A Novel Orthohepadnavirus Identified in a Dead Maxwell’s Duiker (Philantomba maxwellii) in Taï National Park, Côte d’Ivoire

 ,
,
Abstract
1. Introduction
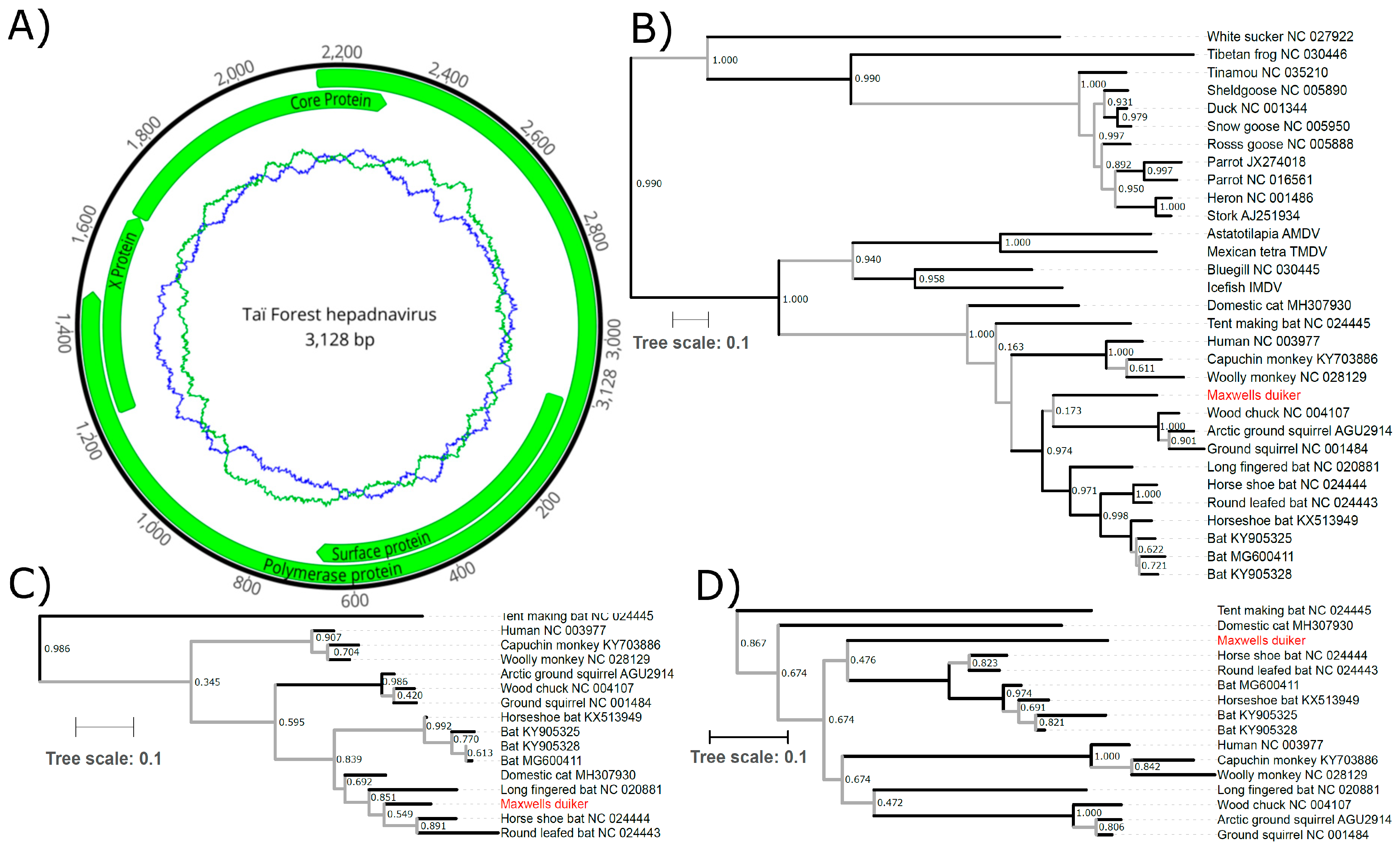
2. Methods and Results
3. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lanford, R.E.; Chavez, D.; Brasky, K.M.; Burns, R.B.; Rico-Hesse, R. Isolation of a hepadnavirus from the woolly monkey, a New World primate. Proc. Natl. Acad. Sci. USA 1998, 95, 5757–5761. [Google Scholar] [CrossRef]
- Souza, B.F.d.C.D.; König, A.; Rasche, A.; de Oliveira Carneiro, I.; Stephan, N.; Corman, V.M.; Roppert, P.L.; Goldmann, N.; Kepper, R.; Müller, S.F. A novel hepatitis B virus species discovered in capuchin monkeys sheds new light on the evolution of primate hepadnaviruses. J. Hepatol. 2018, 68, 1114–1122. [Google Scholar]
- Grethe, S.; Heckel, J.O.; Rietschel, W.; Hufert, F.T. Molecular epidemiology of hepatitis B virus variants in nonhuman primates. J. Virol. 2000, 74, 5377–5381. [Google Scholar] [CrossRef] [PubMed]
- Starkman, S.; MacDonald, D.; Lewis, J.; Holmes, E.; Simmonds, P. Geographic and species association of hepatitis B virus genotypes in non-human primates. Virology 2003, 314, 381–393. [Google Scholar] [CrossRef]
- He, B.; Zhang, F.; Xia, L.; Hu, T.; Chen, G.; Qiu, W.; Fan, Q.; Feng, Y.; Guo, H.; Tu, C. Identification of a novel Orthohepadnavirus in pomona roundleaf bats in China. Arch. Virol. 2015, 160, 335–337. [Google Scholar] [CrossRef]
- He, B.; Fan, Q.; Yang, F.; Hu, T.; Qiu, W.; Feng, Y.; Li, Z.; Li, Y.; Zhang, F.; Guo, H. Hepatitis virus in long-fingered bats, Myanmar. Emerg. Infect. Dis. 2013, 19, 638. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, X.L.; Li, W.; Zhu, Y.; Ge, X.Y.; Zhang, L.B.; Zhang, Y.Z.; Bock, C.T.; Shi, Z.L. Detection and genome characterization of four novel bat hepadnaviruses and a hepevirus in China. Virol. J. 2017, 14, 40. [Google Scholar] [CrossRef] [PubMed]
- Nie, F.Y.; Lin, X.D.; Hao, Z.Y.; Chen, X.N.; Wang, Z.X.; Wang, M.R.; Wu, J.; Wang, H.W.; Zhao, G.; Ma, R.Z. Extensive diversity and evolution of hepadnaviruses in bats in China. Virology 2018, 514, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Summers, J.; Smolec, J.M.; Snyder, R. A virus similar to human hepatitis B virus associated with hepatitis and hepatoma in woodchucks. Proc. Natl. Acad. Sci. USA 1978, 75, 4533–4537. [Google Scholar] [CrossRef]
- Testut, P.; Renard, C.A.; Terradillos, O.; Vitvitski-Trepo, L.; Tekaia, F.; Degott, C.; Blake, J.; Boyer, B.; Buendia, M.A. A new hepadnavirus endemic in arctic ground squirrels in Alaska. J. Virol. 1996, 70, 4210–4219. [Google Scholar]
- Seeger, C.; Ganem, D.; Varmus, H.E. Nucleotide sequence of an infectious molecularly cloned genome of ground squirrel hepatitis virus. J. Virol. 1984, 51, 367–375. [Google Scholar]
- Marion, P.L.; Oshiro, L.S.; Regnery, D.C.; Scullard, G.H.; Robinson, W.S. A virus in Beechey ground squirrels that is related to hepatitis B virus of humans. Proc. Natl. Acad. Sci. USA 1980, 77, 2941–2945. [Google Scholar] [CrossRef]
- Aghazadeh, M.; Shi, M.; Barrs, V.R.; McLuckie, A.J.; Lindsay, S.A.; Jameson, B.; Hampson, B.; Holmes, E.C.; Beatty, J.A. A Novel Hepadnavirus Identified in an Immunocompromised Domestic Cat in Australia. Viruses 2018, 10, 269. [Google Scholar] [CrossRef]
- World Health Organization. Disease Burden and Mortality Estimates: Cause-Specific Mortality, 2000–2016; World Health Organization: Geneva, Switzerland; Available online: https://www.who.int/healthinfo/global_burden_disease/estimates/en/ (accessed on 15 January 2019).
- Drexler, J.F.; Geipel, A.; König, A.; Corman, V.M.; van Riel, D.; Leijten, L.M.; Bremer, C.M.; Rasche, A.; Cottontail, V.M.; Maganga, G.D. Bats carry pathogenic hepadnaviruses antigenically related to hepatitis B virus and capable of infecting human hepatocytes. Proc. Natl. Acad. Sci. USA 2013, 110, 16151–16156. [Google Scholar] [CrossRef]
- Takahashi, K.; Brotman, B.; Usuda, S.; Mishiro, S.; Prince, A.M. Full-genome sequence analyses of hepatitis B virus (HBV) strains recovered from chimpanzees infected in the wild: Implications for an origin of HBV. Virology 2000, 267, 58–64. [Google Scholar] [CrossRef]
- Littlejohn, M.; Locarnini, S.; Yuen, L. Origins and evolution of hepatitis B virus and hepatitis D virus. Cold Spring Harb. Perspect. Med. 2016, 6, a021360. [Google Scholar] [CrossRef]
- Gogarten, J.F.; Akoua-Koffi, C.; Calvignac-Spencer, S.; Leendertz, S.A.J.; Weiss, S.; Couacy-Hymann, E.; Koné, I.; Peeters, M.; Wittig, R.M.; Boesch, C. The ecology of primate retroviruses–an assessment of 12 years of retroviral studies in the Taï national park area, Côte d׳ Ivoire. Virology 2014, 460, 147–153. [Google Scholar] [CrossRef]
- Grützmacher, K.S.; Köndgen, S.; Keil, V.; Todd, A.; Feistner, A.; Herbinger, I.; Petrzelkova, K.; Fuh, T.; Leendertz, S.A.; Calvignac-Spencer, S. Codetection of respiratory syncytial virus in habituated wild western lowland gorillas and humans during a respiratory disease outbreak. EcoHealth 2016, 13, 499–510. [Google Scholar] [CrossRef]
- Hoffmann, C.; Zimmermann, F.; Biek, R.; Kuehl, H.; Nowak, K.; Mundry, R.; Agbor, A.; Angedakin, S.; Arandjelovic, M.; Blankenburg, A.; et al. Persistent anthrax as a major driver of wildlife mortality in a tropical rainforest. Nature 2017, 548, 82. [Google Scholar] [CrossRef]
- Radonić, A.; Metzger, S.; Dabrowski, P.W.; Couacy-Hymann, E.; Schuenadel, L.; Kurth, A.; Mätz-Rensing, K.; Boesch, C.; Leendertz, F.H.; Nitsche, A. Fatal monkeypox in wild-living sooty mangabey, Cote d’Ivoire, 2012. Emerg. Infect. Dis. 2014, 20, 1009. [Google Scholar] [CrossRef]
- Schroeder, K.; Nitsche, A. Multicolour, multiplex real-time PCR assay for the detection of human-pathogenic poxviruses. Mol. Cell. Probes 2010, 24, 110–113. [Google Scholar] [CrossRef]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkin, W.I. Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. MBio 2015, 6, e01491–e041515. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, 2013; arXiv:1303.3997. [Google Scholar]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2009, 27, 221–224. [Google Scholar] [CrossRef]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart model selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Schaefer, S.; Glebe, D.; Wend, U.C.; Oyunbileg, J.; Gerlich, W.H. Universal primers for real-time amplification of DNA from all known Orthohepadnavirus species. J. Clin. Virol. 2003, 27, 30–37. [Google Scholar] [CrossRef]
- Mommeja-Marin, H.; Mondou, E.; Blum, M.R.; Rousseau, F. Serum HBV DNA as a marker of efficacy during therapy for chronic HBV infection: Analysis and review of the literature. Hepatology 2003, 37, 1309–1319. [Google Scholar] [CrossRef]
- Le Guenno, B.; Formenty, P.; Wyers, M.; Gounon, P.; Walker, F.; Boesch, C. Isolation and partial characterisation of a new strain of Ebola virus. Lancet 1995, 345, 1271–1274. [Google Scholar] [CrossRef]
- Korba, B.E.; Wells, F.; Tennant, B.; Cote, P.; Gerin, J. Lymphoid cells in the spleens of woodchuck hepatitis virus-infected woodchucks are a site of active viral replication. J. Virol. 1987, 61, 1318–1324. [Google Scholar]
- Michalak, T.I.; Hodgson, P.D.; Churchill, N.D. Posttranscriptional inhibition of class I major histocompatibility complex presentation on hepatocytes and lymphoid cells in chronic woodchuck hepatitis virus infection. J. Virol. 2000, 74, 4483–4494. [Google Scholar] [CrossRef]
- Rasche, A.; Souza, B.F.D.C.D.; Drexler, J.F. Bat hepadnaviruses and the origins of primate hepatitis B viruses. Curr. Opin. Virol. 2016, 16, 86–94. [Google Scholar] [CrossRef]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Börold, J.; Salzburger, W.; Kaderali, L.; Briggs, J.A. Deciphering the origin and evolution of hepatitis B viruses by means of a family of non-enveloped fish viruses. Cell Host Microbe 2017, 22, 387–399. e6. [Google Scholar] [CrossRef]
- Dill, J.A.; Camus, A.C.; Leary, J.H.; Di Giallonardo, F.; Holmes, E.C.; Ng, T.F.F. Distinct viral lineages from fish and amphibians reveal the complex evolutionary history of hepadnaviruses. J. Virol. 2016, 90, 7920–7933. [Google Scholar] [CrossRef]
- Leroy, E.M.; Rouquet, P.; Formenty, P.; Souquière, S.; Kilbourne, A.; Froment, J.M.; Bermejo, M.; Smit, S.; Karesh, W.; Swanepoel, R. Multiple Ebola virus transmission events and rapid decline of central African wildlife. Science 2004, 303, 387–390. [Google Scholar] [CrossRef]
- Rouquet, P.; Froment, J.M.; Bermejo, M.; Kilbourn, A.; Karesh, W.; Reed, P.; Kumulungui, B.; Yaba, P.; Délicat, A.; Rollin, P.E. Wild animal mortality monitoring and human Ebola outbreaks, Gabon and Republic of Congo, 2001–2003. Emerg. Infect. Dis. 2005, 11, 283. [Google Scholar] [CrossRef]
- Zimmermann, F.; Köhler, S.M.; Nowak, K.; Dupke, S.; Barduhn, A.; Düx, A.; Lang, A.; De Nys, H.M.; Gogarten, J.F.; Grunow, R. Low antibody prevalence against Bacillus cereus biovar anthracis in Taï National Park, Côte d’Ivoire, indicates high rate of lethal infections in wildlife. PLoS Negl. Trop. Dis. 2017, 11, e0005960. [Google Scholar] [CrossRef]


{kind=link}
| Tissue | Total Reads | Reads after Filtering | Reads after Host Subtraction | Reads with Top Blast Hit to a Orthohepadnavirus |
|---|---|---|---|---|
| Liver | 10,548,728 | 10,111,516 | 9,105,066 | 2,475,043 |
| Duodenum | 9,776,183 | 9,082,857 | 6,249,455 | 964 |
| Primer Name | Sequence (5’ -> 3’) | Annealing Temperature (°C) |
|---|---|---|
| Orthohep_1F | TGGTGGACTTCTCTCAGTTTTCC | 56 |
| Orthohep_1R | TGATAAAACGCCGCAGACAC | |
| Orthohep_1F | TGGTGGACTTCTCTCAGTTTTCC | 57 |
| Orthohep_1Rb | AGATGAGGCATAGAACCAGGA | |
| Orthohep_2F | TGCTTAGCCATCCCTTCGTCA | 59 |
| Orthohep_2R | GGCCCCCAGTACCACATCAT | |
| Orthohep_4F | CACAGGTGAAGCGAAGGACA | 58 |
| Orthohep_4R | CCCCAAWACCAVATCATCCATATA |
| Tissue | Extract Conc. (ng/µL) | Duiker Genomes/µL | Viral Copies/µL | Viral Copies/Duiker Genome |
|---|---|---|---|---|
| Heart blood | 5.9 | 801.4 | 4019.4 | 5.0 |
| Heart | 3.9 | 520.3 | 831.4 | 1.6 |
| Liver | 7.8 | 1059.5 | 3205.0 | 3.0 |
| Spleen | 2.2 | 290.5 | 50,203.6 | 172.8 |
| Lung | 1.8 | 243.2 | 4903.6 | 20.2 |
| Kidney | 9.1 | 1232.4 | 283.8 | 0.2 |
| Duodenum | 17.7 | 2391.9 | 939.1 | 0.4 |
| Jejunum | 12.0 | 1621.6 | 0.0 | 0.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gogarten, J.F.; Ulrich, M.; Bhuva, N.; Garcia, J.; Jain, K.; Lee, B.; Löhrich, T.; Oleynik, A.; Couacy-Hymann, E.; Fuh Neba, T.; et al. A Novel Orthohepadnavirus Identified in a Dead Maxwell’s Duiker (Philantomba maxwellii) in Taï National Park, Côte d’Ivoire. Viruses 2019, 11, 279. https://doi.org/10.3390/v11030279
Gogarten JF, Ulrich M, Bhuva N, Garcia J, Jain K, Lee B, Löhrich T, Oleynik A, Couacy-Hymann E, Fuh Neba T, et al. A Novel Orthohepadnavirus Identified in a Dead Maxwell’s Duiker (Philantomba maxwellii) in Taï National Park, Côte d’Ivoire. Viruses. 2019; 11(3):279. https://doi.org/10.3390/v11030279
Chicago/Turabian StyleGogarten, Jan F., Markus Ulrich, Nishit Bhuva, Joel Garcia, Komal Jain, Bohyun Lee, Therese Löhrich, Alexandra Oleynik, Emmanuel Couacy-Hymann, Terence Fuh Neba, and et al. 2019. "A Novel Orthohepadnavirus Identified in a Dead Maxwell’s Duiker (Philantomba maxwellii) in Taï National Park, Côte d’Ivoire" Viruses 11, no. 3: 279. https://doi.org/10.3390/v11030279
APA StyleGogarten, J. F., Ulrich, M., Bhuva, N., Garcia, J., Jain, K., Lee, B., Löhrich, T., Oleynik, A., Couacy-Hymann, E., Fuh Neba, T., Mishra, N., Briese, T., Calvignac-Spencer, S., Lipkin, W. I., & Leendertz, F. H. (2019). A Novel Orthohepadnavirus Identified in a Dead Maxwell’s Duiker (Philantomba maxwellii) in Taï National Park, Côte d’Ivoire. Viruses, 11(3), 279. https://doi.org/10.3390/v11030279