Development and Validation of a Novel Dual Luciferase Reporter Gene Assay to Quantify Ebola Virus VP24 Inhibition of IFN Signaling

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Construction of EBOV VP24 Mammalian Expression Plasmid
2.3. Luciferase Reporter Gene Assay
2.4. EBOV VP24 Inhibition Assay
2.5. Immunoblot Analysis
2.6. Data Analysis
3. Results
3.1. Establishment of a Miniaturized Cell-Based Assay for Evaluating VP24 Inhibition of JAK/STAT Cascade Activation
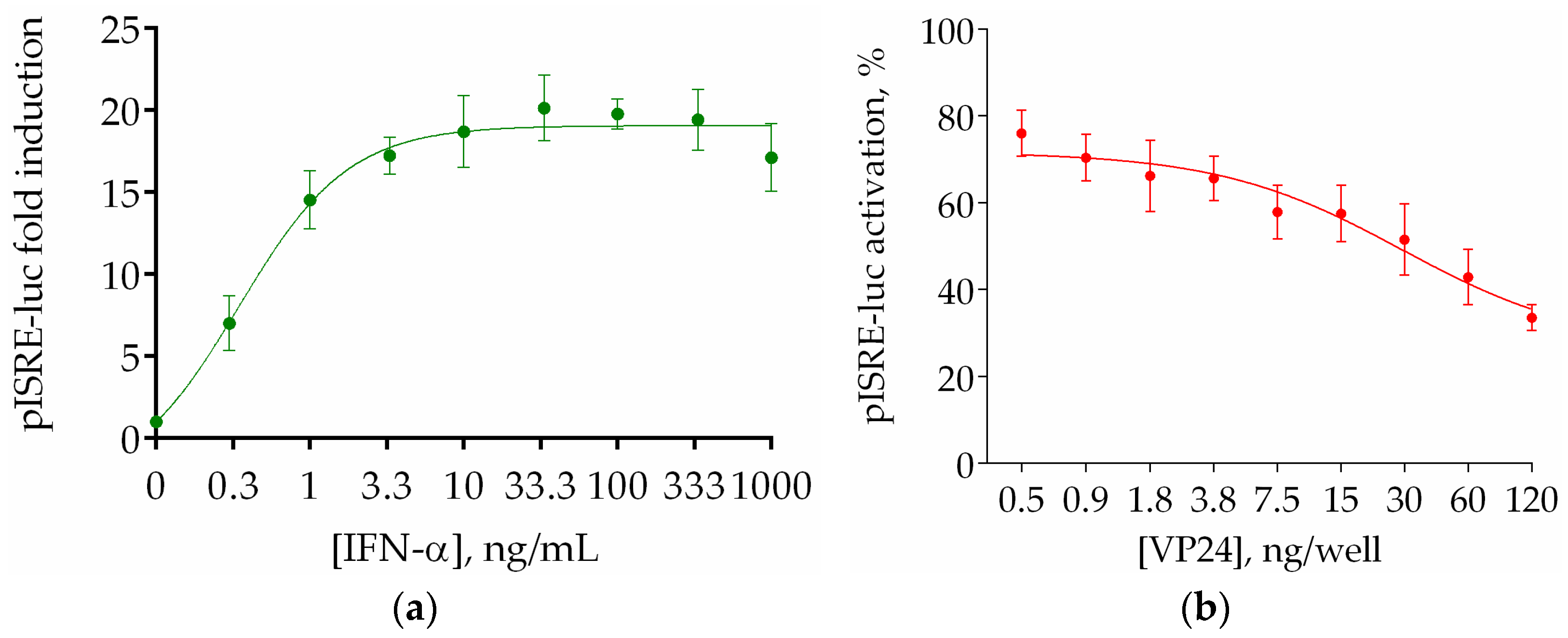
3.2. Optimization of Stimulation Conditions and Evaluation of VP24 IC50
3.3. Luciferase Assay Signal Stability
3.4. Assay Validation
3.5. Designing the Drug Screening Assay
3.6. IFN-α Partially Reverts VP24 Inhibition of JAK/STAT Cascade
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization Ebola Situation Report. Available online: http://apps.who.int/iris/bitstream/10665/208883/1/ebolasitrep_10Jun2016_eng.pdf?ua=1 (accessed on 20 December 2016).
- Mahanty, S.; Bray, M. Pathogenesis of filoviral haemorrhagic fevers. Lancet Infect. Dis. 2004, 4, 487–498. [Google Scholar] [CrossRef]
- Kash, J.C.; Mühlberger, E.; Carter, V.; Grosch, M.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Weber, F.; Klenk, H.-D.; Katze, M.G. Global Suppression of the Host Antiviral Response by Ebola-and Marburgviruses: Increased Antagonism of the Type I Interferon Response Is Associated with Enhanced Virulence. J. Virol. 2006, 80, 3009–3020. [Google Scholar] [CrossRef] [PubMed]
- Prins, K.C.; Delpeut, S.; Leung, D.W.; Reynard, O.; Volchkova, V.A.; Reid, S.P.; Ramanan, P.; Cárdenas, W.B.; Amarasinghe, G.K.; Volchkov, V.E.; et al. Mutations Abrogating VP35 Interaction with Double-Stranded RNA Render Ebola Virus Avirulent in Guinea Pigs. J. Virol. 2010, 84, 3004–3015. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H.H.; Schneider, W.M.; Rice, C.M. Interferons and viruses: An evolutionary arms race of molecular interactions. Trends Immunol. 2015, 36, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, W.B.; Loo, Y.-M.; Gale, M.; Hartman, A.L.; Kimberlin, C.R.; Martínez-Sobrido, L.; Saphire, E.O.; Basler, C.F. Ebola virus VP35 protein binds double-stranded RNA and inhibits α/β interferon production induced by RIG-I signaling. J. Virol. 2006, 80, 5168–5178. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, P.; Shabman, R.S.; Brown, C.S.; Amarasinghe, G.K.; Basler, C.F.; Leung, D.W.; Amarasinghe, G.K. Filoviral immune evasion mechanisms. Viruses 2011, 3, 1634–1649. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F.; Mikulasova, A.; Martinez-Sobrido, L.; Paragas, J.; Mühlberger, E.; Bray, M.; Klenk, H.-D.; Palese, P.; García-Sastre, A. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 2003, 77, 7945–7956. [Google Scholar] [CrossRef] [PubMed]
- Prins, K.C.; Ca, W.B.; Basler, C.F. Ebola Virus Protein VP35 Impairs the Function of Interferon Regulatory Factor-Activating Kinases IKK ε and TBK-1. J. Virol. 2009, 83, 3069–3077. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.R.; Liu, G.; Mire, C.E.; Sureshchandra, S.; Luthra, P.; Amarasinghe, G.K.; Basler, C.F. HHS Public Access. J. Virol. 2016, 14, 1632–1640. [Google Scholar]
- Yen, B.C.; Basler, C.F. Effects of Filovirus Interferon Antagonists on Responses of Human Monocyte-Derived Dendritic Cells to RNA Virus Infection. J. Virol. 2016, 90, 5108–5118. [Google Scholar] [CrossRef] [PubMed]
- Luthra, P.; Ramanan, P.; Mire, C.E.; Weisend, C.; Tsuda, Y.; Yen, B.; Liu, G.; Leung, D.W.; Geisbert, T.W.; Ebihara, H.; et al. Mutual Antagonism between the Ebola Virus VP35 Protein and the RIG-I Activator PACT Determines Infection Outcome. Cell Host Microbe 2013, 14, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Zinzula, L.; Tramontano, E. Strategies of highly pathogenic RNA viruses to block dsRNA detection by RIG-I-like receptors: Hide, mask, hit. Antivir. Res. 2013, 100, 615–635. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.P.; Leung, L.W.; Hartman, A.L.; Martinez, O.; Shaw, M.L.; Carbonnelle, C.; Volchkov, V.E.; Nichol, S.T.; Basler, C.F.D. Ebola virus VP24 binds kaiyopherin α1 and blocks STAT1 nuclear accumulation. J. Virol. 2006, 80, 5156–5167. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.P.; Valmas, C.; Martinez, O.; Mauricio Sanchez, F.; Basler, C.F. Ebola Virus VP24 Proteins Inhibit the Interaction of NPI-1 Subfamily Karyopherin α Proteins with Activated STAT1. J. Virol. 2007, 81, 13469–13477. [Google Scholar] [CrossRef] [PubMed]
- Mateo, M.; Reid, S.P.; Leung, L.W.; Basler, C.F.; Volchkov, V.E. Ebolavirus VP24 binding to karyopherins is required for inhibition of interferon signaling. J. Virol. 2010, 84, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.P.P.; Bornholdt, Z.A.; Liu, T.; Abelson, D.M.; Lee, D.E.; Li, S.; Woods, V.L.; Saphire, E.O. The ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold. PLoS Pathog. 2012, 8, e1002550. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, H.; Takada, A.; Kobasa, D.; Jones, S.; Neumann, G.; Theriault, S. Molecular Determinants of Ebola Virus Virulence in Mice. PLoS Pathog. 2006, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Sebaugh, J.L. Guidelines for accurate EC50/IC50 estimation. Pharm. Stat. 2011, 10, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.Y.; Oldenburg, K.R. A simple statistical Parameter for use in Evaluation and Validation of High Throughput Screening Assay. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Subba-Reddy, C.V.; Goodfellow, I.; Kao, C.C. VPg-Primed RNA Synthesis of Norovirus RNA-Dependent RNA Polymerases by Using a Novel Cell-Based Assay. J. Virol. 2011, 85, 13027–13037. [Google Scholar] [CrossRef] [PubMed]
- Strnadova, P.; Ren, H.; Valentine, R.; Mazzon, M.; Sweeney, T.R.; Brierley, I.; Smith, G.L. Inhibition of Translation Initiation by Protein 169: A Vaccinia Virus Strategy to Suppress Innate and Adaptive Immunity and Alter Virus Virulence. PLoS Pathog. 2015, 11, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Mateo, M.; Carbonnelle, C.; Reynard, O.; Kolesnikova, L.; Nemirov, K.; Page, A.; Volchkova, V.A.; Volchkov, V.E. VP24 Is a molecular determinant of Ebola virus virulence in guinea pigs. J. Infect. Dis. 2011, 204, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Rahbar, R.; Chan, R.W.Y.; Lee, S.M.Y.; Chan, M.C.W.; Xuhao, B.; Baker, D.P.; Sun, B.; Peiris, J.S.M.; Nicholls, J.M.; Fish, E.N. Influenza Virus Non-Structural Protein 1 (NS1) Disrupts Interferon Signaling. PLoS Pathog. 2010, 5, e13927. [Google Scholar] [CrossRef] [PubMed]
- Iversen, P.W.; Beck, B.; Chen, Y.F.; Dere, W.; Devanarayan, V.; Eastwood, B.J.; Farmen, M.W.; Iturria, S.J.; Montrose, C.; Moore, R.A.; et al. HTS Assay Validation. In Assay Guidance Manual [Internet]; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2012; pp. 875–905. [Google Scholar]
- Wong, G.; Kobinger, G.P.; Qiu, X.; Kobinger, G.P. Characterization of host immune responses in Ebola virus infections. Expert Rev. Clin. Immunol. 2014, 10, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F.; Amarasinghe, G.K. Evasion of interferon responses by Ebola and Marburg viruses. J. Interferon Cytokine Res. 2009, 29, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Edwards, M.R.; Borek, D.M.; Feagins, A.R.; Mittal, A.; Pappu, R.V.; Leung, D.W.; Basler, C.F.; Amarasinghe, G.K. Ebola virus VP24 targets a unique NLS binding site on karyopherin α 5 to selectively compete with nuclear import of phosphorylated STAT1. Cell Host Microbe 2015, 16, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dorival, I.; Wu, W.; Dowall, S.; Armstrong, S.; Touzelet, O.; Wastling, J.; Barr, J.N.; Matthews, D.; Carroll, M.; Hewson, R.; et al. Elucidation of the Ebola Virus VP24 Cellular Interactome and Disruption of Virus Biology through Targeted Inhibition of Host-Cell Protein Function. J. Proteome Res. 2014, 13, 5120–5135. [Google Scholar] [CrossRef] [PubMed]
- Cannas, V.; Daino, G.L.; Corona, A.; Esposito, F.; Tramontano, E. A Luciferase Reporter Gene Assay to Measure Ebola Virus Viral Protein 35—Associated Inhibition of Double-Stranded RNA—Stimulated, Retinoic Acid—Inducible Gene 1—Mediated Induction of Interferon β. J. Infect. Dis. 2015, 212, S277–S281. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schwarz, T.M.; Edwards, M.R.; Diederichs, A.; Alinger, J.B.; Leung, D.W.; Amarasinghe, G.K.; Basler, C.F. VP24-Karyopherin α Binding Affinities Differ between Ebolavirus Species, Influencing Interferon Inhibition and VP24 Stability. J. Virol. 2017, 91, e01715-16. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Melén, K.; Maljanen, S.; Lundberg, R.; Jiang, M.; Österlund, P.; Kakkola, L.; Julkunen, I. Ebolavirus protein VP24 interferes with innate immune responses by inhibiting interferon-λ1 gene expression. Virology 2017, 509, 23–34. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Signal | Coefficients of Variation, CV (%) |
|---|---|
| Min | 6.1 |
| Mid | 4.6 |
| Max | 3.3 |
| Inter-Plate Test | pISRE-luc Activation (%) | Inter-Day Test | pISRE-luc Activation (%) |
|---|---|---|---|
| Plate 1 | 52.4 | Plate 1 | 49.3 |
| Plate 2 | 50.8 | Plate 2 | 49.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fanunza, E.; Frau, A.; Sgarbanti, M.; Orsatti, R.; Corona, A.; Tramontano, E. Development and Validation of a Novel Dual Luciferase Reporter Gene Assay to Quantify Ebola Virus VP24 Inhibition of IFN Signaling. Viruses 2018, 10, 98. https://doi.org/10.3390/v10020098
Fanunza E, Frau A, Sgarbanti M, Orsatti R, Corona A, Tramontano E. Development and Validation of a Novel Dual Luciferase Reporter Gene Assay to Quantify Ebola Virus VP24 Inhibition of IFN Signaling. Viruses. 2018; 10(2):98. https://doi.org/10.3390/v10020098
Chicago/Turabian StyleFanunza, Elisa, Aldo Frau, Marco Sgarbanti, Roberto Orsatti, Angela Corona, and Enzo Tramontano. 2018. "Development and Validation of a Novel Dual Luciferase Reporter Gene Assay to Quantify Ebola Virus VP24 Inhibition of IFN Signaling" Viruses 10, no. 2: 98. https://doi.org/10.3390/v10020098
APA StyleFanunza, E., Frau, A., Sgarbanti, M., Orsatti, R., Corona, A., & Tramontano, E. (2018). Development and Validation of a Novel Dual Luciferase Reporter Gene Assay to Quantify Ebola Virus VP24 Inhibition of IFN Signaling. Viruses, 10(2), 98. https://doi.org/10.3390/v10020098