Oncotargeting by Vesicular Stomatitis Virus (VSV): Advances in Cancer Therapy

 and
and
Abstract
1. Introduction
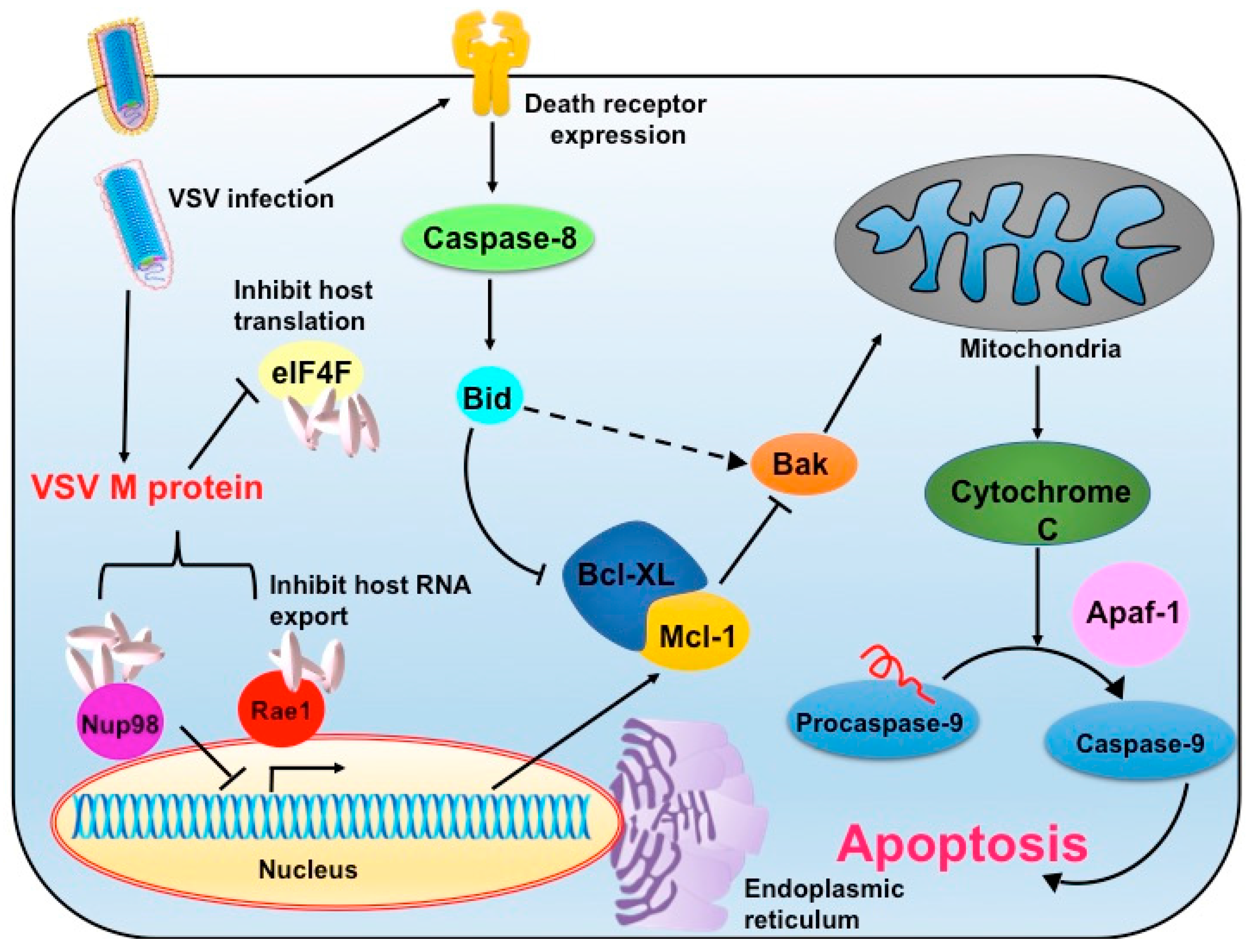
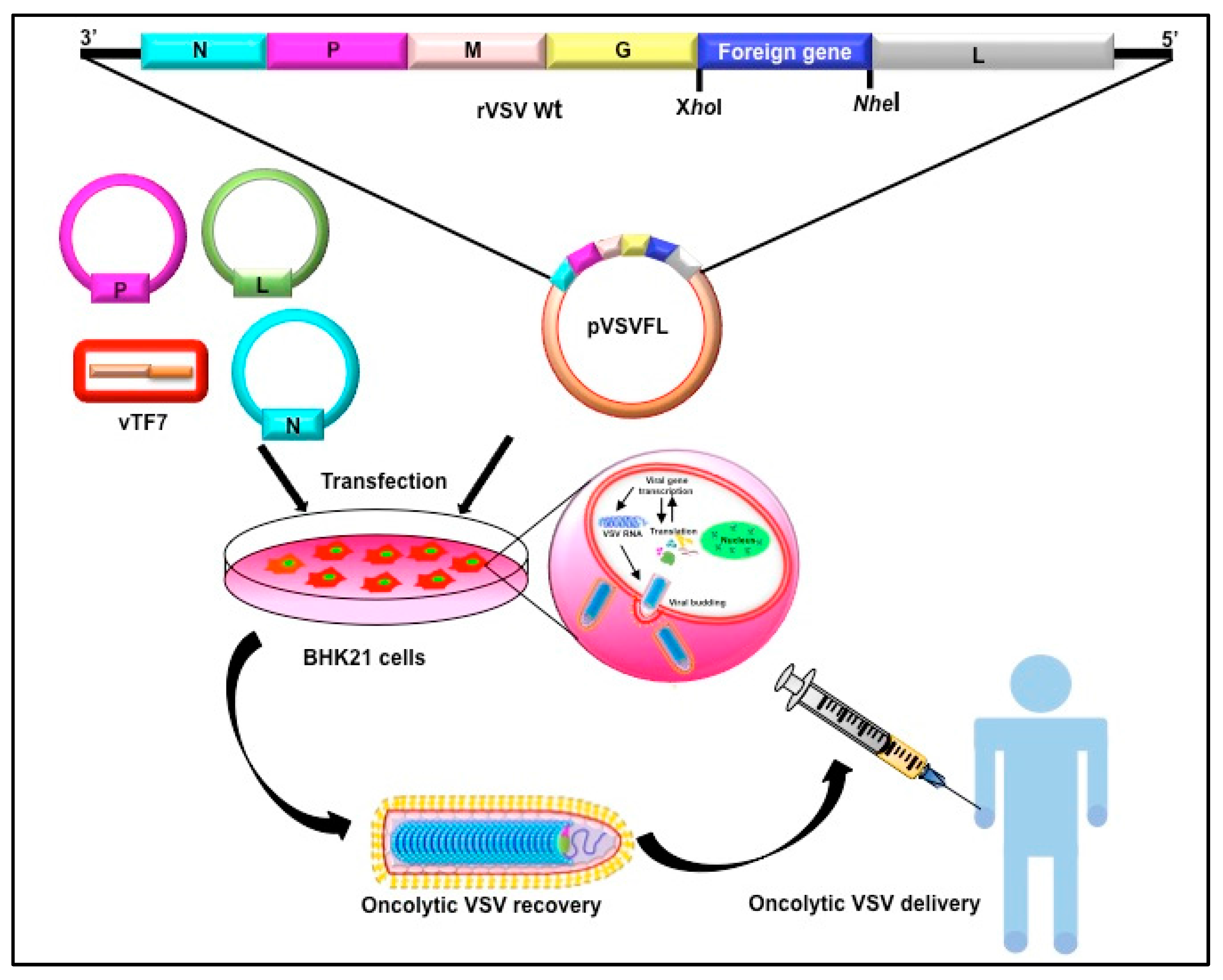
2. Vesicular Stomatitis Virus as an Oncolytic Virus
3. Vesicular Stomatitis Virus and Type I Interferon Signaling
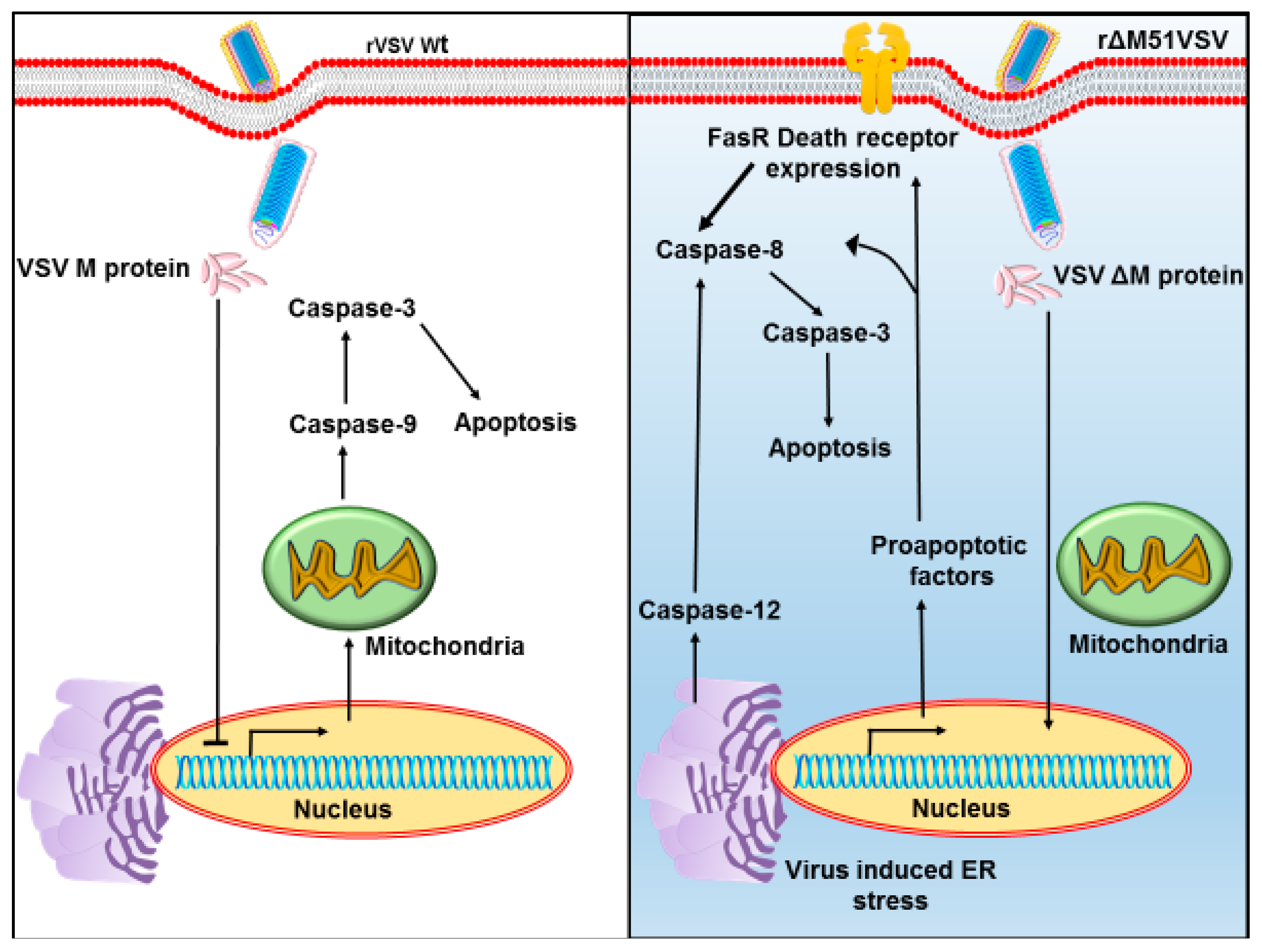
4. Advantages and Limitations of Vesicular Stomatitis Virus as Oncolytic Virotherapy
5. Amelioration of Vesicular Stomatitis Virus-Associated Neuropathogenesis: Different Approaches
5.1. MicroRNA Targeting
5.2. Modifications of M
5.3. Modification of G
5.4. Modulating Viral Replication
6. Advancement in the Tumor-Specific Targeting of Vesicular Stomatitis Virus
6.1. Increasing Tumor Lysing Efficacy
6.2. Oncolytic Vesicular Stomatitis Viruses Expressing a Suicide Gene
7. Next Generation Vesicular Stomatitis Virus as Oncolytic Virotherapy: Immunomodulatory Function
Working with Immune System for Oncolytic Virotherapy Efficacy
8. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W. Viruses as anticancer drugs. Trends Pharmacol. Sci. 2007, 28, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D.; Martuza, R.L.; Zwiebel, J. Replication-selective virotherapy for cancer: Biological principles, risk management and future directions. Nat. Med. 2001, 7, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Pelner, L.; Fowler, G.A.; Nauts, H.C. Effect of Concurrent Infections and Their Toxins on the Course of Leukemia. Acta Med. Scand. 1958, 162, 5–24. [Google Scholar] [CrossRef]
- Gross, S. Measles and leukaemia. Lancet 1971, 1, 397–398. [Google Scholar] [CrossRef]
- Kelly, E.; Russell, S.J. History of Oncolytic Viruses: Genesis to Genetic Engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Still, G.F. On a form of chronic joint disease in children. Med.-Chir. Trans. 1897, 80, 47. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, A.W.; Street, J.C. The need for comprehensive diet studies to assess the relation of lipids to cancer. Cancer Res. 1981, 41, 3748–3749. [Google Scholar] [PubMed]
- Fukuhara, H.; Homma, Y.; Todo, T. Oncolytic virus therapy for prostate cancer. Int. J. Urol. 2010, 17, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Gorman, M.J.; McKenzie, L.D.; Chai, J.N.; Hubert, C.G.; Prager, B.C.; Fernandez, E.; Richner, J.M.; Zhang, R.; Shan, C.; et al. Zika virus has oncolytic activity against glioblastoma stem cells. J. Exp. Med. 2017. [Google Scholar] [CrossRef]
- Betancourt, D.; Ramos, J.C.; Barber, G.N. Retargeting Oncolytic Vesicular Stomatitis Virus to Human T-Cell Lymphotropic Virus Type 1-Associated Adult T-Cell Leukemia. J. Virol. 2015, 89, 11786–11800. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.K. The transcription complex of vesicular stomatitis virus. Cell 1987, 48, 363–364. [Google Scholar] [CrossRef]
- Ahmed, M.; Lyles, D.S. Effect of vesicular stomatitis virus matrix protein on transcription directed by host RNA polymerases I, II, and III. J. Virol. 1998, 72, 8413–8419. [Google Scholar] [PubMed]
- Enninga, J.; Levy, D.E.; Blobel, G.; Fontoura, B.M.A. Role of nucleoporin induction in releasing an mRNA nuclear export block. Science 2002, 295, 1523–1525. [Google Scholar] [CrossRef] [PubMed]
- Her, L.S.; Lund, E.; Dahlberg, J.E. Inhibition of Ran guanosine triphosphatase-dependent nuclear transport by the matrix protein of vesicular stomatitis virus. Science 1997, 276, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Faria, P.A.; Chakraborty, P.; Levay, A.; Barber, G.N.; Ezelle, H.J.; Enninga, J.; Arana, C.; van Deursen, J.; Fontoura, B.M.A. VSV disrupts the Rae1/mrnp41 mRNA nuclear export pathway. Mol. Cell 2005, 17, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Rajani, K.R.; Pettit Kneller, E.L.; McKenzie, M.O.; Horita, D.A.; Chou, J.W.; Lyles, D.S. Complexes of Vesicular Stomatitis Virus Matrix Protein with Host Rae1 and Nup98 Involved in Inhibition of Host Transcription. PLoS Pathog. 2012, 8, e1002929. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.-Y.; Burdeinick-Kerr, R.; Whelan, S.P.J. A ribosome-specialized translation initiation pathway is required for cap-dependent translation of vesicular stomatitis virus mRNAs. Proc. Natl. Acad. Sci. USA 2013, 110, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Whitlow, Z.W.; Connor, J.H.; Lyles, D.S. Preferential translation of vesicular stomatitis virus mRNAs is conferred by transcription from the viral genome. J. Virol. 2006, 80, 11733–11742. [Google Scholar] [CrossRef] [PubMed]
- Black, B.L.; Brewer, G.; Lyles, D.S. Effect of vesicular stomatitis virus matrix protein on host-directed translation in vivo. J. Virol. 1994, 68, 555–560. [Google Scholar] [PubMed]
- Koyama, A.H. Induction of apoptotic DNA fragmentation by the infection of vesicular stomatitis virus. Virus Res. 1995, 37, 85–90. [Google Scholar]
- Black, B.L.; Lyles, D.S.; Carolina, N. Vesicular Stomatitis Virus Matrix Protein Inhibits Host Cell-Directed Transcription of Target Genes In Vivo. J. Virol. 1992, 66, 4058–4064. [Google Scholar] [PubMed]
- Blondel, D.; Harmison, G.G.; Schubert, M. Role of matrix protein in cytopathogenesis of vesicular stomatitis virus. J. Virol. 1990, 64, 1716–1725. [Google Scholar] [PubMed]
- Gaddy, D.F.; Lyles, D.S. Vesicular stomatitis viruses expressing wild-type or mutant M proteins activate apoptosis through distinct pathways. J. Virol. 2005, 79, 4170–4179. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Roberts, P.C.; Kipperman, T.; Bhalla, K.N.; Compans, R.W.; Archer, D.R.; Barber, G.N. Alpha/beta interferons potentiate virus-induced apoptosis through activation of the FADD/Caspase-8 death signaling pathway. J. Virol. 2000, 74, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Stojdl, D.F.; Lichty, B.D.; TenOever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 2003, 4, 263–275. [Google Scholar] [CrossRef]
- Gaddy, D.F.; Lyles, D.S. Oncolytic Vesicular Stomatitis Virus Induces Apoptosis via Signaling. J. Virol. 2007, 81, 2792–2804. [Google Scholar] [CrossRef] [PubMed]
- Schache, P.; Gürlevik, E.; Strüver, N.; Woller, N.; Malek, N.; Zender, L.; Manns, M.; Wirth, T.; Kühnel, F.; Kubicka, S. VSV virotherapy improves chemotherapy by triggering apoptosis due to proteasomal degradation of Mcl-1. Gene Ther. 2009, 16, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Du, L.; Chen, X.; Chen, L.; Yi, T.; Chen, X.; Wen, Y.; Wei, Y.; Zhao, X. VEGF-D-enhanced lymph node metastasis of ovarian cancer is reversed by vesicular stomatitis virus matrix protein. Int. J. Oncol. 2016, 49, 123–132. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mahoney, D.J.; Lefebvre, C.; Allan, K.; Brun, J.; Sanaei, C.A.; Baird, S.; Pearce, N.; Grönberg, S.; Wilson, B.; Prakesh, M.; et al. Virus-Tumor Interactome Screen Reveals ER Stress Response Can Reprogram Resistant Cancers for Oncolytic Virus-Triggered Caspase-2 Cell Death. Cancer Cell 2011, 20, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Rosebeck, S.; Sudini, K.; Chen, T.; Leaman, D.W. Involvement of Noxa in mediating cellular ER stress responses to lytic virus infection. Virology 2011, 417, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Paterson, J.M.; Lemay, C.G.; Falls, T.J.; McGuire, A.; Parato, K.A.; Stojdl, D.F.; Daneshmand, M.; Speth, K.; Kirn, D.; et al. Targeted Inflammation During Oncolytic Virus Therapy Severely Compromises Tumor Blood Flow. Mol. Ther. 2007, 15, 1686–1693. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Silva, N.S.; de Falls, T.J.; Aladl, U.; Evgin, L.; Paterson, J.; Sun, Y.Y.; Roy, D.G.; Rintoul, J.L.; Daneshmand, M.; et al. Targeting Tumor Vasculature With an Oncolytic Virus. Mol. Ther. 2011, 19, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, C.; Berra, E.; Pouysségur, J. Hypoxia: The tumor’s gateway to progression along the angiogenic pathway. Trends Cell Biol. 2001, 11, 32–36. [Google Scholar] [CrossRef]
- Probst, G.; Riedinger, H.J.; Martin, P.; Engelcke, M.; Probst, H. Fast control of DNA replication in response to hypoxia and to inhibited protein synthesis in CCRF-CEM and HeLa cells. Biol. Chem. 1999, 380, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M. Tumor microenvironment and the response to anticancer therapy. Cancer Biol. Ther. 2002, 1, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.H.; Naczki, C.; Koumenis, C.; Lyles, D.S. Replication and Cytopathic Effect of Oncolytic Vesicular Stomatitis Virus in Hypoxic Tumor Cells In Vitro and In Vivo. J. Virol. 2004, 78, 8960–8970. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.R.; Walters, K.F.; Port, G.R. Behaviour of the adult seven spot ladybird, Coccinella septempunctata (Coleoptera: Coccinellidae), in response to dimethoate residue on bean plants in the laboratory. Bull. Entomol. Res. 2001, 91, 221–226. [Google Scholar] [PubMed]
- Stark, G.R.; Kerr, I.M.; Williams, B.R.; Silverman, R.H.; Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Barber, G.N. Vesicular Stomatitis Virus (VSV) Therapy of Tumors. IUBMB Life 2000, 50, 135–138. [Google Scholar] [CrossRef] [PubMed]
- D’agostino, P.M.; Amenta, J.J.; Reiss, C.S. IFN-β-induced alteration of VSV protein phosphorylation in neuronal cells. Viral Immunol. 2009, 22, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Stojdl, D.; Lichty, B.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825. [Google Scholar] [PubMed]
- Zhang, K.X.; Matsui, Y.; Hadaschik, B.A.; Lee, C.; Jia, W.; Bell, J.C.; Fazli, L.; So, A.I.; Rennie, P.S. Down-regulation of type I interferon receptor sensitizes bladder cancer cells to vesicular stomatitis virus-induced cell death. Int. J. Cancer 2010, 127, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Westcott, M.M.; Liu, J.; Rajani, K.; D’Agostino, R.; Lyles, D.S.; Porosnicu, M. Interferon β and Interferon α2a Differentially Protect Head and Neck Cancer Cells from Vesicular Stomatitis Virus-Induced Oncolysis. J. Virol. 2015, 89, 7944–7954. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Porosnicu, M.; Barber, G.N. Oncolytic activity of vesicular stomatitis virus is effective against tumors exhibiting aberrant p53, Ras, or myc function and involves the induction of apoptosis. J. Virol. 2001, 75, 3474–3479. [Google Scholar] [CrossRef] [PubMed]
- Noser, J.A.; Mael, A.A.; Sakuma, R.; Ohmine, S.; Marcato, P.; WK Lee, P.; Ikeda, Y. The RAS/Raf1/MEK/ERK Signaling Pathway Facilitates VSV-mediated Oncolysis: Implication for the Defective Interferon Response in Cancer Cells. Mol. Ther. 2007, 15, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.C.; Friedmann, T.; Drievert, W.; Burrascano, M.; Yee, J.-K. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: Concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells (gene therapy/zebrafish). Genetics 1993, 90, 8033–8037. [Google Scholar] [CrossRef]
- Van den Pol, A.N.; Dalton, K.P.; Rose, J.K. Relative neurotropism of a recombinant rhabdovirus expressing a green fluorescent envelope glycoprotein. J. Virol. 2002, 76, 1309–1327. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.J.; Nace, R.; Barber, G.N.; Russell, S.J. Attenuation of Vesicular Stomatitis Virus Encephalitis through MicroRNA Targeting. J. Virol. 2010, 84, 1550–1562. [Google Scholar] [CrossRef] [PubMed]
- Takamizawa, J.; Konishi, H.; Yanagisawa, K.; Tomida, S.; Osada, H.; Endoh, H.; Harano, T.; Yatabe, Y.; Nagino, M.; Nimura, Y.; et al. Reduced Expression of the let-7 MicroRNAs in Human Lung Cancers in Association with Shortened Postoperative Survival Advances in Brief Reduced Expression of the let-7 MicroRNAs in Human Lung Cancers in Association with Shortened Postoperative Survival. Cancer Res. 2004, 64, 3753–3756. [Google Scholar] [CrossRef] [PubMed]
- Edge, R.E.; Falls, T.J.; Brown, C.W.; Lichty, B.D.; Atkins, H.; Bell, J.C. A let-7 MicroRNA-sensitive Vesicular Stomatitis Virus Demonstrates Tumor-specific Replication. Mol. Ther. 2008, 16, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Lawson, N.D.; Stillman, E.A.; Whitt, M.A.; Rose, J.K. Recombinant vesicular stomatitis viruses from DNA. Proc. Natl. Acad. Sci. USA 1995, 92, 4477–4481. [Google Scholar] [CrossRef] [PubMed]
- Publicover, J.; Ramsburg, E.; Robek, M.; Rose, J.K. Rapid Pathogenesis Induced by a Vesicular Stomatitis Virus Matrix Protein Mutant: Viral Pathogenesis Is Linked to Induction of Tumor Necrosis Factor α. J. Virol. 2006, 80, 7028–7036. [Google Scholar] [CrossRef] [PubMed]
- Ammayappan, A.; Nace, R.; Peng, K.-W.; Russell, S.J. Neuroattenuation of Vesicular Stomatitis Virus through Picornaviral Internal Ribosome Entry Sites. J. Virol. 2013, 87, 3217–3228. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Janelle, V.; Brassard, F.; Lapierre, P.; Lamarre, A.; Poliquin, L. Mutations in the Glycoprotein of Vesicular Stomatitis Virus Affect Cytopathogenicity: Potential for Oncolytic Virotherapy. J. Virol. 2011, 85, 6513–6520. [Google Scholar] [CrossRef] [PubMed]
- Publicover, J.; Ramsburg, E.; Rose, J.K. Characterization of nonpathogenic, live, viral vaccine vectors inducing potent cellular immune responses. J. Virol. 2004, 78, 9317–9324. [Google Scholar] [CrossRef] [PubMed]
- Duntsch, C.D.; Zhou, Q.; Jayakar, H.R.; Weimar, J.D.; Robertson, J.H.; Pfeffer, L.M.; Wang, L.; Xiang, Z.; Whitt, M.A. Recombinant vesicular stomatitis virus vectors as oncolytic agents in the treatment of high-grade gliomas in an organotypic brain tissue slice-glioma coculture model. J. Neurosurg. 2004, 100, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Puckett, S.; Lyles, D.S. Susceptibility of breast cancer cells to an oncolytic matrix (M) protein mutant of vesicular stomatitis virus. Cancer Gene Ther. 2010, 17, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Muik, A.; Stubbert, L.J.; Jahedi, R.Z.; Geib, Y.; Kimpel, J.; Dold, C.; Tober, R.; Volk, A.; Klein, S.; Dietrich, U.; et al. Re-engineering vesicular stomatitis virus to abrogate neurotoxicity, circumvent humoral immunity, and enhance oncolytic potency. Cancer Res. 2014, 74, 3567–3578. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.B.; Barra, N.G.; Davies, E.; Ashkar, A.A.; Lichty, B.D. Expressing human interleukin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther. 2012, 19, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Tesfay, M.Z.; Kirk, A.C.; Hadac, E.M.; Griesmann, G.E.; Federspiel, M.J.; Barber, G.N.; Henry, S.M.; Peng, K.; Russell, J. PEGylation of Vesicular Stomatitis Virus Extends Virus Persistence in Blood Circulation of Passively Immunized Mice. J. Virol. 2013, 87, 3752–3759. [Google Scholar] [CrossRef] [PubMed]
- Muik, A.; Dold, C.; Geiß, Y.; Volk, A.; Werbizki, M.; Dietrich, U.; von Laer, D. Semireplication-competent vesicular stomatitis virus as a novel platform for oncolytic virotherapy. J. Mol. Med. 2012, 90, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Van den Pol, A.N.; Davis, J.N. Highly Attenuated Recombinant Vesicular Stomatitis Virus VSV-12’GFP Displays Immunogenic and Oncolytic Activity. J. Virol. 2013, 87, 1019–1034. [Google Scholar] [CrossRef] [PubMed]
- Wollmann, G.; Rogulin, V.; Simon, I.; Rose, J.K.; van den Pol, A.N. Some Attenuated Variants of Vesicular Stomatitis Virus Show Enhanced Oncolytic Activity against Human Glioblastoma Cells relative to Normal Brain Cells. J. Virol. 2010, 84, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Dreja, H.; Piechaczyk, M. The effects of N-terminal insertion into VSV-G of an scFv peptide. Virol. J. 2006, 8, 1–8. [Google Scholar]
- Guibinga, G.H.; Hall, F.L.; Gordon, E.M.; Ruoslahti, E.; Friedmann, T. Ligand-modified vesicular stomatitis virus glycoprotein displays a temperature-sensitive intracellular trafficking and virus assembly phenotype. Mol. Ther. 2004, 9, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Ammayappan, A.; Peng, K.-W.; Russell, S.J. Characteristics of oncolytic vesicular stomatitis virus displaying tumor-targeting ligands. J. Virol. 2013, 87, 13543–13555. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Padmashali, R.M.; Andreadis, S.T. Engineering fibrinogen-binding VSV-G envelope for spatially- and cell-controlled lentivirus delivery through fibrin hydrogels. Biomaterials 2011, 32, 3330–3339. [Google Scholar] [CrossRef] [PubMed]
- Bergman, I.; Whitaker-dowling, P.; Gao, Y.; Griffin, J.A.; Watkins, S.C. Vesicular stomatitis virus expressing a chimeric Sindbis glycoprotein containing an Fc antibody binding domain targets to Her2/neu overexpressing breast cancer cells. Virology 2003, 316, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Whitaker-Dowling, P.; Griffin, J.A.; Barmada, M.A.; Bergman, I. Recombinant vesicular stomatitis virus targeted to Her2/neu combined with anti-CTLA4 antibody eliminates implanted mammary tumors. Cancer Gene Ther. 2009, 16, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Whitaker-Dowling, P.; Griffin, J.A.; Bergman, I. Treatment with targeted vesicular stomatitis virus generates therapeutic multifunctional anti-tumor memory CD4 T cells. Cancer Gene Ther. 2012, 19, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Amariglio, N.; Rechavi, G.; Jakob-Hirsch, J.; Kela, I.; Kaminski, N.; Getz, G.; Domany, E.; Givol, D. DNA microarrays identification of primary and secondary target genes regulated by p53. Oncogene 2001, 20, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Heiber, J.F.; Barber, G.N. Vesicular Stomatitis Virus Expressing Tumor Suppressor p53 Is a Highly Attenuated, Potent Oncolytic Agent. J. Virol. 2011, 85, 10440–10450. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Peng, K.-W.; Vongpunsawad, S.; Harvey, M.; Mizuguchi, H.; Hayakawa, T.; Cattaneo, R.; Russell, S.J. Antibody-targeted cell fusion. Nat. Biotechnol. 2004, 22, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Kleinlützum, D.; Hanauer, J.D.S.; Muik, A.; Hanschmann, K.-M.; Kays, S.-K.; Ayala-Breton, C.; Peng, K.-W.; Mühlebach, M.D.; Abel, T.; Buchholz, C.J. Enhancing the Oncolytic Activity of CD133-Targeted Measles Virus: Receptor Extension or Chimerism with Vesicular Stomatitis Virus Are Most Effective. Front. Oncol. 2017, 7, 127. [Google Scholar] [CrossRef] [PubMed]
- Altomonte, J.; Wu, L.; Meseck, M.; Chen, L.; Ebert, O.; Garcia-Sastre, A.; Fallon, J.; Mandeli, J.; Woo, S.L.C. Enhanced Oncolytic Potency of Vesicular Stomatitis Virus via Vector-Mediated Inhibition of NK and NKT Cells. Cancer Gene Ther. 2009, 16, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Huang, T.; Meseck, M.; Altomonte, J.; Ebert, O.; Shinozaki, K.; García-Sastre, A.; Fallon, J.; Mandeli, J.; Woo, S.L.C. rVSV(M Delta 51)-M3 is an effective and safe oncolytic virus for cancer therapy. Hum. Gene Ther. 2008, 19, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Ebert, O.; Shinozaki, K.; Kournioti, C.; Ebert, O.; Shinozaki, K.; Kournioti, C.; Park, M.; Garcı, A. Syncytia Induction Enhances the Oncolytic Potential of Vesicular Stomatitis Virus in Virotherapy for Cancer Syncytia Induction Enhances the Oncolytic Potential of Vesicular Stomatitis Virus in Virotherapy for Cancer. Cancer Res. 2004, 64, 3265–3270. [Google Scholar] [CrossRef] [PubMed]
- Le Boeuf, F.; Gebremeskel, S.; McMullen, N.; He, H.; Greenshields, A.L.; Hoskin, D.W.; Bell, J.C.; Johnston, B.; Pan, C.; Duncan, R. Reovirus FAST Protein Enhances Vesicular Stomatitis Virus Oncolytic Virotherapy in Primary and Metastatic Tumor Models. Mol. Ther. Oncolytics 2017, 6, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Porosnicu, M.; Markovic, D.; Barber, G.N. Genetically engineered vesicular stomatitis virus in gene therapy: Application for treatment of malignant disease. J. Virol. 2002, 76, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Lauer, U.M.; Niethammer, D.; Beck, J.F.; Schlegel, P.G. Suicide genes: Past, present and future perspectives. Rev. Immunol. Today 2000, 21, 48–54. [Google Scholar] [CrossRef]
- Porosnicu, M.; Mian, A.; Barber, G.N. The Oncolytic Effect of Recombinant Vesicular Stomatitis Virus Is Enhanced by Expression of the Fusion Cytosine Deaminase/Uracil Phosphoribosyltransferase Suicide Gene. Cancer Res. 2003, 8366–8376. [Google Scholar]
- Leveille, S.; Samuel, S.; Goulet, M.; Hiscott, J. Enhancing VSV oncolytic activity with an improved cytosine deaminase suicide gene strategy. Cancer Gene Ther. 2011, 18, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Miest, T.S.; Cattaneo, R. New viruses for cancer therapy: Meeting clinical needs. Nat. Rev. Microbiol. 2013, 12, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, S.; Pirola, B.; Pollo, B.; Magrassi, L.; Bruzzone, M.G.; Rigamonti, D.; Galli, R.; Selleri, S.; di Meco, F.; de Fraja, C.; et al. Gene therapy of experimental brain tumors using neural progenitor cells. Nat. Med. 2000, 6, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Tepper, R.I.; Pattengale, P.K.; Leder, P. Murine interleukin-4 displays potent anti-tumor activity in vivo. Cell 1989, 57, 503–512. [Google Scholar] [CrossRef]
- Shin, E.J.; Wanna, G.B.; Choi, B.; Iii, D.A.; Ebert, O.; Genden, E.M.; Woo, S.L. Interleukin-12 Expression Enhances Vesicular Stomatitis Virus Oncolytic Therapy in Murine Squamous Cell Carcinoma. Laryngoscope 2007, 117, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Ramsburg, E.; Publicover, J.; Buonocore, L.; Poholek, A.; Robek, M.; Palin, A.; John, K.; Rose, J.K. A Vesicular Stomatitis Virus Recombinant Expressing Granulocyte-Macrophage Colony-Stimulating Factor Induces Enhanced T-Cell Responses and Is Highly Attenuated for Replication in Animals A Vesicular Stomatitis Virus Recombinant Expressing Granulocyte-Macr. J. Virol. 2005, 79, 15043–15053. [Google Scholar] [CrossRef] [PubMed]
- Bergman, I.; Griffin, J.A.; Gao, Y.; Whitaker-dowling, P. Treatment of implanted mammary tumors with recombinant vesicular stomatitis virus targeted to Her2/neu. Int. J. Cancer 2007, 430, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; Bidula, S.M.; Jensen, T.M.; Reiss, C.S. Cytokine-modified VSV is attenuated for neural pathology, but is both highly immunogenic and oncolytic. Int. J. Interferon Cytokine Mediat. Res. 2009, 1, 15–32. [Google Scholar] [PubMed]
- Wongthida, P.; Diaz, R.M.; Galivo, F.; Kottke, T.; Thompson, J.; Pulido, J.; Pavelko, K.; Pease, L.; Melcher, A.; Vile, R. Type III IFN interleukin-28 mediates the antitumor efficacy of oncolytic virus VSV in immune-competent mouse models of cancer. Cancer Res. 2010, 70, 4539–4549. [Google Scholar] [CrossRef] [PubMed]
- Obuchi, M.; Fernandez, M.; Barber, G.N. Development of Recombinant Vesicular Stomatitis Viruses That Exploit Defects in Host Defense To Augment Specific Oncolytic Activity. J. Virol. 2003, 77, 8843–8856. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Salinas, L.; Naik, S.; Pauszek, S.J.; Peng, K.-W.; Russell, S.J.; Rodriguez, L.L. Oncolytic Recombinant Vesicular Stomatitis Virus (VSV) Is Nonpathogenic and Nontransmissible in Pigs, a Natural Host of VSV. Hum. Gene Ther. Clin. Dev. 2017, 28, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Steele, M.B.; Jenks, N.; Grell, J.; Suksanpaisan, L.; Naik, S.; Federspiel, M.J.; Lacy, M.Q.; Russell, S.J.; Peng, K.-W. Safety Studies in Tumor and Non-Tumor-Bearing Mice in Support of Clinical Trials Using Oncolytic VSV-IFNβ-NIS. Hum. Gene Ther. Clin. Dev. 2016, 27, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.; Galyon, G.D.; Jenks, N.J.; Steele, M.B.; Miller, A.C.; Allstadt, S.D.; Suksanpaisan, L.; Peng, K.W.; Federspiel, M.J.; Russell, S.J.; et al. Comparative Oncology Evaluation of Intravenous Recombinant Oncolytic Vesicular Stomatitis Virus Therapy in Spontaneous Canine Cancer. Mol. Cancer Ther. 2018, 17, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Galivo, F.; Diaz, R.M.; Thanarajasingam, U.; Jevremovic, D.; Wongthida, P.; Thompson, J.; Kottke, T.; Barber, G.N.; Melcher, A.; Vile, R.G. Interference of CD40L-mediated tumor immunotherapy by oncolytic vesicular stomatitis virus. Hum. Gene Ther. 2010, 21, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Wongthida, P.; Diaz, R.M.; Galivo, F.; Kottke, T.; Thompson, J.; Melcher, A.; Vile, R. VSV Oncolytic Virotherapy in the B16 Model Depends Upon Intact MyD88 Signaling. Mol. Ther. 2011, 19, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. VSV-tumor selective replication and protein translation. Oncogene 2005, 7710–7719. [Google Scholar] [CrossRef] [PubMed]
- Power, A.T.; Wang, J.; Falls, T.J.; Paterson, J.M.; Parato, K.A.; Lichty, B.D.; Stojdl, D.F.; Forsyth, P.A.J.; Atkins, H.; Bell, J.C. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol. Ther. 2007, 15, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Diaz, R.M.; Galivo, F.; Kottke, T.; Wongthida, P.; Qiao, J.; Thompson, J.; Valdes, M.; Barber, G.; Vile, R.G. Oncolytic Immunovirotherapy for Melanoma Using Vesicular Stomatitis Virus. Cancer Res. 2007, 67, 2840–2849. [Google Scholar] [CrossRef] [PubMed]
- Olagnier, D.; Lababidi, R.R.; Hadj, S.B.; Sze, A.; Liu, Y.; Naidu, S.D.; Ferrari, M.; Jiang, Y.; Chiang, C.; Beljanski, V.; et al. Activation of Nrf2 Signaling Augments Vesicular Stomatitis Virus Oncolysis via Autophagy-Driven Suppression of Antiviral Immunity. Mol. Ther. 2017, 25, 1900–1916. [Google Scholar] [CrossRef] [PubMed]
- Bridle, B.W.; Nguyen, A.; Salem, O.; Zhang, L.; Koshy, S.; Clouthier, D.; Chen, L.; Pol, J.; Swift, S.L.; Bowdish, D.M.E.; et al. Privileged Antigen Presentation in Splenic B Cell Follicles Maximizes T Cell Responses in Prime-Boost Vaccination. J. Immunol. 2016, 196, 4587. [Google Scholar] [CrossRef] [PubMed]
- Altomonte, J.; Wu, L.; Chen, L.; Meseck, M.; Ebert, O.; Garcia-Sastre, A.; Fallon, J.; Woo, S.L. Exponential enhancement of oncolytic vesicular stomatitis virus potency by vector-mediated suppression of inflammatory responses in vivo. Mol. Ther. 2008, 16, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, J.E.; Bridle, B.W.; Stephenson, K.B.; Jenkins, K.M.; Brunellière, J.; Bramson, J.L.; Lichty, B.D.; Wan, Y. Recombinant Vesicular Stomatitis Virus Transduction of Dendritic Cells Enhances Their Ability to Prime Innate and Adaptive Antitumor Immunity. Mol. Ther. 2009, 17, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Galivo, F.; Diaz, R.M.; Wongthida, P.; Thompson, J.; Kottke, T.; Barber, G.; Melcher, A.; Vile, R. Single-cycle viral gene expression, rather than progressive replication and oncolysis, is required for VSV therapy of B16 melanoma. Gene Ther. 2009, 17, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Leveille, S.; Goulet, M.L.; Lichty, B.D.; Hiscott, J. Vesicular stomatitis virus oncolytic treatment interferes with tumor-associated dendritic cell functions and abrogates tumor antigen presentation. J. Virol. 2011, 85, 12160–12169. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois-Daigneault, M.-C.; Roy, D.G.; Falls, T.; Twumasi-Boateng, K.; St-Germain, L.E.; Marguerie, M.; Garcia, V.; Selman, M.; Jennings, V.A.; Pettigrew, J.; et al. Oncolytic vesicular stomatitis virus expressing interferon-γ has enhanced therapeutic activity. Mol. Ther. Oncol. 2016, 3, 16001. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Patnaik, M.M.; Ruiz, A.; Russell, S.J.; Peng, K.W. Immunovirotherapy with vesicular stomatitis virus and PD-L1 blockade enhances therapeutic outcome in murine acute myeloid leukemia. Blood 2016, 127, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Carlson, S.K.; Classic, K.L.; Greiner, S.; Naik, S.; Power, A.T.; Bell, J.C.; Russell, S.J. Radioiodide imaging and radiovirotherapy of multiple myeloma using VSV(Delta51)-NIS, an attenuated vesicular stomatitis virus encoding the sodium iodide symporter gene. Blood 2007, 110, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Dastidar, H.; Zhang, C.; Zemp, F.J.; Lau, K.; Ernst, M.; Rakic, A.; Sikdar, S.; Rajwani, J.; Naumenko, V.; et al. Smac mimetics and oncolytic viruses synergize in driving anticancer T-cell responses through complementary mechanisms. Nat. Commun. 2017, 8, 344. [Google Scholar] [CrossRef] [PubMed]
- Fehl, D.J.; Ahmed, M. Curcumin promotes the oncoltyic capacity of vesicular stomatitis virus for the treatment of prostate cancers. Virus Res. 2017, 228, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Felt, S.A.; Droby, G.N.; Grdzelishvili, V.Z. Ruxolitinib and Polycation Combination Treatment Overcomes Multiple Mechanisms of Resistance of Pancreatic Cancer Cells to Oncolytic Vesicular Stomatitis Virus. J. Virol. 2017, 91, e00461-17. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| VSV Modification | Virus Description | Reference |
|---|---|---|
| VSV-IL4 | rVSV expressing IL-4 cytokine with enhanced oncolytic activity | [81] |
| VSV-IFNβ | rVSV expressing IFN-β gene, show oncolytic activity against metastatic lung disease, and able to generate T cell response | [93] |
| VSV-IL12 | rVSV is expressing murine IL-12 gene show oncolytic activity against squamous cell carcinoma. | [88] |
| rVSV-gG | rVSV expressing equine herpes virus-1 glycoprotein G, which acts as a broad-spectrum viral chemokine binding protein | [104] |
| rVSV-UL141 | rVSV expressing a protein from human cytomegalovirus which down regulates the natural killer (NK) cell-activating ligand CD155 and inhibits the function of NK cell | [77] |
| rVSV(MΔ51)-M3 | rVSV expressing the murine gammaherpesvirus-68 chemokine-binding protein M3 in modified matrix protein backbone with enhanced tumor necrosis | [78] |
| ΔM51-VSV | ΔM51-VSV infection activated DCs to produce proinflammatory cytokines (IL-12 and IFNs) | [105] |
| VSV-CD40L | rVSV expressing CD40L, a member of the TNF family expressed on the surface of activated Th cells. | [106] |
| VSV-p14 | rVSV expressing p14 FAST protein increase oncolytic property | [80] |
| VSV-CD133 | rVSV expressing CD133 (a marker for cancer stem cells) increase specificity for CD133 expressing tumours. | [75] |
| VSV-IL15 | rVSV expressing secreted version of human interleukin15, it enhances both NK cell and T cell response | [60] |
| VSV-IL28 | rVSV expressing IL-28, a member of type 3 IFN | [92] |
| VSV-rFlt3L | rVSV expressing the Fms-like tyrosine kinase 3 ligand (rFlt3L). rFlt3L is a growth factor which promotes the differentiation and proliferation of DC. | [107] |
| VSV-IFNγ | rVSV expressing IFNγ which slows tumor growth | [108] |
| VSV-mIFNβ-NIS | rVSV expressing IFNβ and the NIS reporter, in the presence of anti-PD-L1 antibody, it shows higher anti tumor activity | [109] |
| VSV expressing suicide gene | ||
| VSV-TK | rVSV expressing thymidine kinase of herpes virus, increase oncolytic property | [81] |
| VSV(ΔM51) NIS | rVSV expressing human NIS gene, shows specific oncolytic activity against myeloma. | [110] |
| VSV-C:U | rVSV expressing the fusion suicide gene Escherichia coli cytosine deaminase (CD)/uracil phosphoribosyltransferase (UPRT), catalyzing the modification of 5-fluorocytosine into chemotherapeutic 5-fluorouracil | [83] |
| VSV-mp53 and VSV-ΔM-mp53 | VSV-mp53 and VSV-ΔM-mp53 both expressing high level of functional p53 in respective backbone VSV with chemical compounds | [74] |
| LCL161 and VSV-ΔM51 | SMC and OV therapies combination also synergize in vivo by promoting anticancer immunity through an increase in CD8+ T-cell response | [111] |
| Curcumin and VSV | Cumulative decrease in the expression of the anti-apoptotic protein, Bcl-XL, and in the phosphorylation of NF-κB and increase in the number of virus infected cells | [112] |
| Ruxolitinib and Polycation with VSV | Ruxolitinib and polycation improve VSV attachment and replication in HPAF-II cells | [113] |
| SFN (antioxidant compound sulforaphane) and VSV | SFN enhances VSVΔ51 spread in oncolytic virus-resistant cancer cells | [102] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bishnoi, S.; Tiwari, R.; Gupta, S.; Byrareddy, S.N.; Nayak, D. Oncotargeting by Vesicular Stomatitis Virus (VSV): Advances in Cancer Therapy. Viruses 2018, 10, 90. https://doi.org/10.3390/v10020090
Bishnoi S, Tiwari R, Gupta S, Byrareddy SN, Nayak D. Oncotargeting by Vesicular Stomatitis Virus (VSV): Advances in Cancer Therapy. Viruses. 2018; 10(2):90. https://doi.org/10.3390/v10020090
Chicago/Turabian StyleBishnoi, Suman, Ritudhwaj Tiwari, Sharad Gupta, Siddappa N. Byrareddy, and Debasis Nayak. 2018. "Oncotargeting by Vesicular Stomatitis Virus (VSV): Advances in Cancer Therapy" Viruses 10, no. 2: 90. https://doi.org/10.3390/v10020090
APA StyleBishnoi, S., Tiwari, R., Gupta, S., Byrareddy, S. N., & Nayak, D. (2018). Oncotargeting by Vesicular Stomatitis Virus (VSV): Advances in Cancer Therapy. Viruses, 10(2), 90. https://doi.org/10.3390/v10020090