Expression Profiling in Pinus pinaster in Response to Infection with the Pine Wood Nematode Bursaphelenchus xylophilus

 , , , , , ,
, , , , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Biological Material, Pine Wood Nematode Inoculation and Sampling
2.2. RNA Extraction, cDNA Synthesis, Library Preparation and Sequencing
2.3. Pre-Processing RNA-Sequencing Data and Transcriptome Assembly
2.4. Prediction of Candidate Coding Regions
2.5. Mapping and Differential Expression Analysis
2.6. qPCR Validation
2.7. Transcriptome Annotation
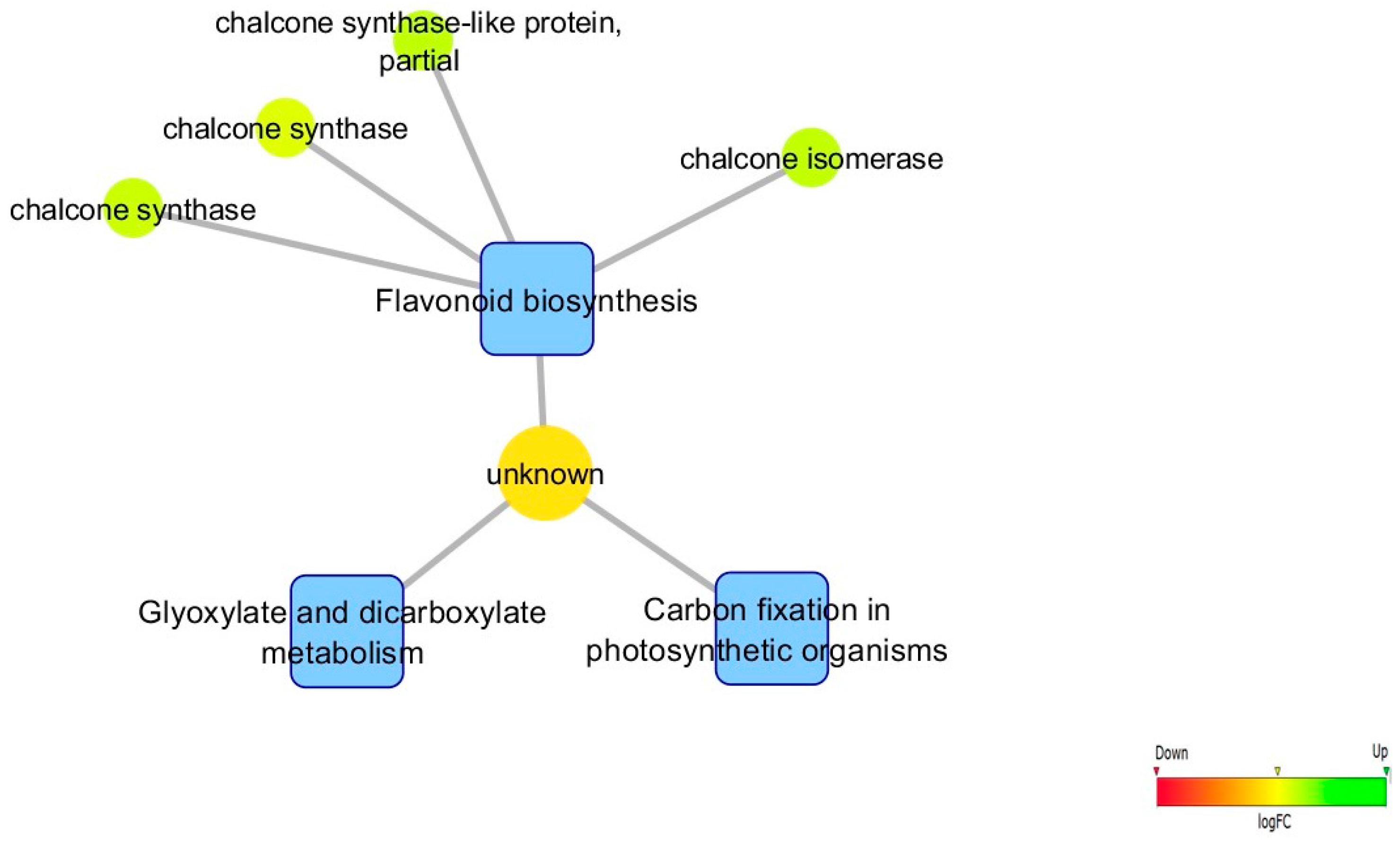
2.8. Biological Networks Analysis
2.9. SNP Calling
2.10. Data Archiving Statement
3. Results
3.1. Pre-Processing of RNA-Sequencing Data and Transcriptome Assembly
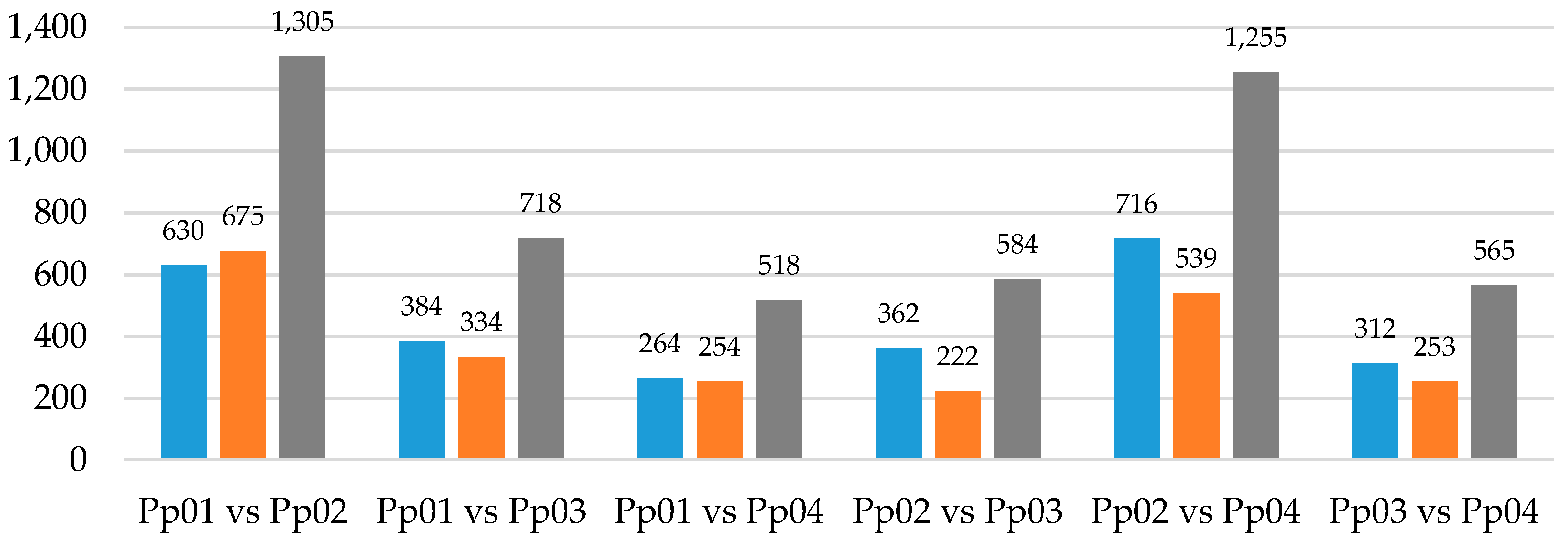
3.2. Mapping and Differential Expression Analysis
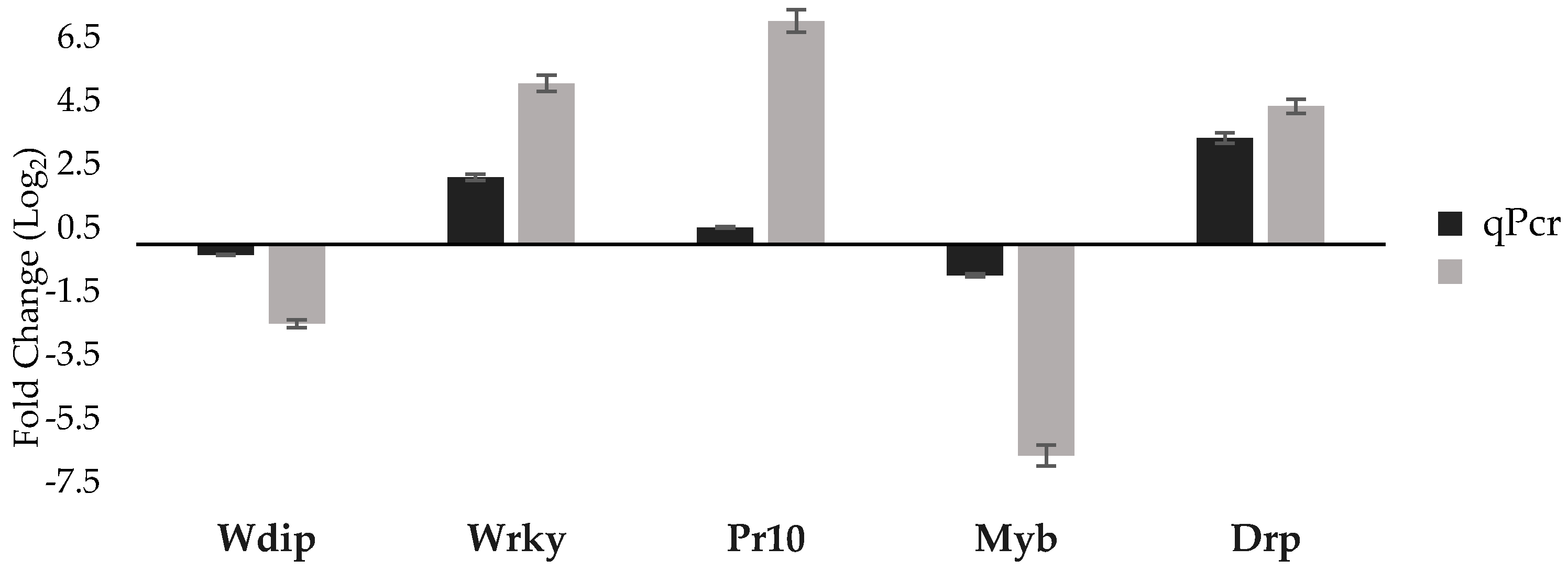
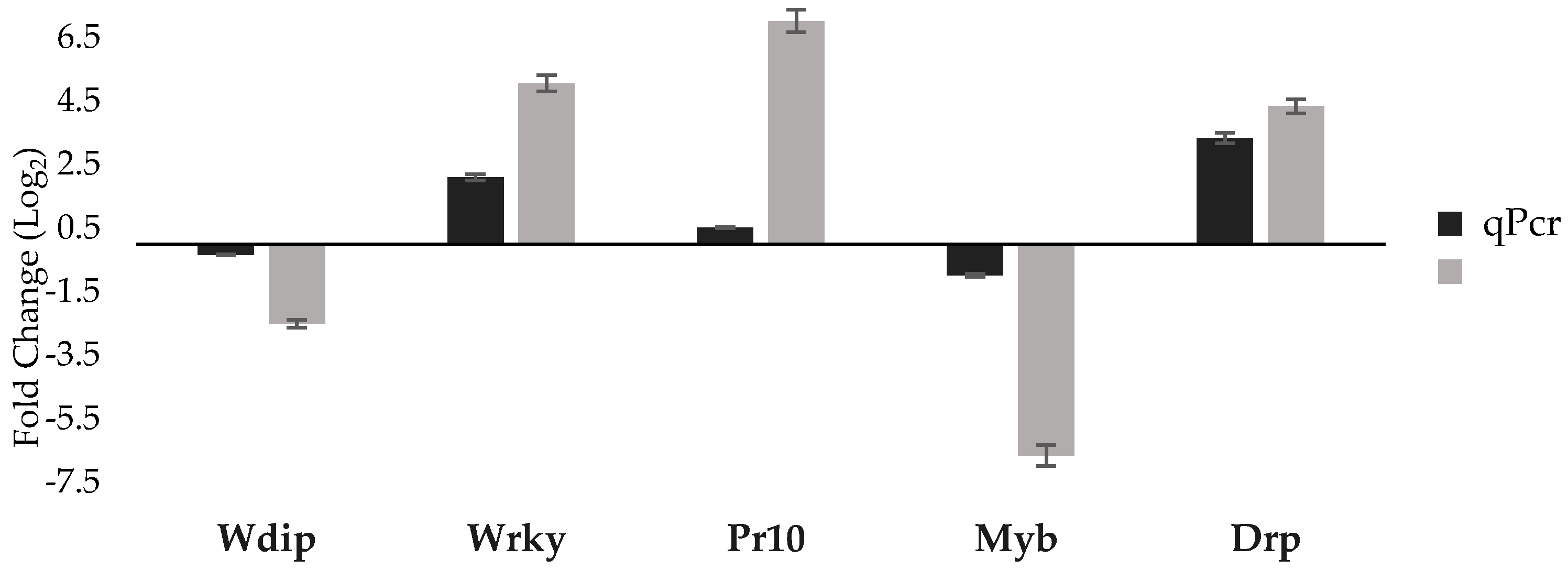
3.3. qPCR Validation
3.4. Transcriptome Annotation
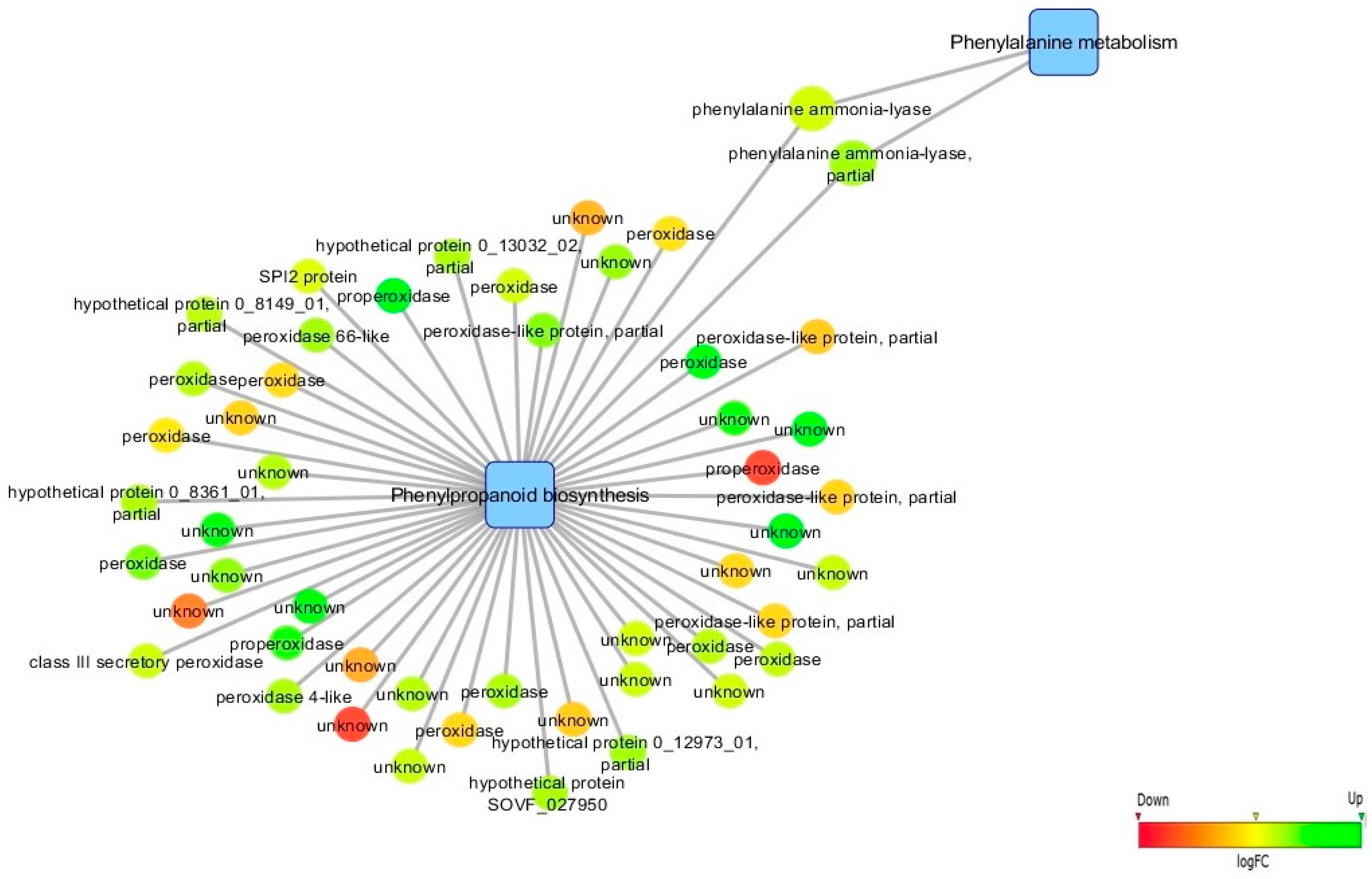
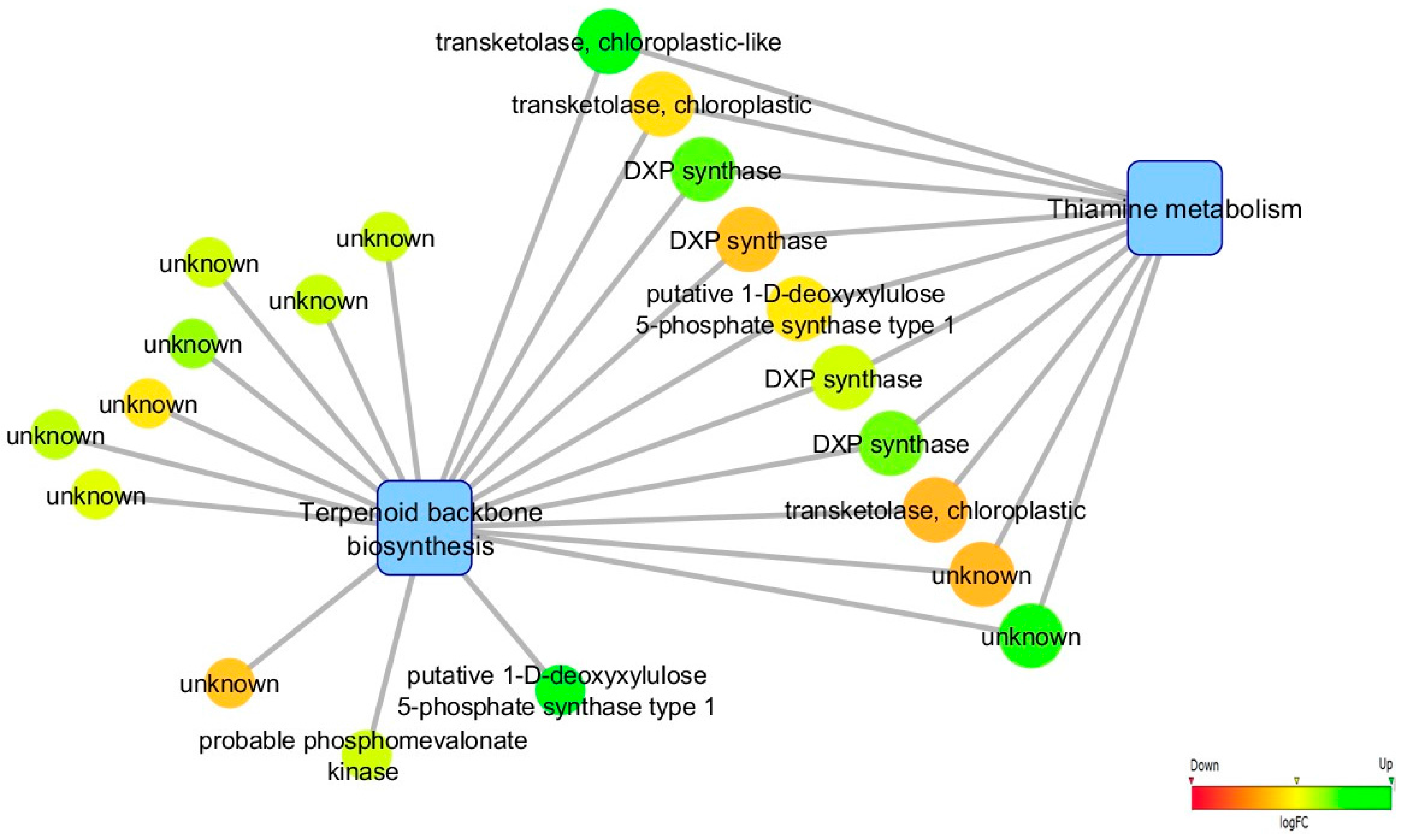
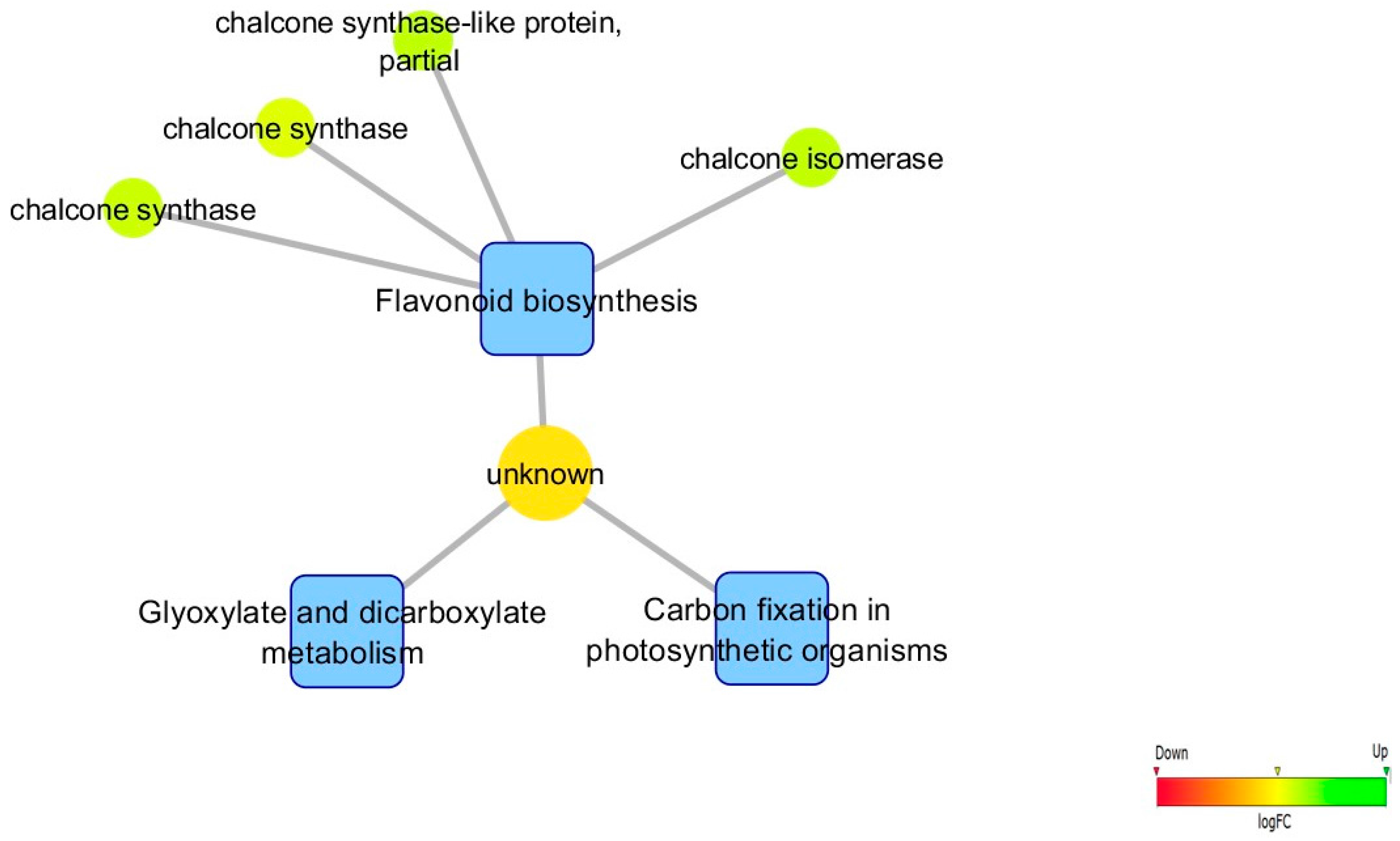
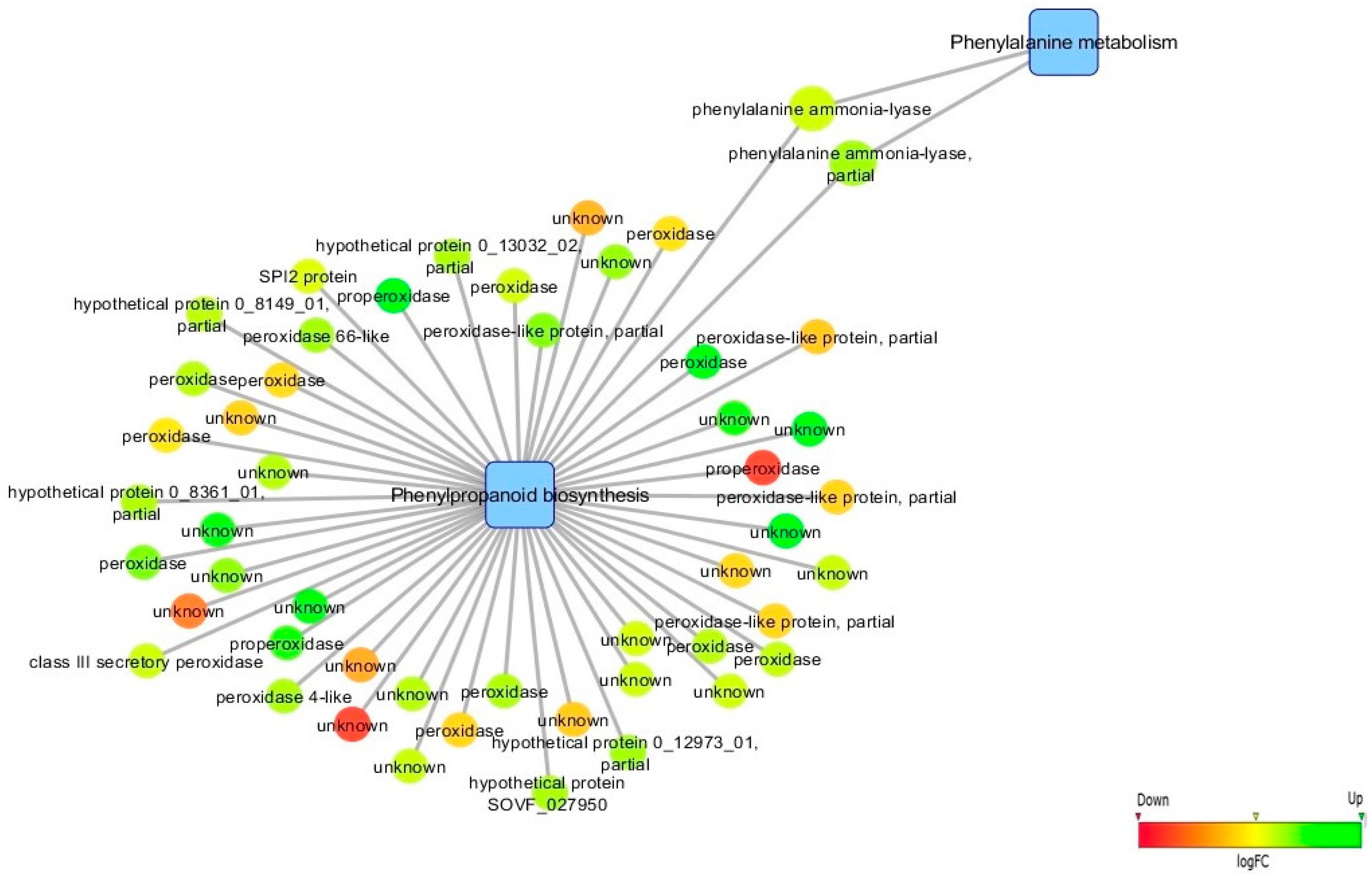
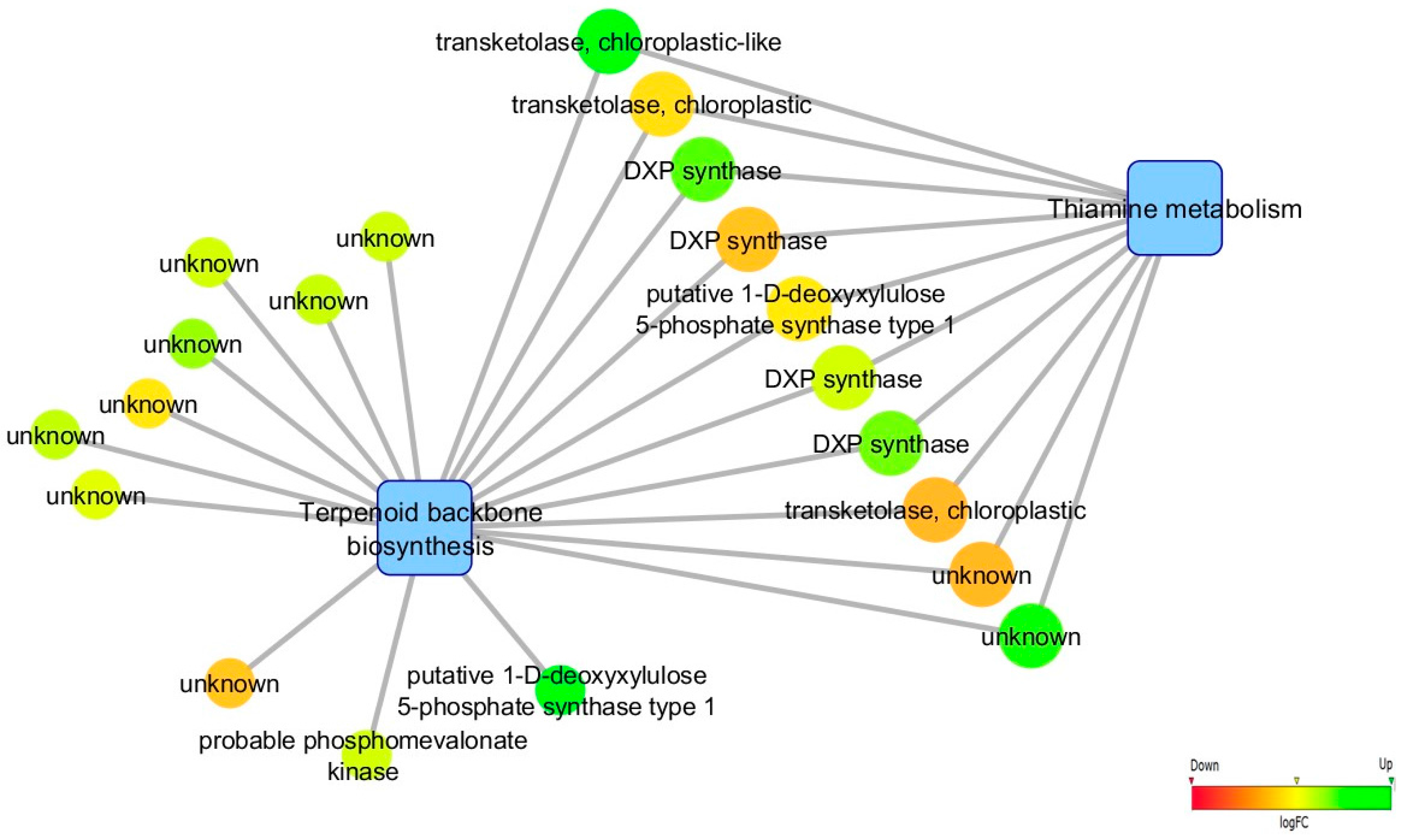
3.5. Biological Networks Analysis
3.6. SNP Calling Analysis
4. Discussion
4.1. Infection Leads to de novo Transcription of Genes Involved in Biotic Stress Response, Phenylpropanoid/Terpenoid Metabolisms and Hormonal Regulation
4.2. Infection Leads to a Reprogramming of Cell Wall Metabolism Putatively Involved in Cell Wall Reinforcement
4.3. Late Responses to Infection Seem to Be Involved in the Mitigation of Stress Caused by an Inefficient Early Response
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Plomion, C.; Pionneau, C.; Brach, J.; Costa, P.; Baillères, H. Compression wood-responsive proteins in developing xylem of maritime pine (Pinus pinaster Ait.). Plant Physiol. 2000, 123, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Uva, J.S. IFN6—Áreas dos usos do solo e das espécies florestais de Portugal continental. Inst. Conserv. Nat. Florestas I.P. 2013, 1, 1–35. [Google Scholar]
- Mota, M.M.; Futai, K.; Vieira, P. Pine Wilt Disease And The Pinewood Nematode, Bursaphelenchus Xylophilus. In Integrated Management of Fruit Crops Nematodes; Springer: Dordrecht, The Netherlands, 2009; pp. 253–274. [Google Scholar]
- Futai, K.; Sutherland, J.R.; Takeuchi, Y. Pine Wilt Disease; Springer: Tokyo, Japan, 2008; ISBN 978-4-431-75655-2. [Google Scholar]
- Mota, M.; Braasch, H.; Bravo, M.A.; Penas, A.C.; Burgermeister, W.; Metge, K.; Sousa, E. First report of Bursaphelenchus xylophilus in Portugal and in Europe. Nematology 1999, 1, 727–734. [Google Scholar] [CrossRef]
- Farjon, A. A Handbook of the World’s Conifers; Brill: Leiden, The Netherlands, 2010; ISBN 9789004177185. [Google Scholar]
- Sousa, E.; Bravo, M.A.; Pires, J.; Naves, P.; Penas, A.C.; Bonifácio, L.M.M. Bursaphelenchus xylophilus (Nematoda: Aphelenchoididae) associated with Monochamus galloprovincialis (Coleoptera: Cerambycidae) in Portugal. Nematology 2001, 3, 89–91. [Google Scholar] [CrossRef]
- Naves, P.M.; Camacho, S.; De Sousa, E.; Quartau, J.A. Transmission of the pine wood nematode Bursaphelenchus xylophilus through oviposition activity of Monochamus galloprovincialis (Coleoptera: Cerambycidae). Entomol. Fenn. 2007, 18, 193–198. [Google Scholar]
- Fielding, N.J.; Evans, H.F. The pine wood nematode Bursaphelenchus xylophilus (Steiner and Buhrer) Nickle (= B. lignicolus Mamiya and Kiyohara): An assessment of the current position. Forestry 1996, 69, 35–46. [Google Scholar] [CrossRef]
- Edwards, O.R.; Linit, M.J. Transmission of Bursaphelenchus xylophilus through Oviposition Wounds of Monochamm carolinensis (Coleoptera: Cerambycidae). J. Nematol. 1992, 24, 133–139. [Google Scholar] [PubMed]
- Naves, P.M.; Camacho, S.; de Sousa, E.M.; Quartau, J.A. Transmission of the pine wood nematode Bursaphelenchus xylophilus through feeding activity of Monochamus galloprovincialis (Col., Cerambycidae). J. Appl. Entomol. 2007, 131, 21–25. [Google Scholar] [CrossRef]
- Fukuda, K. Physiological process of the symptom development and resistance mechanism in pine wilt disease. J. For. Res. 1997, 2, 171–181. [Google Scholar] [CrossRef]
- Jusheng, H. A brief account of forest tree improvment in China. Genet. Resour. Inf. 1985, 14, 2–6. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Parchman, T.L.; Geist, K.S.; Grahnen, J.A.; Benkman, C.W.; Buerkle, C.A. Transcriptome sequencing in an ecologically important tree species: Assembly, annotation, and marker discovery. BMC Genom. 2010, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Mota, M.M.; Takemoto, S.; Takeuchi, Y.; Hara, N.; Futai, K. Comparative Studies between Portuguese and Japanese Isolates of the Pinewood Nematode, Bursaphelenchus xylophilus. J. Nematol. 2006, 38, 429–433. [Google Scholar] [PubMed]
- Naves, P.M.; Sousa, E.; Rodrigues, J.M. Biology of Monochamus galloprovincialis (Coleoptera, Cerambycidae) in the Pine Wilt Disease Affected Zone, Southern Portugal. Silva Lusit. 2008, 16, 133–148. [Google Scholar]
- Valadas, V.; Oliveira, S.; Espada, M.; Laranjo, M.; Mota, M.; Barbosa, P. The pine wood nematode, Bursaphelenchus xylophilus, in Portugal: Possible introductions and spread routes of a serious biological invasion revealed by molecular methods. Nematology 2012, 14, 899–911. [Google Scholar] [CrossRef]
- Vicente, C.S.L.; Nascimento, F.; Espada, M.; Barbosa, P.; Mota, M.; Glick, B.R.; Oliveira, S. Characterization of Bacteria Associated with Pinewood Nematode Bursaphelenchus xylophilus. PLoS ONE 2012, 7, e46661. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.S.; Pinheiro, M.; Silva, A.I.; Egas, C.; Vasconcelos, M.W. Searching for resistance genes to Bursaphelenchus xylophilus using high throughput screening. BMC Genom. 2012, 13, 599. [Google Scholar] [CrossRef] [PubMed]
- Baermann, G. Ein einfache Methode zur Auffindung von Anklyostomum (Nematoden) Larven in Erdproben. Ned. Tijdschr. Geneeskd. 1917, 57, 131–137. [Google Scholar]
- Futai, K.; Furuno, T. The variety of resistances among pine species to pine wood nematode, Bursaphelenchus lignicolus. Bull. Kyoto Univ. For. 1979, 51, 23–36. [Google Scholar]
- Le Provost, G.; Herrera, R.; Paiva, J.A.; Chaumeil, P.; Salin, F.; Plomion, C. A micromethod for high throughput RNA extraction in forest trees. Biol. Res. 2007, 40, 291–297. [Google Scholar] [CrossRef] [PubMed]
- FastQC—A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 20 September 2016).
- Sickle: A Windowed Adaptive Trimming Tool for FASTQ Files Using Quality. Available online: https://github.com/najoshi/sickle (accessed on 20 September 2016).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Haas, B. TransDecoder (Find Coding Regions Within Transcripts). Available online: http://transdecoder.github.io (accessed on 16 May 2016).
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.-C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2015, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Eddy, S.R. Multiple alignment using hidden Markov models. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1995, 3, 114–120. [Google Scholar] [PubMed]
- Srivastava, A.; Sarkar, H.; Gupta, N.; Patro, R. RapMap: A Rapid, Sensitive and Accurate Tool for Mapping RNA-seq Reads to Transcriptomes. Bioinformatics 2016, 32, i192–i200. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [PubMed]
- Sebastiana, M.; Vieira, B.; Lino-Neto, T.; Monteiro, F.; Figueiredo, A.; Sousa, L.; Pais, M.S.; Tavares, R.; Paulo, O.S. Oak Root Response to Ectomycorrhizal Symbiosis Establishment: RNA-Seq Derived Transcript Identification and Expression Profiling. PLoS ONE 2014, 9, e98376. [Google Scholar] [CrossRef] [PubMed]
- Yoav, B.; Yosef, H. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [PubMed]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Zhi-Liang, H.; Jie, B.; James, M.R. CateGOrizer: A Web-Based Program to Batch Analyze Gene Ontology Classification Categories. Online J. Bioinform. 2008, 9, 108–112. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [PubMed]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Vogt, T. Phenylpropanoid Biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Vásquez, R.; Day, R.; Buschmann, H.; Randles, S.; Beeching, J.R.; Cooper, R.M. Phenylpropanoids, phenylalanine ammonia lyase and peroxidases in elicitor-challenged cassava (Manihot esculenta) suspension cells and leaves. Ann. Bot. 2004, 94, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Sattler, S.E.; Funnell-Harris, D.L. Modifying lignin to improve bioenergy feedstocks: Strengthening the barrier against pathogens? Front. Plant Sci. 2013, 4, 70. [Google Scholar] [CrossRef] [PubMed]
- Stermer, B.A.; Bianchini, G.M.; Korth, K.L. Regulation of HMG-CoA reductase activity in plants. J. Lipid Res. 1994, 35, 1133–1140. [Google Scholar] [PubMed]
- Wright, L.P.; Phillips, M.A. Measuring the activity of 1-deoxy-d-xylulose 5-phosphate synthase, the first enzyme in the MEP pathway, in plant extracts. Methods Mol. Biol. 2014, 1153, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.T.H.; Linthorst, H.J.M.; Verpoorte, R. Chalcone synthase and its functions in plant resistance. Phytochem. Rev. 2011, 10, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Gensheimer, M.; Mushegian, A. Chalcone isomerase family and fold: No longer unique to plants. Protein Sci. 2004, 13, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Guo, J.; Bai, P.; Duan, Y.; Wang, X.; Cheng, Y.; Feng, H.; Huang, L.; Kang, Z. Wheat TaRab7 GTPase Is Part of the Signaling Pathway in Responses to Stripe Rust and Abiotic Stimuli. PLoS ONE 2012, 7, e37146. [Google Scholar] [CrossRef] [PubMed]
- Navrot, N.; Collin, V.; Gualberto, J.; Gelhaye, E.; Hirasawa, M.; Rey, P.; Knaff, D.B.; Issakidis, E.; Jacquot, J.-P.; Rouhier, N. Plant Glutathione Peroxidases Are Functional Peroxiredoxins Distributed in Several Subcellular Compartments and Regulated during Biotic and Abiotic Stresses. Plant Physiol. 2006, 142, 1364–1379. [Google Scholar] [CrossRef] [PubMed]
- Hanin, M.; Brini, F.; Ebel, C.; Toda, Y.; Takeda, S.; Masmoudi, K. Plant dehydrins and stress tolerance: Versatile proteins for complex mechanisms. Plant Signal. Behav. 2011, 6, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.L.; Lee, J.M.; Tearse, B.R.; Neale, D.B. TreeGenes: A forest tree genome database. Int. J. Plant Genom. 2008, 412875. [Google Scholar] [CrossRef] [PubMed]
- Bassingthwaighte, J.B. Strategies for the Physiome Project. Ann. Biomed. Eng. 2000, 28, 1043–1058. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, B.A.; Ashihara, H. Purine and pyrimidine nucleotide synthesis and metabolism. Arabidopsis Book 2002, 1, e0018. [Google Scholar] [CrossRef] [PubMed]
- Ling, H. Sequence analysis of GDSL lipase gene family in Arabidopsis thaliana. Pak. J. Biol. Sci. 2008, 11, 763–767. [Google Scholar] [CrossRef]
- Oh, S., II; Park, A.R.; Bae, M.S.; Kwon, S.J.; Kim, Y.S.; Lee, J.E.; Kang, N.Y.; Lee, S.; Cheong, H.; Park, O.K. Secretome Analysis Reveals an Arabidopsis Lipase Involved in Defense against Alternaria brassicicola. Plant Cell 2005, 17, 2832–2847. [Google Scholar] [CrossRef] [PubMed]
- Bommer, U.A.; Thiele, B.J. The translationally controlled tumour protein (TCTP). Int. J. Biochem. Cell Biol. 2004, 36, 379–385. [Google Scholar] [CrossRef]
- Xiang, Y.; Song, M.; Wei, Z.; Tong, J.; Zhang, L.; Xiao, L.; Ma, Z.; Wang, Y. A jacalin-related lectin-like gene in wheat is a component of the plant defence system. J. Exp. Bot. 2011, 62, 5471–5483. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, Z.-Y.; Zhang, K.; Lu, Q.; Liang, J.; Zhang, X.-Y. Characterization of the Pinus massoniana transcriptional response to Bursaphelenchus xylophilus infection using suppression subtractive hybridization. Int. J. Mol. Sci. 2013, 14, 11356–11375. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Lv, Y.; Hou, Z.; Li, X.; Ding, L. Expansion and evolution of thaumatin-like protein (TLP) gene family in six plants. Plant Growth Regul. 2016, 79, 299–307. [Google Scholar] [CrossRef]
- Liu, J.-J.; Sturrock, R.; Ekramoddoullah, A.K.M. The superfamily of thaumatin-like proteins: Its origin, evolution, and expression towards biological function. Plant Cell Rep. 2010, 29, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Zalabák, D.; Pospíšilová, H.; Šmehilová, M.; Mrízová, K.; Frébort, I.; Galuszka, P. Genetic engineering of cytokinin metabolism: Prospective way to improve agricultural traits of crop plants. Biotechnol. Adv. 2013, 31, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Wani, S.H.; Kumar, V.; Shriram, V.; Sah, S.K. Phytohormones and their metabolic engineering for abiotic stress tolerance in crop plants. Crop J. 2016, 4, 162–176. [Google Scholar] [CrossRef]
- Shin, H.; Lee, H.; Woo, K.S.; Noh, E.W.; Koo, Y.B.; Lee, K.J. Identification of genes upregulated by pinewood nematode inoculation in Japanese red pine. Tree Physiol. 2009, 29, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Bouché, N.; Yellin, A.; Snedden, W.A.; Fromm, H. Plant-specific calmodulin-binding proteins. Annu. Rev. Plant Biol. 2005, 56, 435–466. [Google Scholar] [CrossRef] [PubMed]
- Nairn, C.J.; Lennon, D.M.; Wood-Jones, A.; Nairn, A.V.; Dean, J.F.D. Carbohydrate-related genes and cell wall biosynthesis in vascular tissues of loblolly pine (Pinus taeda). Tree Physiol. 2008, 28, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Parrotta, L.; Faleri, C.; Cresti, M.; Cai, G. Heat stress affects the cytoskeleton and the delivery of sucrose synthase in tobacco pollen tubes. Planta 2016, 243, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Belkhadir, Y.; Subramaniam, R.; Dangl, J.L. Plant disease resistance protein signaling: NBS–LRR proteins and their partners. Curr. Opin. Plant Biol. 2004, 7, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Doi, R.H.; Kosugi, A. Cellulosomes: Plant-cell-wall-degrading enzyme complexes. Nat. Rev. Microbiol. 2004, 2, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Christopher, L.P.; Yao, B.; Ji, Y. Lignin Biodegradation with Laccase-Mediator Systems. Front. Energy Res. 2014, 2, 12. [Google Scholar] [CrossRef]
- Dubos, C. Drought differentially affects expression of a PR-10 protein, in needles of maritime pine (Pinus pinaster Ait.) seedlings. J. Exp. Bot. 2001, 52, 1143–1144. [Google Scholar] [CrossRef] [PubMed]
- McHale, L.; Tan, X.; Koehl, P.; Michelmore, R.W. Plant NBS-LRR proteins: Adaptable guards. Genome Biol. 2006, 7, 212. [Google Scholar] [CrossRef] [PubMed]
- Carna, M.; Repka, V.; Skupa, P.; Sturdik, E. Auxins in defense strategies. Biologia (Bratisl) 2014, 69, 1255–1263. [Google Scholar] [CrossRef]
- Canonne, J.; Froidure-Nicolas, S.; Rivas, S. Phospholipases in action during plant defense signaling. Plant Signal. Behav. 2011, 6, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Kilili, K.G.; Atanassova, N.; Vardanyan, A.; Clatot, N.; Al-Sabarna, K.; Kanellopoulos, P.N.; Makris, A.M.; Kampranis, S.C. Differential roles of tau class glutathione S-transferases in oxidative stress. J. Biol. Chem. 2004, 279, 24540–24551. [Google Scholar] [CrossRef] [PubMed]
- Preisig-müller, R.; Schwekendiek, A.; Brehm, I.; Reif, H.-J.; Kindl, H. Characterization of a pine multigene family containing elicitor-responsive stilbene synthase genes. Plant Mol. Biol. 1999, 39, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Kodan, A.; Kuroda, H.; Sakai, F. A stilbene synthase from Japanese red pine (Pinus densiflora): Implications for phytoalexin accumulation and down-regulation of flavonoid biosynthesis. Proc. Natl. Acad. Sci. USA 2002, 99, 3335–3339. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.; Stenlid, J.; Olson, Å. Identification of a superoxide dismutase gene from the conifer pathogen Heterobasidion annosum. Physiol. Mol. Plant Pathol. 2005, 66, 99–107. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Number of Sequenced Reads | Average Read Length (bp) | Number of Reads after QC | % Reads after QC |
|---|---|---|---|---|
| Pp01—Control (0 h) | 47,903,109 | 122 | 39,091,399 | 81.6 |
| Pp02—6 h + 24 h | 38,483,969 | 119 | 30,863,177 | 80.2 |
| Pp03—48 h | 44,943,925 | 122 | 37,186,370 | 82.7 |
| Pp04—7 days | 44,951,165 | 121 | 37,281,261 | 82.9 |
| Total | 176,282,168 | 121 | 144,422,207 | 81.9 |
| Sample | Number of Reads Mapped | Number of Unique Mapped Reads | % of Mapped Reads | % of Unique Mapped Reads |
|---|---|---|---|---|
| Pp01—Control | 27,683,922 | 14,432,190 | 70.8 | 36.9 |
| Pp02—6 h + 24 h | 22,608,382 | 12,162,427 | 73.3 | 39.4 |
| Pp03—48 h | 26,553,659 | 14,077,794 | 71.4 | 37.9 |
| Pp04—7 days | 26,423,215 | 13,828,813 | 70.9 | 37.1 |
| Total | 103,269,178 | 54,501,224 | 71.6 | 37.7 |
| Gene Annotation | Condition | Comparison | Log Fold-Change |
|---|---|---|---|
| GDSL esterase/lipase | Up | Control vs. Pp02 | 12.4 |
| Translationally-controlled tumor protein homolog | Up | Control vs. Pp02 | 11.8 |
| Jacalin-related lectin 3 protein | Up | Control vs. Pp02 | 6.9 |
| Cytokinin dehydrogenase 6-like | Up | Control vs. Pp02 | 6.9 |
| Endoglucanase | Up | Control vs. Pp02 | 9.4 |
| Acyl-CoA oxidase | Up | Control vs. Pp02 | 4.4 |
| Thaumatin-like protein | Up | Control vs. Pp02 | 4.3 |
| Nucleotide-binding site leucine-rich repeat (NBS-LRR) | Down | Control vs. Pp02 | −3.1 |
| 12-oxophytodienoate reductase 3-like | Up | Control vs. Pp02 | 2.9 |
| Iron superoxide dismutase | Up | Control vs. Pp02 | 1.9 |
| Auxin-induced cell wall protein | Up | Control vs. Pp02 | 7.7 |
| Multifunctional protein (MFP) | Up | Control vs. Pp02 | 1.3 |
| Mildew resistance locus 6 calmodulin binding protein | Up | Control vs. Pp02 | 2.7 |
| Sucrose synthase | Up | Control vs. Pp02 | 2.5 |
| TMV resistance protein N-like | Down | Control vs. Pp02 | −7.1 |
| Phenylalanine ammonia-lyase | Up | Control vs. Pp02 | 2.1 |
| Peroxidase | Up | Control vs. Pp02 | 7.7 |
| GDSL esterase/lipase | Up | Control vs. Pp03 | 9.9 |
| Translationally-controlled tumor protein homolog | Up | Control vs. Pp03 | 10.4 |
| Endoglucanase | Up | Control vs. Pp03 | 9.7 |
| Thaumatin-like protein | Up | Control vs. Pp03 | 2.5 |
| (E)-4-hydroxy-3-methylbut-2-enyl diphosphate synthase | Up | Control vs. Pp03 | 4.3 |
| Nucleotide-binding site leucine-rich repeat (NBS-LRR) | Down | Control vs. Pp03 | −12.5 |
| GDSL esterase/lipase | Up | Control vs. Pp04 | 8.1 |
| Translationally-controlled tumor protein homolog | Up | Control vs. Pp04 | 9.6 |
| Jacalin-related lectin 3 protein | Up | Control vs. Pp04 | 8.6 |
| Cytokinin dehydrogenase 6-like | Up | Control vs. Pp04 | 6.9 |
| Endoglucanase | Up | Control vs. Pp04 | 9.1 |
| Acyl-CoA oxidase | Up | Control vs. Pp04 | 4.9 |
| Thaumatin-like protein | Up | Control vs. Pp04 | 3.1 |
| (E)-4-hydroxy-3-methylbut-2-enyl diphosphate synthase | Up | Control vs. Pp04 | 3.7 |
| Pinosylvin synthase | Up | Control vs. Pp04 | 2.8 |
| Nucleotide-binding site leucine-rich repeat (NBS-LRR) | Down | Control vs. Pp04 | −8.5 |
| Auxin-induced protein 1 | Down | Pp02 vs. Pp03 | −1.9 |
| Laccase | Up | Pp02 vs. Pp03 | 6.7 |
| Dehydrin | Down | Pp02 vs. Pp04 | −11.0 |
| Pathogenesis related 10 | Down | Pp02 vs. Pp04 | −3.0 |
| Pinosylvin synthase | Up | Pp02 vs. Pp04 | 2.6 |
| Heat shock protein | Up | Pp02 vs. Pp04 | 7.6 |
| Light harvesting complex protein | Up | Pp02 vs. Pp04 | 7.9 |
| Nucleotide-binding site leucine-rich repeat (NBS-LRR) | Up | Pp02 vs. Pp04 | 8.8 |
| Phospholipase D alpha 1-like | Up | Pp03 vs. Pp04 | 2.7 |
| Tau class glutathione S-transferase | Up | Pp03 vs. Pp04 | 3.6 |
| Pinosylvin synthase | Up | Pp03 vs. Pp04 | 3.5 |
| Pathways | Enzymes in All Set of Genes | Enzymes in DE Genes |
|---|---|---|
| Purine metabolism | 37 | 24 |
| Pyrimidine metabolism | 26 | 18 |
| Aminoacyl-tRNA biosynthesis | 21 | 15 |
| Cysteine and methionine metabolism | 20 | 16 |
| Starch and sucrose metabolism | 19 | 16 |
| Porphyrin and chlorophyll metabolism | 20 | 12 |
| Phenylalanine, tyrosine and tryptophan biosynthesis | 18 | 14 |
| Pyruvate metabolism | 17 | 14 |
| Glycolysis/Gluconeogenesis | 17 | 15 |
| Carbon fixation in photosynthetic organisms | 13 | 12 |
| Terpenoid backbone biosynthesis | 17 | 7 |
| Region | Count | Percentage |
|---|---|---|
| Exon | 15,232 | 31.9% |
| Intergenic | 14,600 | 30.6% |
| Splice site region | 1 | <0.1% |
| Transcript | 31 | 0.1% |
| UTR 3 Prime | 9072 | 19.0% |
| UTR 5 Prime | 8718 | 18.3% |
| Type | Count | Percentage |
|---|---|---|
| MISSENSE | 7410 | 48.5% |
| NONSENSE | 121 | 0.8% |
| SILENT | 7732 | 50.7% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaspar, D.; Trindade, C.; Usié, A.; Meireles, B.; Barbosa, P.; Fortes, A.M.; Pesquita, C.; Costa, R.L.; Ramos, A.M. Expression Profiling in Pinus pinaster in Response to Infection with the Pine Wood Nematode Bursaphelenchus xylophilus. Forests 2017, 8, 279. https://doi.org/10.3390/f8080279
Gaspar D, Trindade C, Usié A, Meireles B, Barbosa P, Fortes AM, Pesquita C, Costa RL, Ramos AM. Expression Profiling in Pinus pinaster in Response to Infection with the Pine Wood Nematode Bursaphelenchus xylophilus. Forests. 2017; 8(8):279. https://doi.org/10.3390/f8080279
Chicago/Turabian StyleGaspar, Daniel, Cândida Trindade, Ana Usié, Brígida Meireles, Pedro Barbosa, Ana M. Fortes, Cátia Pesquita, Rita L. Costa, and António M. Ramos. 2017. "Expression Profiling in Pinus pinaster in Response to Infection with the Pine Wood Nematode Bursaphelenchus xylophilus" Forests 8, no. 8: 279. https://doi.org/10.3390/f8080279
APA StyleGaspar, D., Trindade, C., Usié, A., Meireles, B., Barbosa, P., Fortes, A. M., Pesquita, C., Costa, R. L., & Ramos, A. M. (2017). Expression Profiling in Pinus pinaster in Response to Infection with the Pine Wood Nematode Bursaphelenchus xylophilus. Forests, 8(8), 279. https://doi.org/10.3390/f8080279