar]
- Feldmann, H.; Geisbert, T.W. Ebola haemorrhagic fever. Lancet 2011, 377, 849–862. [Google Scholar] [CrossRef]
- World Health Organization. Ebola haemorrhagic fever in Zaire, 1976. Bull. World Health Organ. 1978, 56, 271–293. [Google Scholar]
- WHO Ebola Response Team. After Ebola in west africa—Unpredictable risks, preventable epidemics. N. Engl. J. Med. 2016, 375, 587–596. [Google Scholar] [CrossRef] [PubMed]
- CDC. 2017 Democratic Republic of the Congo, Bas Uélé District. Available online: https://www.cdc.gov/vhf/ebola/outbreaks/drc/2017-may.html (accessed on 17 July 2018).
- CDC. Years of Ebola Virus Disease Outbreaks. Available online: https://www.cdc.gov/vhf/ebola/history/chronology.html (accessed on 17 July 2018).
- WHO. Ebola Situation Reports: Democratic Republic of the Congo. Available online: http://www.who.int/ebola/situation-reports/drc-2018/en/ (accessed on 10 August 2019).
- Haque, A.; Hober, D.; Blondiaux, J. Addressing therapeutic options for ebola virus infection in current and future outbreaks. Antimicrob. Agents Chemother. 2015, 59, 5892–5902. [Google Scholar] [CrossRef]
- Espeland, E.M.; Tsai, C.W.; Larsen, J.; Disbrow, G.L. Safeguarding against Ebola: Vaccines and therapeutics to be stockpiled for future outbreaks. PLoS Negl. Trop. Dis. 2018, 12, e0006275. [Google Scholar] [CrossRef] [PubMed]
- Timmins, J.; Schoehn, G.; Ricard-Blum, S.; Scianimanico, S.; Vernet, T.; Ruigrok, R.W.H.; Weissenhorn, W. Ebola virus matrix protein VP40 interaction with human cellular factors Tsg101 and Nedd4. J. Mol. Biol. 2003, 326, 493–502. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Peterson, M.; Yang, Z.Y.; Kong, W.P.; Duckers, H.; Nabel, E.; Nabel, G.J. Ebola virus glycoprotein toxicity is mediated by a dynamin-dependent protein-trafficking pathway. J. Virol. 2005, 79, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.P.; Leung, L.W.; Hartman, A.L.; Martinez, O.; Shaw, M.L.; Carbonnelle, C.; Volchkov, V.E.; Nichol, S.T.; Basler, C.F. Ebola virus VP24 binds karyopherin alpha1 and blocks stat1 nuclear accumulation. J. Virol. 2006, 80, 5156–5167. [Google Scholar] [CrossRef]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter niemann-pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Ilinykh, P.A.; Tigabu, B.; Ivanov, A.; Ammosova, T.; Obukhov, Y.; Garron, T.; Kumari, N.; Kovalskyy, D.; Platonov, M.O.; Naumchik, V.S.; et al. Role of protein phosphatase 1 in dephosphorylation of Ebola virus VP30 protein and its targeting for the inhibition of viral transcription. J. Biol. Chem. 2014, 289, 22723–22738. [Google Scholar] [CrossRef]
- Luthra, P.; Jordan, D.S.; Leung, D.W.; Amarasinghe, G.K.; Basler, C.F. Ebola virus VP35 interaction with dynein LC8 regulates viral RNA synthesis. J. Virol. 2015, 89, 5148–5153. [Google Scholar] [CrossRef]
- Biedenkopf, N.; Lier, C.; Becker, S. Dynamic phosphorylation of VP30 is essential for Ebola virus life cycle. J. Virol. 2016, 90, 4914–4925. [Google Scholar] [CrossRef] [PubMed]
- Batra, J.; Hultquist, J.F.; Liu, D.; Shtanko, O.; Von Dollen, J.; Satkamp, L.; Jang, G.M.; Luthra, P.; Schwarz, T.M.; Small, G.I.; et al. Protein interaction mapping identifies RBBP6 as a negative regulator of Ebola virus replication. Cell 2018, 175, 1917–1930.e13. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, L.; Sun, Y.; Nabel, G.J. The assembly of Ebola virus nucleocapsid requires virion-associated proteins 35 and 24 and posttranslational modification of nucleoprotein. Mol. Cell 2002, 10, 307–316. [Google Scholar] [CrossRef]
- Watanabe, S.; Noda, T.; Kawaoka, Y. Functional mapping of the nucleoprotein of ebola virus. J. Virol. 2006, 80, 3743–3751. [Google Scholar] [CrossRef] [PubMed]
- Kirchdoerfer, R.N.; Abelson, D.M.; Li, S.; Wood, M.R.; Saphire, E.O. Assembly of the Ebola virus nucleoprotein from a chaperoned VP35 complex. Cell Rep. 2015, 12, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Kruse, T.; Biedenkopf, N.; Hertz, E.P.T.; Dietzel, E.; Stalmann, G.; Lopez-Mendez, B.; Davey, N.E.; Nilsson, J.; Becker, S. The Ebola virus nucleoprotein recruits the host PP2A-B56 phosphatase to activate transcriptional support activity of VP30. Mol. Cell 2018, 69, 136–145.e6. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dorival, I.; Wu, W.; Armstrong, S.D.; Barr, J.N.; Carroll, M.W.; Hewson, R.; Hiscox, J.A. Elucidation of the cellular interactome of Ebola virus nucleoprotein and identification of therapeutic targets. J. Proteome. Res. 2016, 15, 4290–4303. [Google Scholar] [CrossRef]
- Fang, J.; Pietzsch, C.; Ramanathan, P.; Santos, R.I.; Ilinykh, P.A.; Garcia-Blanco, M.A.; Bukreyev, A.; Bradrick, S.S. Staufen1 interacts with multiple components of the Ebola virus ribonucleoprotein and enhances viral RNA synthesis. MBio 2018, 9, e01771-18. [Google Scholar] [CrossRef] [PubMed]
- Muhlberger, E. Filovirus replication and transcription. Future Virol. 2007, 2, 205–215. [Google Scholar] [CrossRef]
- Nanbo, A.; Watanabe, S.; Halfmann, P.; Kawaoka, Y. The spatio-temporal distribution dynamics of Ebola virus proteins and RNA in infected cells. Sci. Rep. 2013, 3, 1206. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Hagiwara, K.; Sagara, H.; Kawaoka, Y. Characterization of the ebola virus nucleoprotein-RNA complex. J. Gen. Virol. 2010, 91, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Bharat, T.A.; Noda, T.; Riches, J.D.; Kraehling, V.; Kolesnikova, L.; Becker, S.; Kawaoka, Y.; Briggs, J.A. Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc. Natl. Acad. Sci. USA 2012, 109, 4275–4280. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.-K.; Schlee, M.; et al. 5′-triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, R.W.; Crepin, T.; Kolakofsky, D. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr. Opin. Microbiol. 2011, 14, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Spehner, D.; Kirn, A.; Drillien, R. Assembly of nucleocapsidlike structures in animal cells infected with a vaccinia virus recombinant encoding the measles virus nucleoprotein. J. Virol. 1991, 65, 6296–6300. [Google Scholar] [PubMed]
- Errington, W.; Emmerson, P.T. Assembly of recombinant newcastle disease virus nucleocapsid protein into nucleocapsid-like structures is inhibited by the phosphoprotein. J. Gen. Virol. 1997, 78, 2335–2339. [Google Scholar] [CrossRef]
- Iseni, F.; Barge, A.; Baudin, F.; Blondel, D.; Ruigrok, R.W. Characterization of rabies virus nucleocapsids and recombinant nucleocapsid-like structures. J. Gen. Virol. 1998, 79, 2909–2919. [Google Scholar] [CrossRef]
- Bhella, D.; Ralph, A.; Murphy, L.B.; Yeo, R.P. Significant differences in nucleocapsid morphology within the paramyxoviridae. J. Gen. Virol. 2002, 83, 1831–1839. [Google Scholar] [CrossRef]
- Mühlberger, E.; Weik, M.; Volchkov, V.E.; Klenk, H.D.; Becker, S. Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J. Virol. 1999, 73, 2333–2342. [Google Scholar]
- Jahrling, T.W.; Jahrling, P.B. Differentiation of filoviruses by electron microscopy. Virus Res. 1995, 39, 129–150. [Google Scholar]
- Bhattacharyya, S.; Hope, T.J. Full-length ebola glycoprotein accumulates in the endoplasmic reticulum. Virol. J. 2011, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Meng, Z.; Mason, P.J.; Veenstra, T.D.; Artandi, S.E. Identification of atpases pontin and reptin as telomerase components essential for holoenzyme assembly. Cell 2008, 132, 945–957. [Google Scholar] [CrossRef]
- Nelson, E.V.; Pacheco, J.R.; Hume, A.J.; Cressey, T.N.; Deflube, L.R.; Ruedas, J.B.; Connor, J.H.; Ebihara, H.; Muhlberger, E. An RNA polymerase II-driven Ebola virus minigenome system as an advanced tool for antiviral drug screening. Antiviral Res. 2017, 146, 21–27. [Google Scholar] [CrossRef]
- Tsai, W.L.; Cheng, J.S.; Shu, C.W.; Lai, K.H.; Chan, H.H.; Wu, C.C.; Wu, J.M.; Hsu, P.I.; Chung, R.T.; Chang, T.H. Asunaprevir evokes hepatocytes innate immunity to restrict the replication of hepatitis C and dengue virus. Front. Microbiol. 2017, 8, 668. [Google Scholar] [CrossRef]
- Santamaria, E.; Mora, M.I.; Potel, C.; Fernandez-Irigoyen, J.; Carro-Roldan, E.; Hernandez-Alcoceba, R.; Prieto, J.; Epstein, A.L.; Corrales, F.J. Identification of replication-competent HSV-1 Cgal+ strain signaling targets in human hepatoma cells by functional organelle proteomics. Mol. Cell Proteomics 2009, 8, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Fu, Y.; Zhu, Y.; Wang, X.; Xuan, Y.; Shang, H.; Goff, S.P.; Gao, G. HIV-1 exploits the host factor RuvB-like 2 to balance viral protein expression. Cell Host Microbe. 2015, 18, 233–242. [Google Scholar] [CrossRef]
- Olanubi, O.; Frost, J.R.; Radko, S.; Pelka, P. Suppression of type I interferon signaling by E1A via RuvBL1/pontin. J. Virol. 2017, 91, e02484-16. [Google Scholar] [CrossRef]
- Nishitsuji, H.; Ujino, S.; Harada, K.; Shimotohno, K. TIP60 complex inhibits hepatitis B virus transcription. J. Virol. 2018, 92, e01788-17. [Google Scholar] [CrossRef]
- Nano, N.; Houry, W.A. Chaperone-like activity of the AAA+ proteins Rvb1 and Rvb2 in the assembly of various complexes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20110399. [Google Scholar] [CrossRef]
- Matias, P.M.; Baek, S.H.; Bandeiras, T.M.; Dutta, A.; Houry, W.A.; Llorca, O.; Rosenbaum, J. The AAA+ proteins pontin and reptin enter adult age: From understanding their basic biology to the identification of selective inhibitors. Front. Mol. Biosci. 2015, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Matias, P.M.; Gorynia, S.; Donner, P.; Carrondo, M.A. Crystal structure of the human AAA+ protein RuvBL1. J. Biol. Chem. 2006, 281, 38918–38929. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, H.J.; Kim, B.; Kim, M.H.; Lee, J.M.; Kim, I.S.; Lee, M.H.; Choi, S.J.; Kim, K.I.; Kim, S.I.; et al. Roles of sumoylation of a reptin chromatin-remodelling complex in cancer metastasis. Nat. Cell. Biol. 2006, 8, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Saeki, M.; Xu, L.; Nakahara, H.; Saijo, M.; Tanaka, K.; Kamisaki, Y. RPAP3 interacts with reptin to regulate UV-induced phosphorylation of H2AX and DNA damage. J. Cell. Biochem. 2009, 106, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Gentili, C.; Castor, D.; Kaden, S.; Lauterbach, D.; Gysi, M.; Steigemann, P.; Gerlich, D.W.; Jiricny, J.; Ferrari, S. Chromosome missegregation associated with RuvBL1 deficiency. PLoS ONE 2015, 10, e0133576. [Google Scholar] [CrossRef] [PubMed]
- Izumi, N.; Yamashita, A.; Iwamatsu, A.; Kurata, R.; Nakamura, H.; Saari, B.; Hirano, H.; Anderson, P.; Ohno, S. AAA+ proteins RUVBL1 and RUVBL2 coordinate PIKK activity and function in nonsense-mediated mRNA decay. Sci. Signal. 2010, 3, ra27. [Google Scholar] [CrossRef] [PubMed]
- Haurie, V.; Menard, L.; Nicou, A.; Touriol, C.; Metzler, P.; Fernandez, J.; Taras, D.; Lestienne, P.; Balabaud, C.; Bioulac-Sage, P.; et al. Adenosine triphosphatase pontin is overexpressed in hepatocellular carcinoma and coregulated with reptin through a new posttranslational mechanism. Hepatology 2009, 50, 1871–1883. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, B.; Cai, L.; Choi, H.J.; Ohgi, K.A.; Tran, C.; Chen, C.; Chung, C.H.; Huber, O.; Rose, D.W.; et al. Transcriptional regulation of a metastasis suppressor gene by Tip60 and beta-catenin complexes. Nature 2005, 434, 921–926. [Google Scholar] [CrossRef]
- Diop, S.B.; Bertaux, K.; Vasanthi, D.; Sarkeshik, A.; Goirand, B.; Aragnol, D.; Tolwinski, N.S.; Cole, M.D.; Pradel, J.; Yates, J.R., 3rd; et al. Reptin and pontin function antagonistically with PcG and TrxG complexes to mediate Hox gene control. EMBO Rep. 2008, 9, 260–266. [Google Scholar] [CrossRef]
- Izumi, N.; Yamashita, A.; Ohno, S. Integrated regulation of PIKK-mediated stress responses by AAA+ proteins RUVBL1 and RUVBL2. Nucleus 2012, 3, 29–43. [Google Scholar] [CrossRef]
- Huber, O.; Menard, L.; Haurie, V.; Nicou, A.; Taras, D.; Rosenbaum, J. Pontin and reptin, two related atpases with multiple roles in cancer. Cancer Res. 2008, 68, 6873–6876. [Google Scholar] [CrossRef] [PubMed]
- Takai, H.; Xie, Y.; de Lange, T.; Pavletich, N.P. Tel2 structure and function in the Hsp90-dependent maturation of mTOR and ATR complexes. Genes Dev. 2010, 24, 2019–2030. [Google Scholar] [CrossRef] [PubMed]
- Horejsi, Z.; Takai, H.; Adelman, C.A.; Collis, S.J.; Flynn, H.; Maslen, S.; Skehel, J.M.; de Lange, T.; Boulton, S.J. CK2 phospho-dependent binding of R2TP complex to TEL2 is essential for mTOR and SMG1 stability. Mol. Cell 2010, 39, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Boulon, S.; Pradet-Balade, B.; Verheggen, C.; Molle, D.; Boireau, S.; Georgieva, M.; Azzag, K.; Robert, M.C.; Ahmad, Y.; Neel, H.; et al. HSP90 and its R2TP/Prefoldin-like cochaperone are involved in the cytoplasmic assembly of RNA polymerase II. Mol. Cell 2010, 39, 912–924. [Google Scholar] [CrossRef] [PubMed]
- Forget, D.; Lacombe, A.A.; Cloutier, P.; Al-Khoury, R.; Bouchard, A.; Lavallee-Adam, M.; Faubert, D.; Jeronimo, C.; Blanchette, M.; Coulombe, B. The protein interaction network of the human transcription machinery reveals a role for the conserved GTPase RPAP4/GPN1 and microtubule assembly in nuclear import and biogenesis of RNA polymerase II. Mol. Cell Proteomics 2010, 9, 2827–2839. [Google Scholar] [CrossRef]
- Boulon, S.; Marmier-Gourrier, N.; Pradet-Balade, B.; Wurth, L.; Verheggen, C.; Jady, B.E.; Rothe, B.; Pescia, C.; Robert, M.C.; Kiss, T.; et al. The Hsp90 chaperone controls the biogenesis of L7Ae RNPs through conserved machinery. J. Cell Biol. 2008, 180, 579–595. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Kakihara, Y.; Gribun, A.; Huen, J.; Yang, G.; Khanna, M.; Costanzo, M.; Brost, R.L.; Boone, C.; Hughes, T.R.; et al. Molecular chaperone Hsp90 stabilizes Pih1/Nop17 to maintain R2TP complex activity that regulates snoRNA accumulation. J. Cell Biol. 2008, 180, 563–578. [Google Scholar] [CrossRef]
- Smith, D.R.; McCarthy, S.; Chrovian, A.; Olinger, G.; Stossel, A.; Geisbert, T.W.; Hensley, L.E.; Connor, J.H. Inhibition of heat-shock protein 90 reduces Ebola virus replication. Antiviral Res. 2010, 87, 187–194. [Google Scholar] [CrossRef]
- Cloutier, P.; Poitras, C.; Durand, M.; Hekmat, O.; Fiola-Masson, E.; Bouchard, A.; Faubert, D.; Chabot, B.; Coulombe, B. R2TP/Prefoldin-like component RUVBL1/RUVBL2 directly interacts with ZNHIT2 to regulate assembly of U5 small nuclear ribonucleoprotein. Nat. Commun. 2017, 8, 15615. [Google Scholar] [CrossRef]
- Knowlton, J.J.; Fernández de Castro, I.; Ashbrook, A.W.; Gestaut, D.R.; Zamora, P.F.; Bauer, J.A.; Forrest, J.C.; Frydman, J.; Risco, C.; Dermody, T.S. The TRiC chaperonin controls reovirus replication through outer-capsid folding. Nat. Microbiol. 2018, 3, 481–493. [Google Scholar] [CrossRef]
- Yoshida, M.; Saeki, M.; Egusa, H.; Irie, Y.; Kamano, Y.; Uraguchi, S.; Sotozono, M.; Niwa, H.; Kamisaki, Y. RPAP3 splicing variant isoform 1 interacts with PIH1D1 to compose R2TP complex for cell survival. Biochem. Biophys. Res. Commun. 2013, 430, 320–324. [Google Scholar] [CrossRef]
- King, B.R.; Hershkowitz, D.; Eisenhauer, P.L.; Weir, M.E.; Ziegler, C.M.; Russo, J.; Bruce, E.A.; Ballif, B.A.; Botten, J. A Map of the Arenavirus Nucleoprotein-Host Protein Interactome Reveals that Junín Virus Selectively Impairs the Antiviral Activity of Double-Stranded RNA-Activated Protein Kinase (PKR). J. Virol. 2017, 91, e00763-17. [Google Scholar] [CrossRef]
- Iwasaki, M.; Minder, P.; Cai, Y.; Kuhn, J.H.; Yates, J.R., 3rd; Torbett, B.E.; de la Torre, J.C. Interactome analysis of the lymphocytic choriomeningitis virus nucleoprotein in infected cells reveals ATPase Na+/K+ transporting subunit Alpha 1 and prohibitin as host-cell factors involved in the life cycle of mammarenaviruses. PLoS Pathog. 2018, 14, e1006892. [Google Scholar] [CrossRef]
- Hoenen, T.; Shabman, R.S.; Groseth, A.; Herwig, A.; Weber, M.; Schudt, G.; Dolnik, O.; Basler, C.F.; Becker, S.; Feldmann, H. Inclusion bodies are a site of Ebolavirus replication. J. Virol. 2012, 86, 11779–11788. [Google Scholar] [CrossRef]
- Dolnik, O.; Stevermann, L.; Kolesnikova, L.; Becker, S. Marburg virus inclusions: A virus-induced microcompartment and interface to multivesicular bodies and the late endosomal compartment. Eur. J. Cell Biol. 2015, 94, 323–331. [Google Scholar] [CrossRef]
- Basler, C.F.; Krogan, N.J.; Leung, D.W.; Amarasinghe, G.K. Virus and host interactions critical for filoviral RNA synthesis as therapeutic targets. Antiviral Res. 2019, 162, 90–100. [Google Scholar] [CrossRef]
- Trunschke, M.; Conrad, D.; Enterlein, S.; Olejnik, J.; Brauburger, K.; Muhlberger, E. The L-VP35 and L-L interaction domains reside in the amino terminus of the Ebola virus L protein and are potential targets for antivirals. Virology 2013, 441, 135–145. [Google Scholar] [CrossRef]
- Rivera-Calzada, A.; Pal, M.; Munoz-Hernandez, H.; Luque-Ortega, J.R.; Gil-Carton, D.; Degliesposti, G.; Skehel, J.M.; Prodromou, C.; Pearl, L.H.; Llorca, O. The Structure of the R2TP Complex Defines a Platform for Recruiting Diverse Client Proteins to the HSP90 Molecular Chaperone System. Structure 2017, 25, 1145–1152.e4. [Google Scholar] [CrossRef]
- Kakihara, Y.; Houry, W.A. The R2TP complex: Discovery and functions. Biochim. Biophys. Acta. 2012, 1823, 101–107. [Google Scholar] [CrossRef]
- Connor, J.H.; McKenzie, M.O.; Parks, G.D.; Lyles, D.S. Antiviral activity and RNA polymerase degradation following Hsp90 inhibition in a range of negative strand viruses. Virology 2007, 362, 109–119. [Google Scholar] [CrossRef]
- Kakugawa, S.; Shimojima, M.; Neumann, G.; Goto, H.; Kawaoka, Y. RuvB-like protein 2 is a suppressor of influenza A virus polymerases. J. Virol. 2009, 83, 6429–6434. [Google Scholar] [CrossRef]
- Yasunaga, A.; Hanna, S.L.; Li, J.; Cho, H.; Rose, P.P.; Spiridigliozzi, A.; Gold, B.; Diamond, M.S.; Cherry, S. Genome-wide RNAi screen identifies broadly-acting host factors that inhibit arbovirus infection. PLoS Pathog. 2014, 10, e1003914. [Google Scholar] [CrossRef]
- Ikura, T.; Ogryzko, V.V.; Grigoriev, M.; Groisman, R.; Wang, J.; Horikoshi, M.; Scully, R.; Qin, J.; Nakatani, Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 2000, 102, 463–473. [Google Scholar] [CrossRef]
- Puri, T.; Wendler, P.; Sigala, B.; Saibil, H.; Tsaneva, I.R. Dodecameric structure and ATPase activity of the human TIP48/TIP49 complex. J. Mol. Biol. 2007, 366, 179–192. [Google Scholar] [CrossRef]
- Assimon, V.A.; Tang, Y.; Vargas, J.D.; Lee, G.J.; Wu, Z.Y.; Lou, K.; Yao, B.; Menon, M.K.; Pios, A.; Perez, K.C.; et al. CB-6644 Is a Selective Inhibitor of the RUVBL1/2 Complex with Anticancer Activity. ACS Chem. Biol. 2019, 14, 236–244. [Google Scholar] [CrossRef]
- Horejsi, Z.; Stach, L.; Flower, T.G.; Joshi, D.; Flynn, H.; Skehel, J.M.; O’Reilly, N.J.; Ogrodowicz, R.W.; Smerdon, S.J.; Boulton, S.J. Phosphorylation-dependent PIH1D1 interactions define substrate specificity of the R2TP cochaperone complex. Cell Rep. 2014, 7, 19–26. [Google Scholar] [CrossRef]
- Shi, W.; Huang, Y.; Sutton-Smith, M.; Tissot, B.; Panico, M.; Morris, H.R.; Dell, A.; Haslam, S.M.; Boyington, J.; Graham, B.S.; et al. A filovirus-unique region of Ebola virus nucleoprotein confers aberrant migration and mediates its incorporation into virions. J. Virol. 2008, 82, 6190–6199. [Google Scholar] [CrossRef]
- Baker, L.E.; Ellena, J.F.; Handing, K.B.; Derewenda, U.; Utepbergenov, D.; Engel, D.A.; Derewenda, Z.S. Molecular architecture of the nucleoprotein C-terminal domain from the Ebola and Marburg viruses. Acta Crystallogr. D Struct. Biol. 2016, 72, 49–58. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morwitzer, M.J.; Tritsch, S.R.; Cazares, L.H.; Ward, M.D.; Nuss, J.E.; Bavari, S.; Reid, S.P. Identification of RUVBL1 and RUVBL2 as Novel Cellular Interactors of the Ebola Virus Nucleoprotein. Viruses 2019, 11, 372. https://doi.org/10.3390/v11040372
Morwitzer MJ, Tritsch SR, Cazares LH, Ward MD, Nuss JE, Bavari S, Reid SP. Identification of RUVBL1 and RUVBL2 as Novel Cellular Interactors of the Ebola Virus Nucleoprotein. Viruses. 2019; 11(4):372. https://doi.org/10.3390/v11040372
Chicago/Turabian StyleMorwitzer, M. Jane, Sarah R. Tritsch, Lisa H. Cazares, Michael D. Ward, Jonathan E. Nuss, Sina Bavari, and St Patrick Reid. 2019. "Identification of RUVBL1 and RUVBL2 as Novel Cellular Interactors of the Ebola Virus Nucleoprotein" Viruses 11, no. 4: 372. https://doi.org/10.3390/v11040372
APA StyleMorwitzer, M. J., Tritsch, S. R., Cazares, L. H., Ward, M. D., Nuss, J. E., Bavari, S., & Reid, S. P. (2019). Identification of RUVBL1 and RUVBL2 as Novel Cellular Interactors of the Ebola Virus Nucleoprotein. Viruses, 11(4), 372. https://doi.org/10.3390/v11040372