
De Novo Transcriptome Characterization, Gene Expression Profiling and Ionic Responses of Nitraria sibirica Pall. under Salt Stress


Abstract
:1. Introduction
2. Materials and Methods
2.1. Nitraria sibirica Pall. Materials and Hydroponic Culture
2.2. Salt Treatment and Sampling
2.3. RNA Extraction and Library Preparation for Transcriptome Sequencing
2.4. Analysis of Illumina Sequencing Data
2.5. Digital Gene Expression Library Preparation and Sequencing
2.6. Analysis and Mapping of Digital Gene Expression Tags
2.7. Quantitative PCR (qPCR) Analysis
2.8. Element Profiling Analysis
3. Results
3.1. Sequencing Data Quality and Assembly of Reads
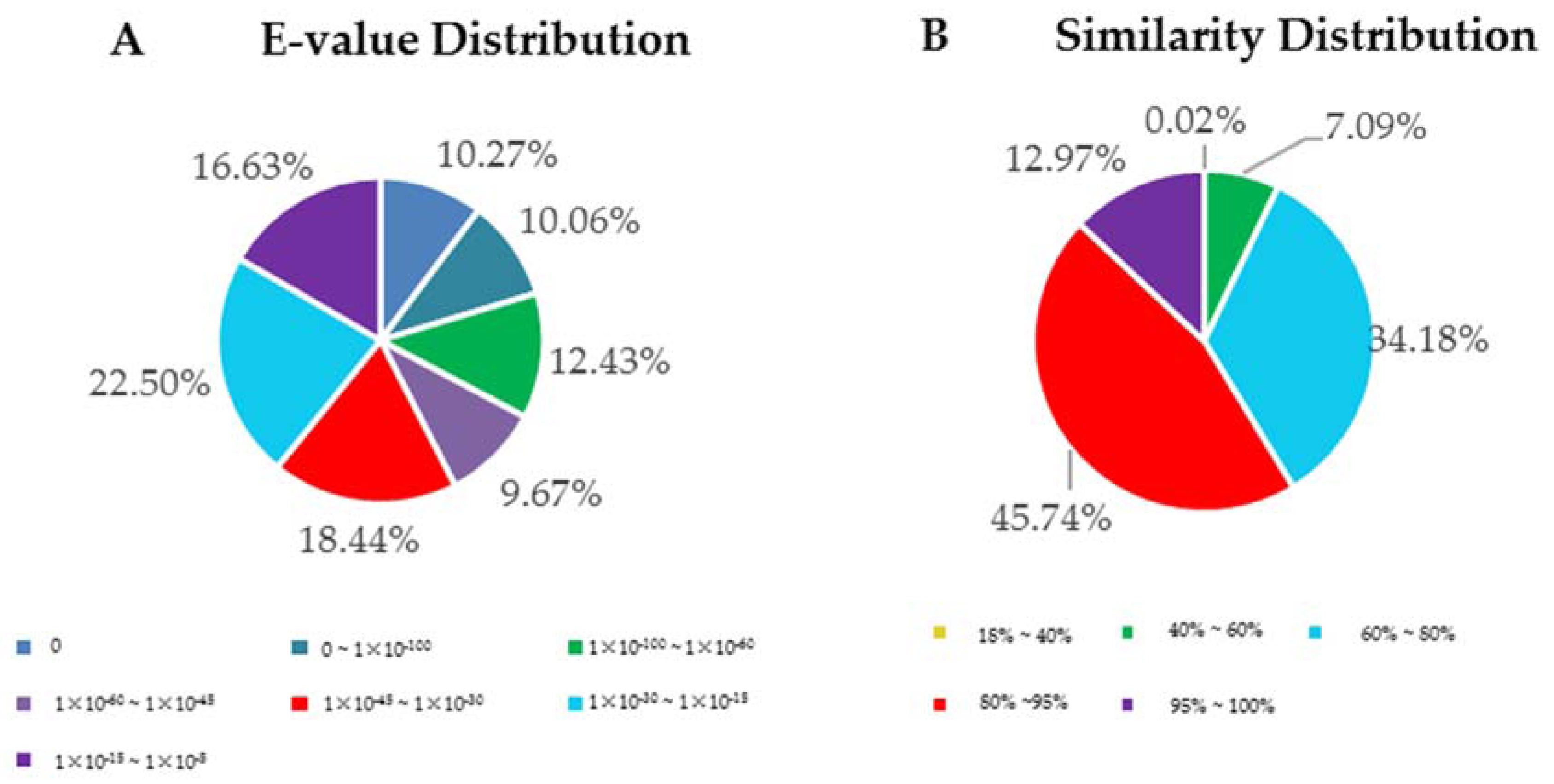
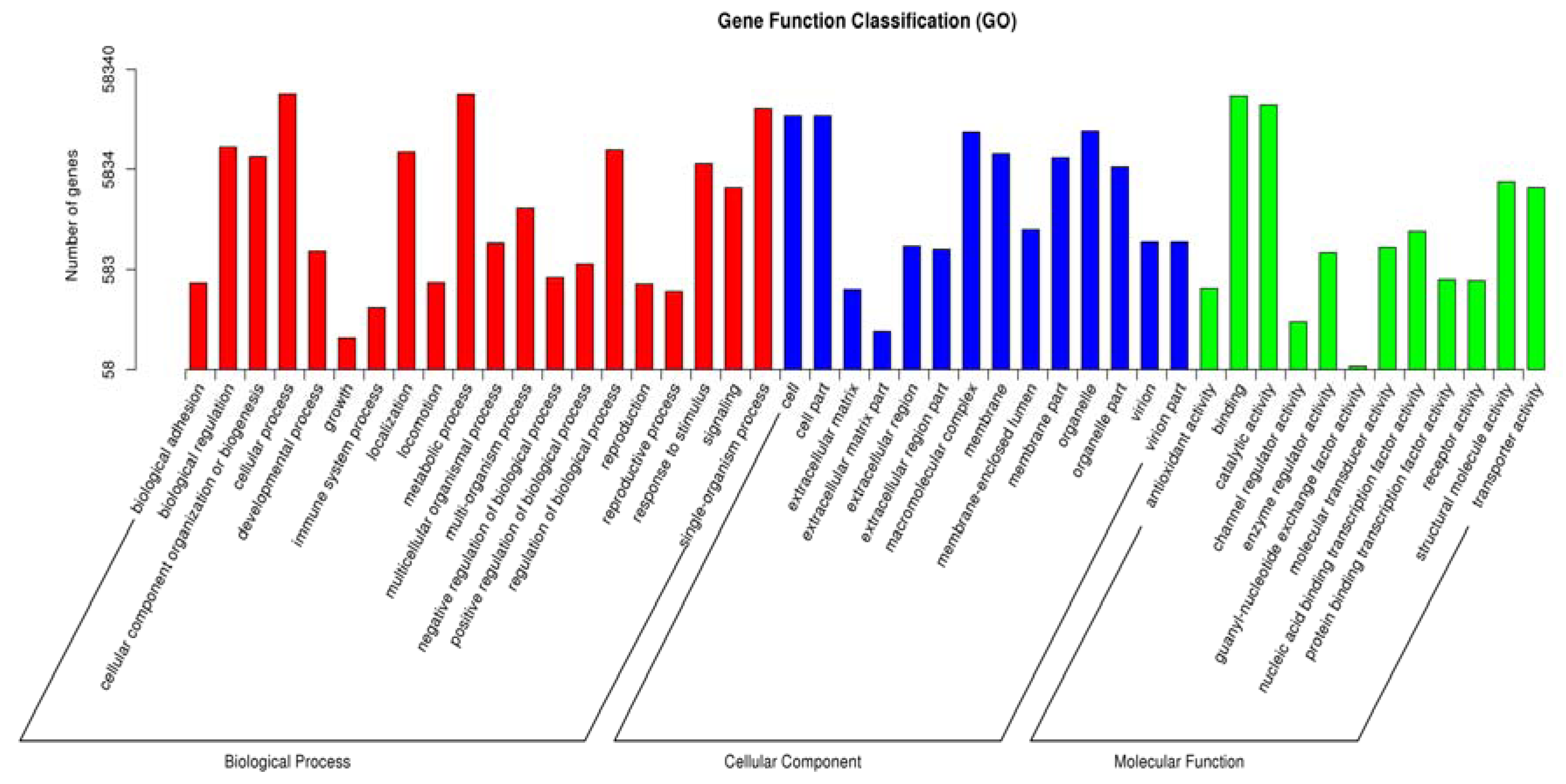
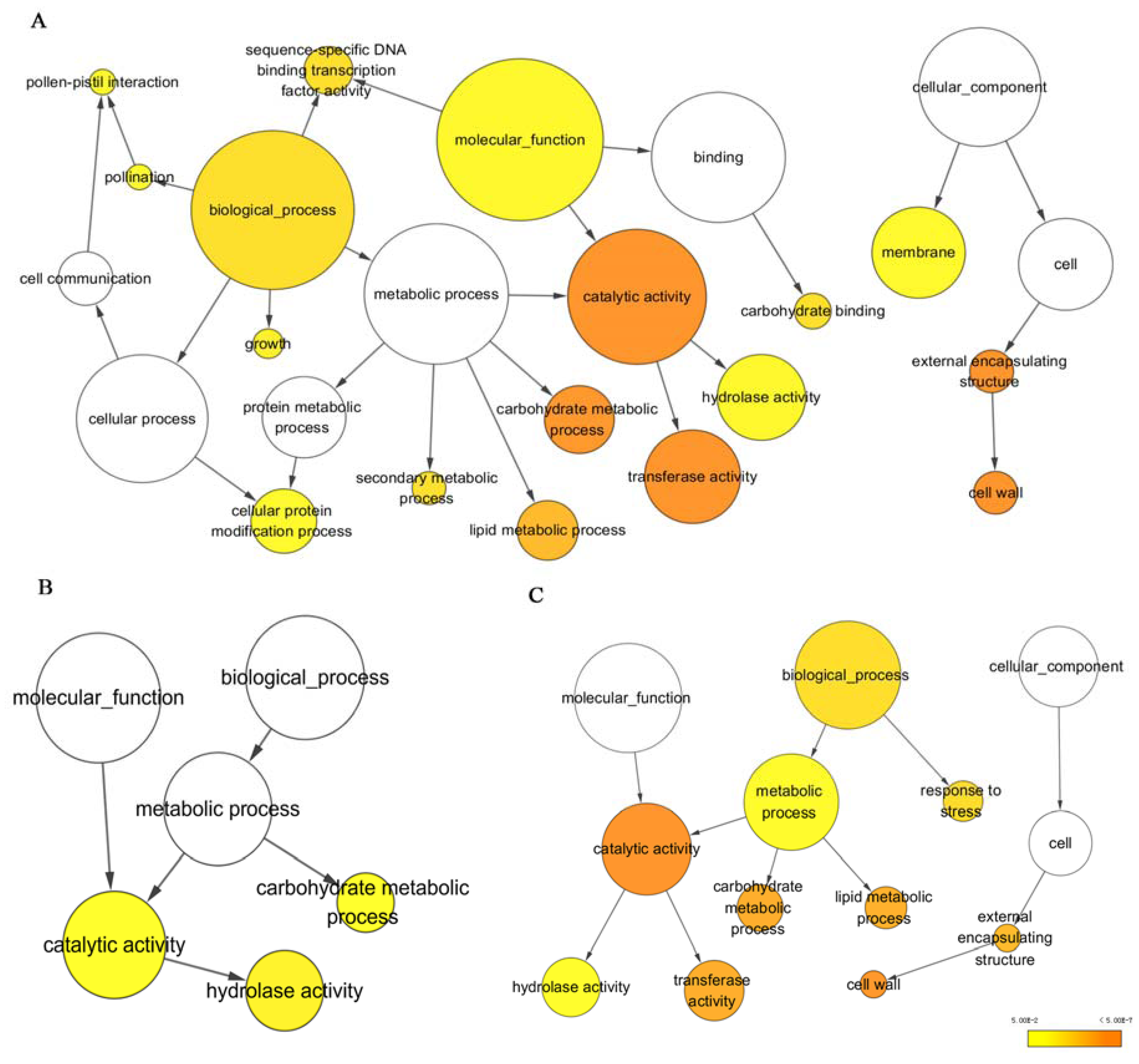
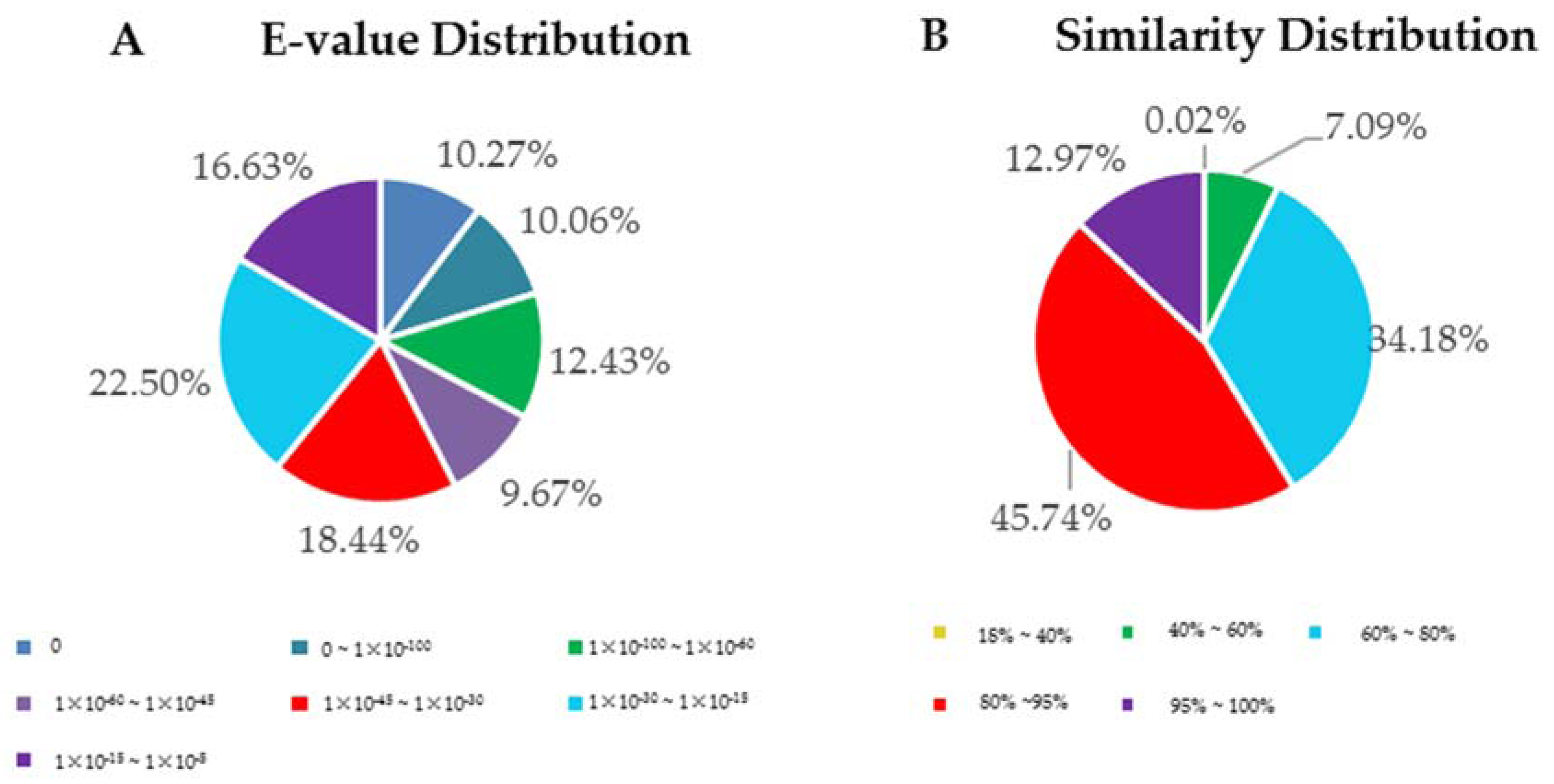
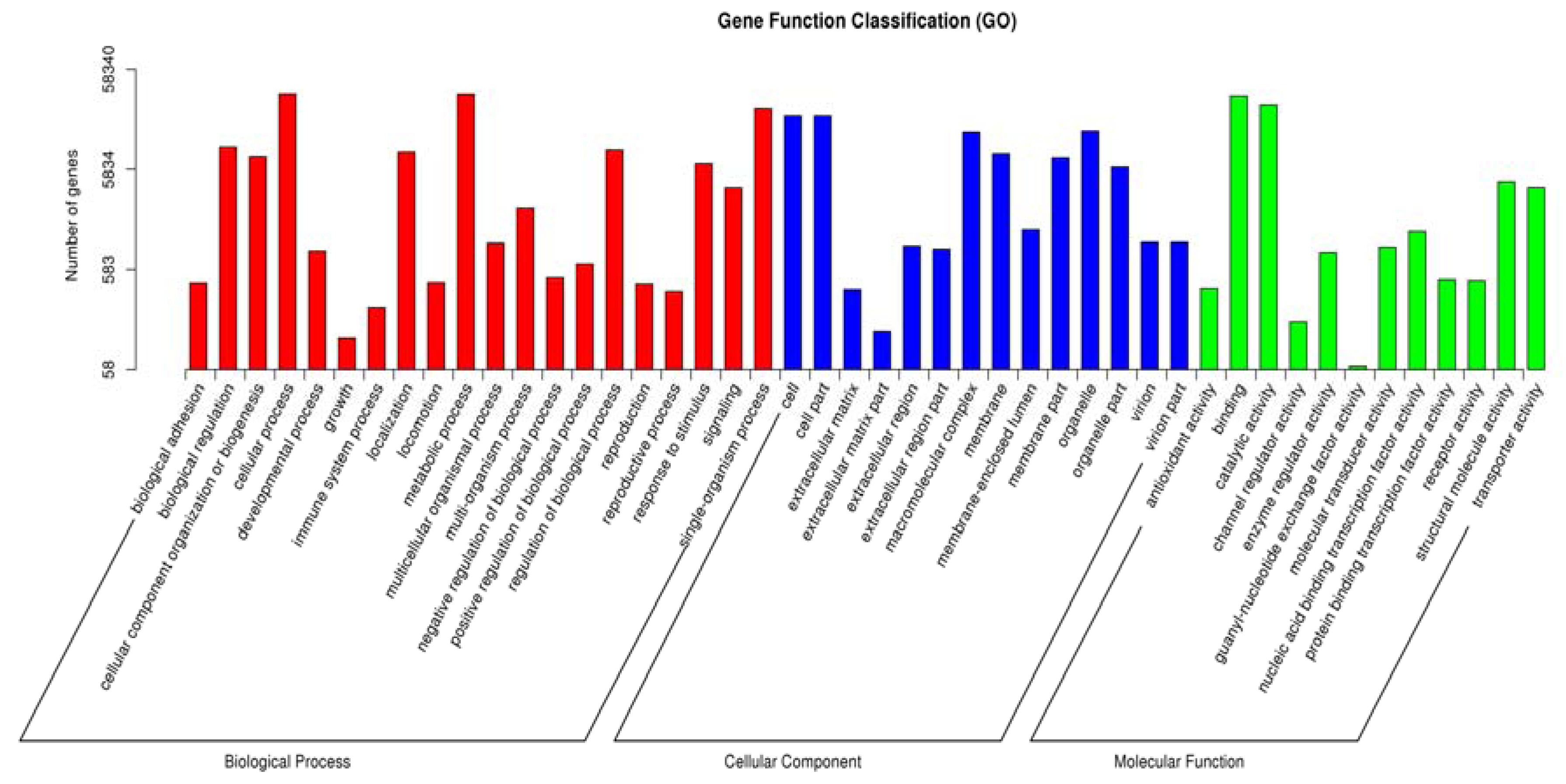
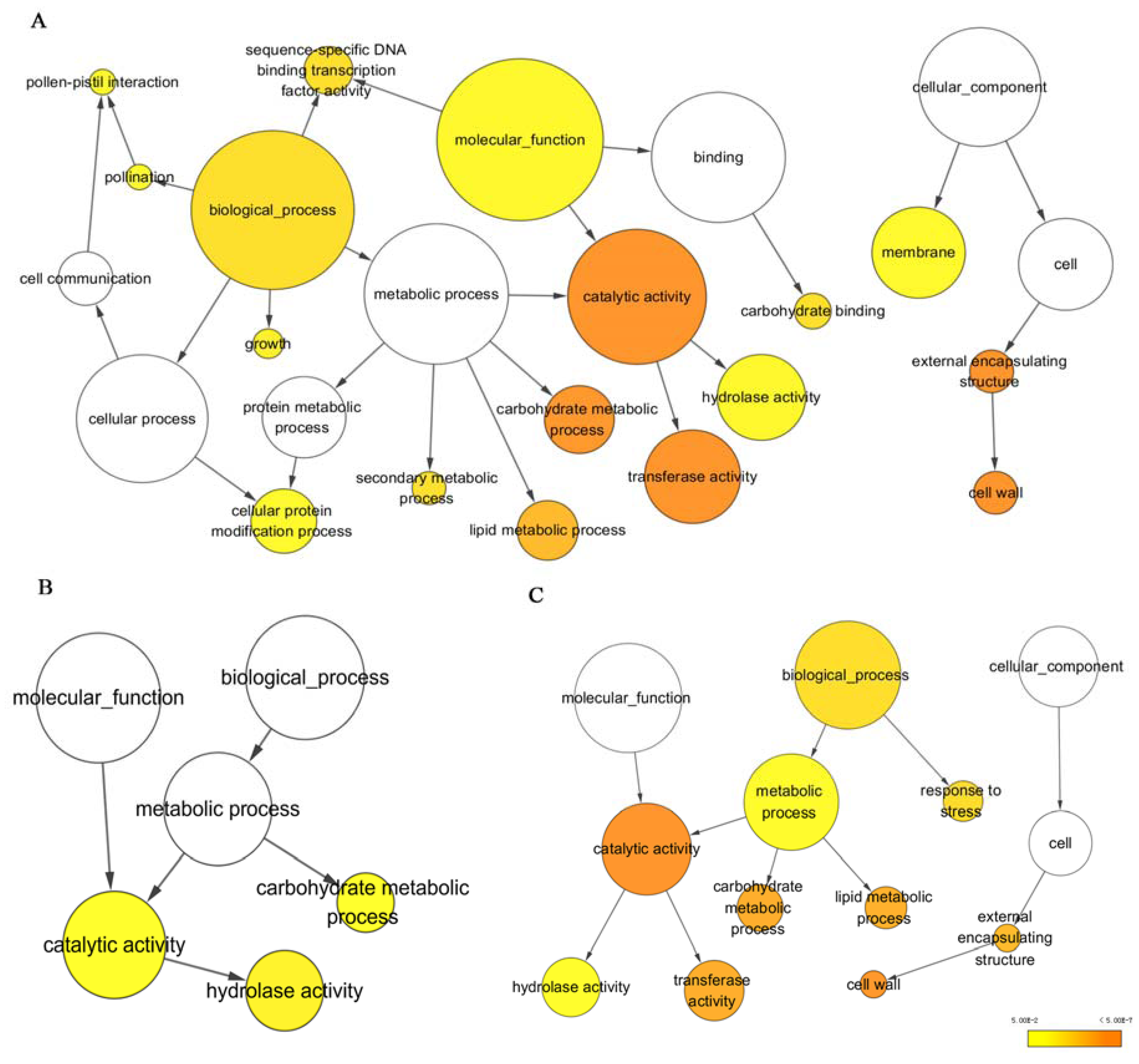
3.2. Functional Annotation
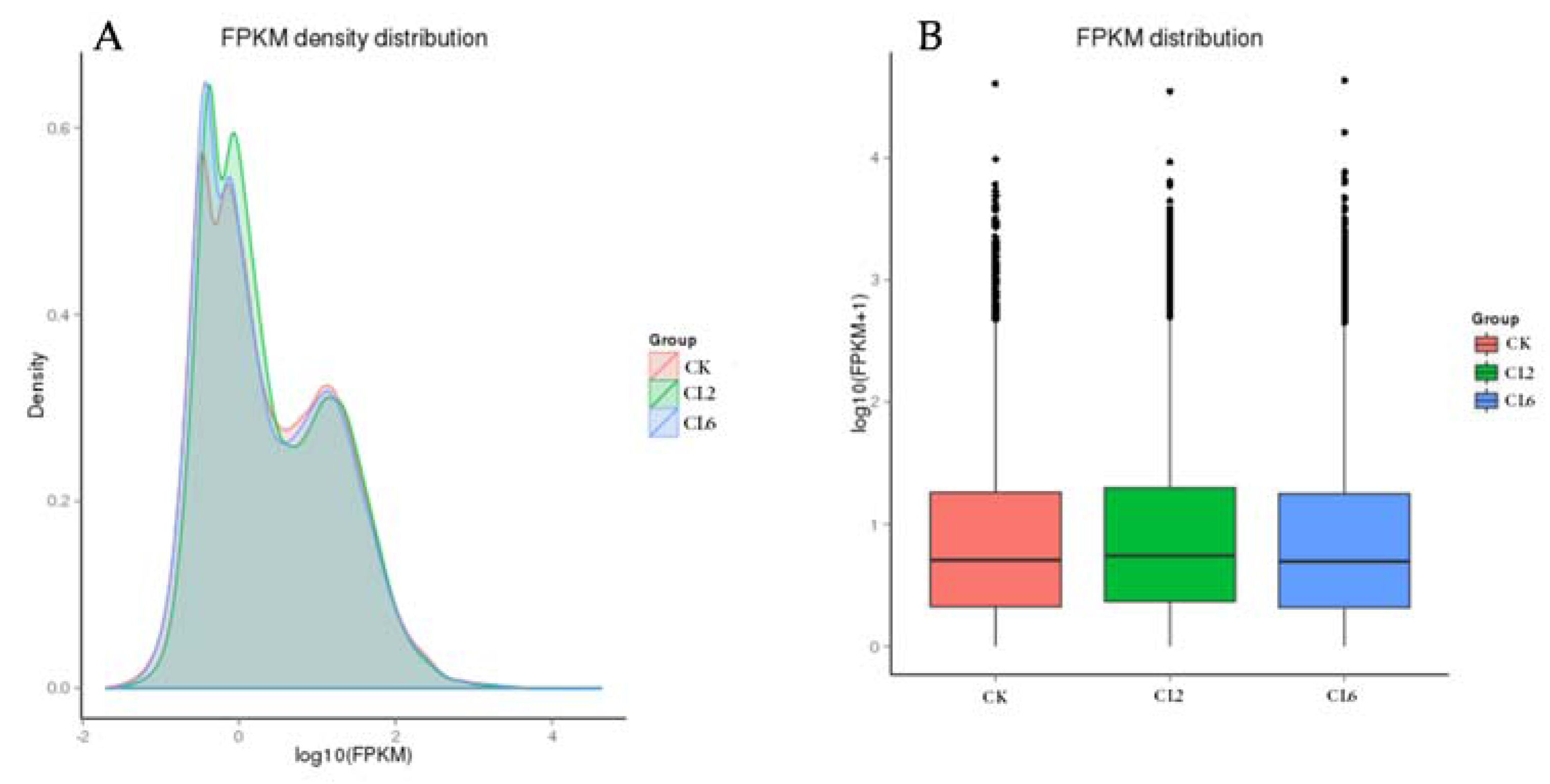
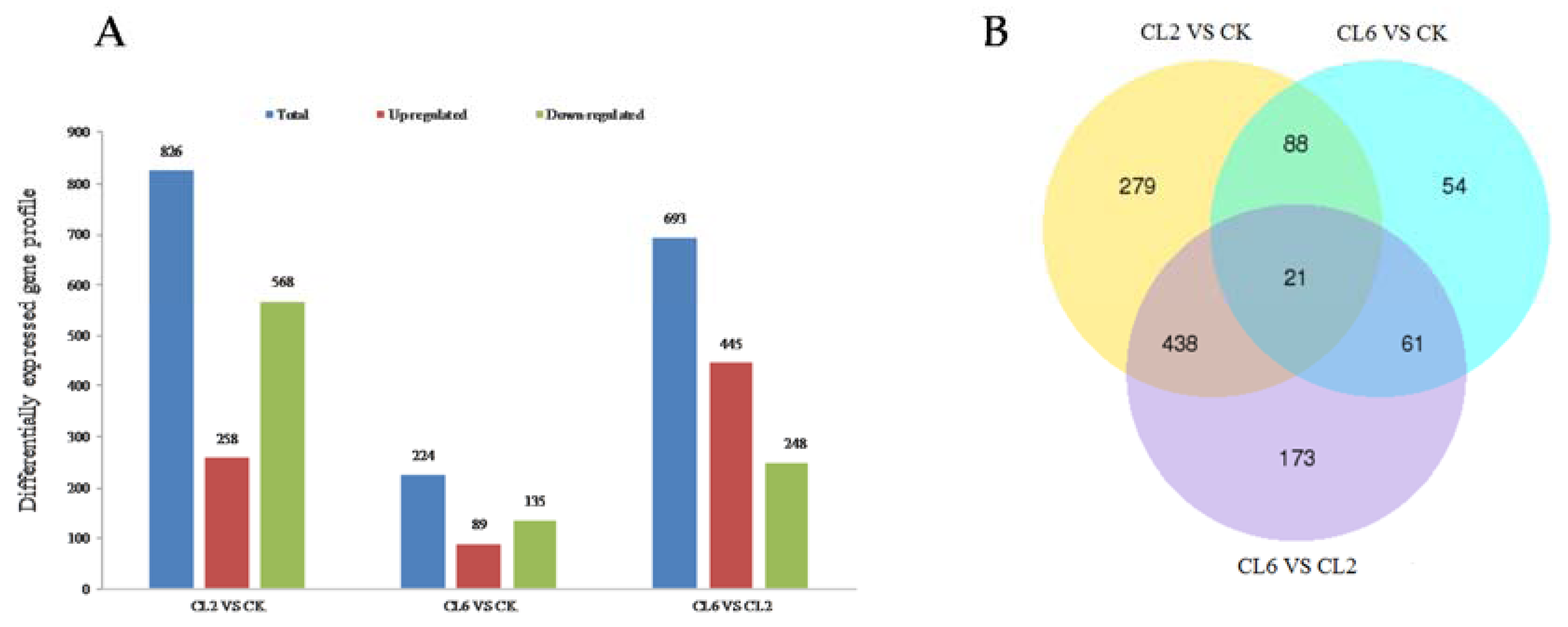
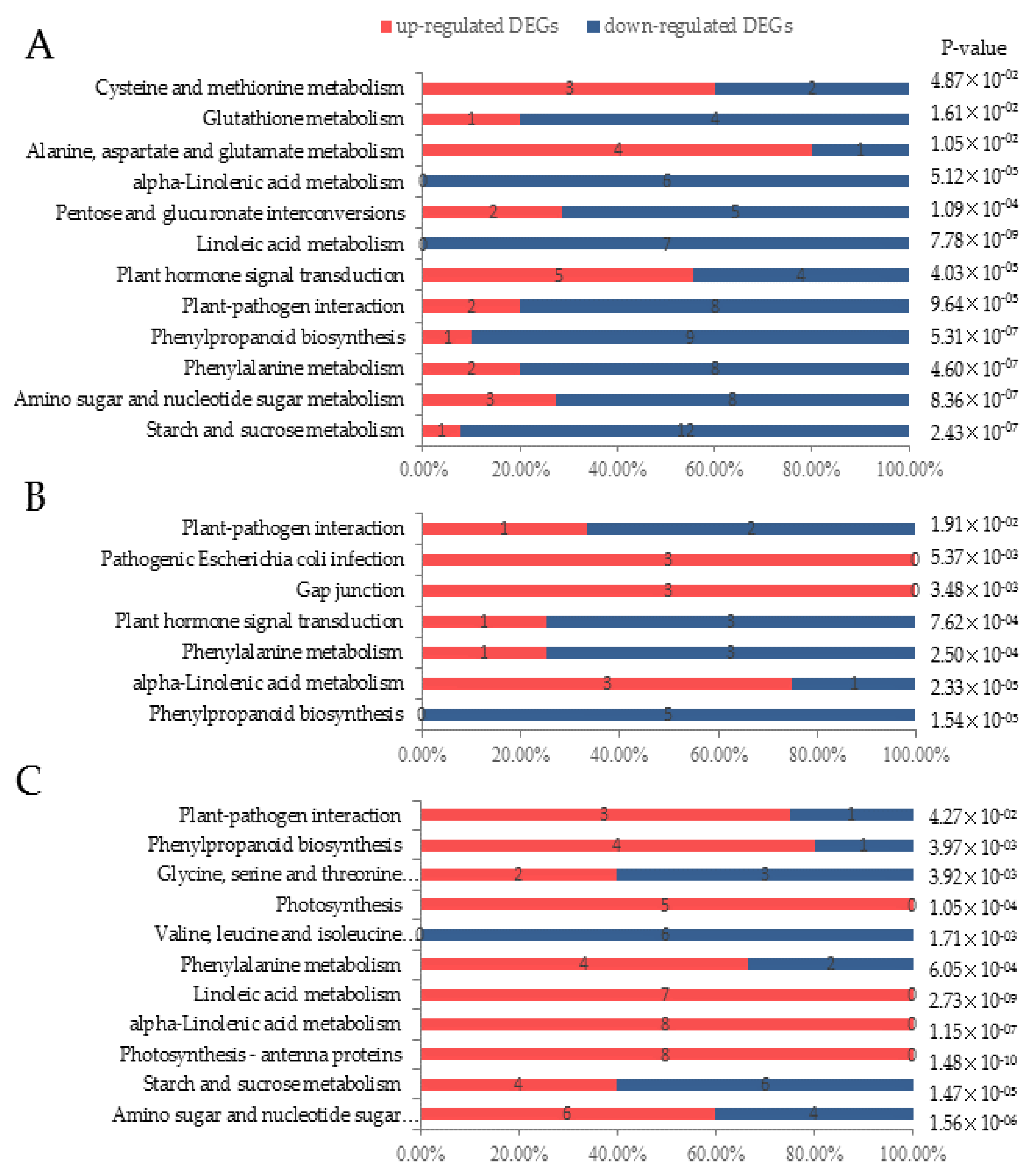
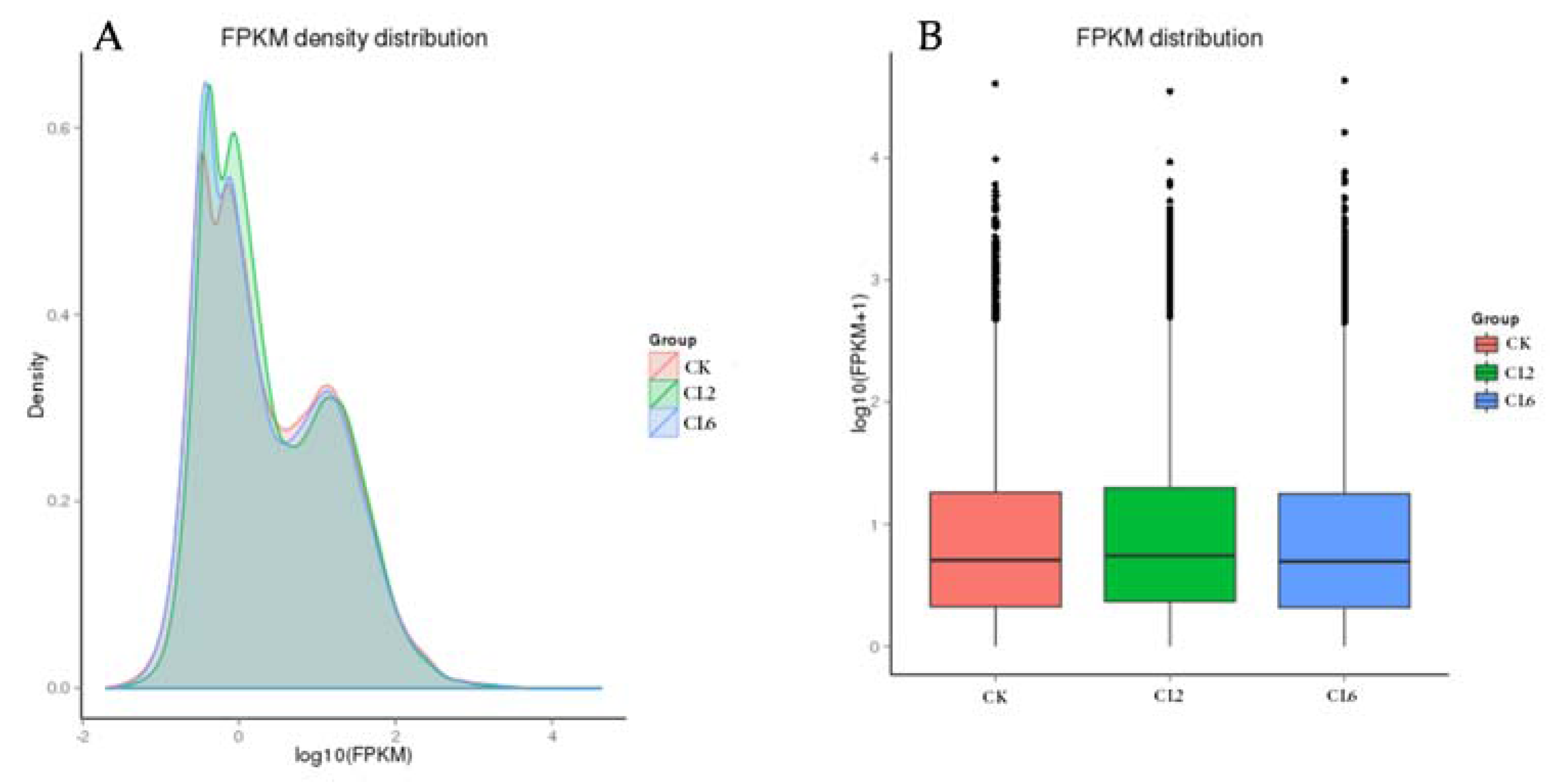
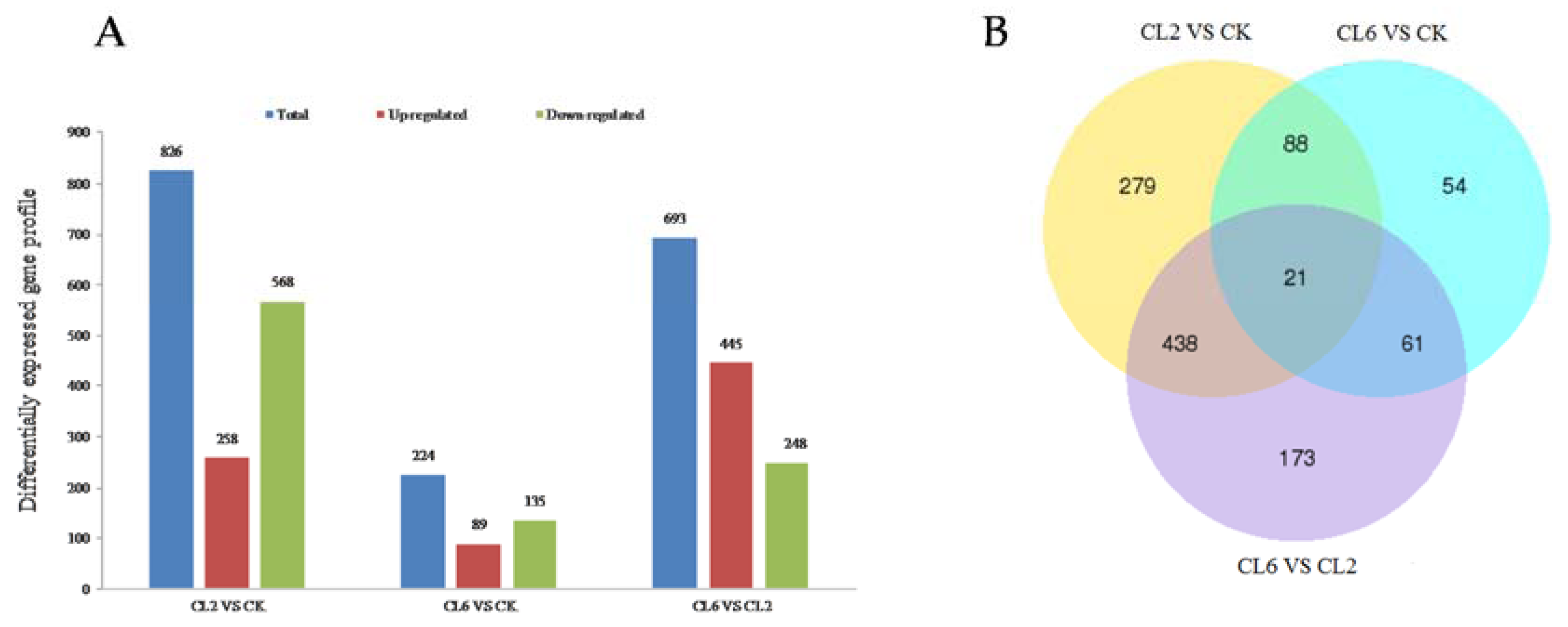
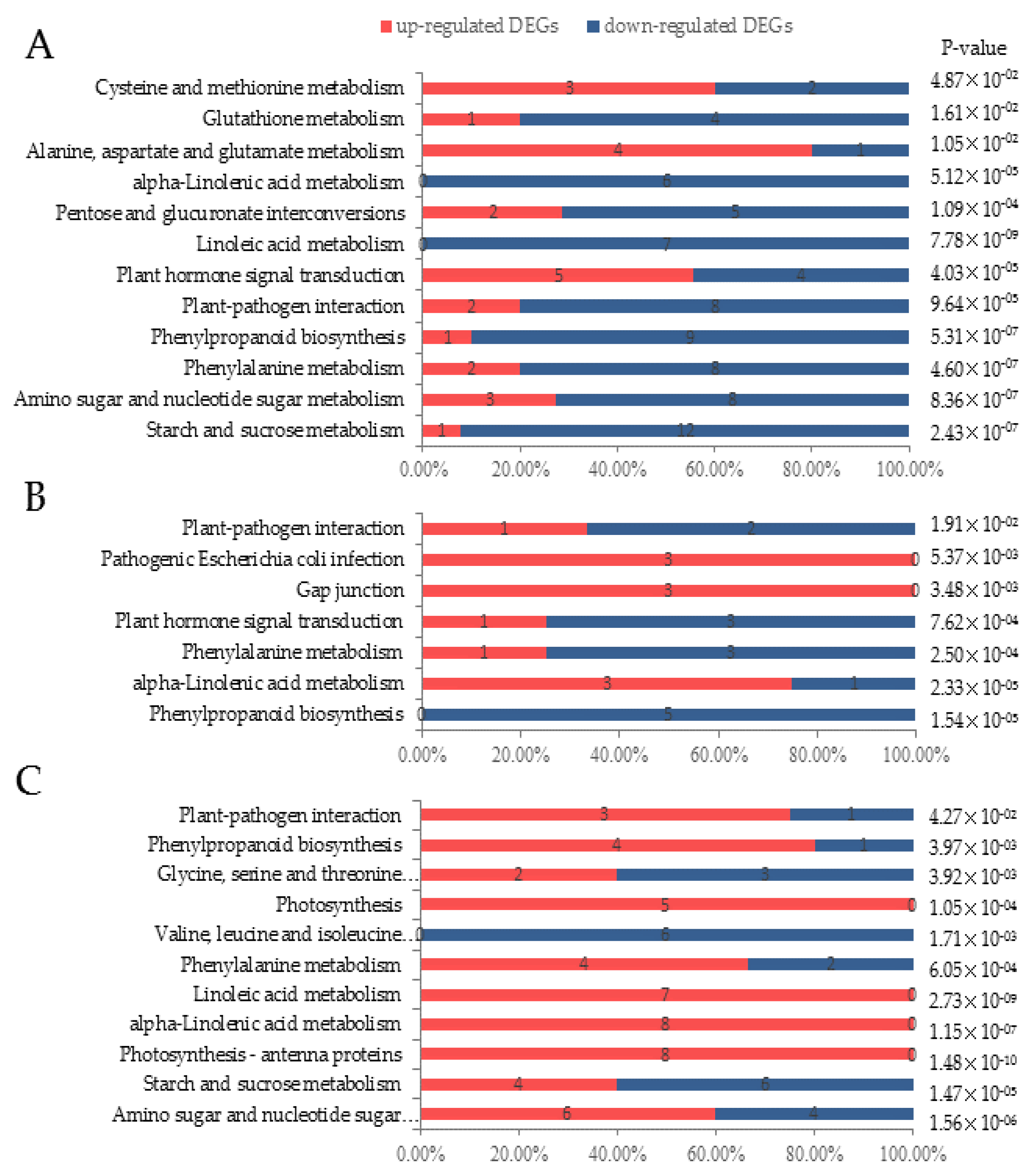
3.3. Digital Gene Expression Library Sequencing
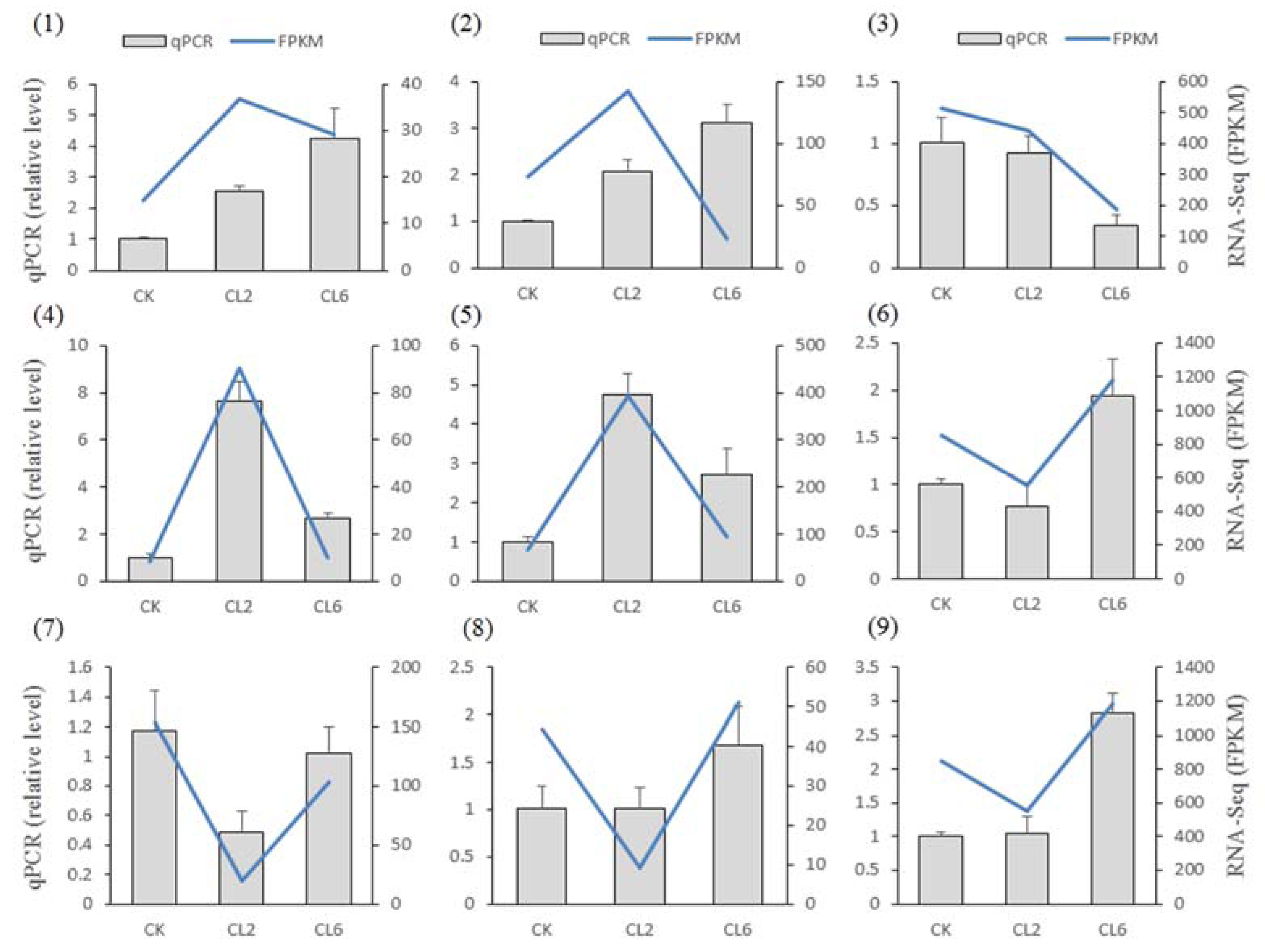
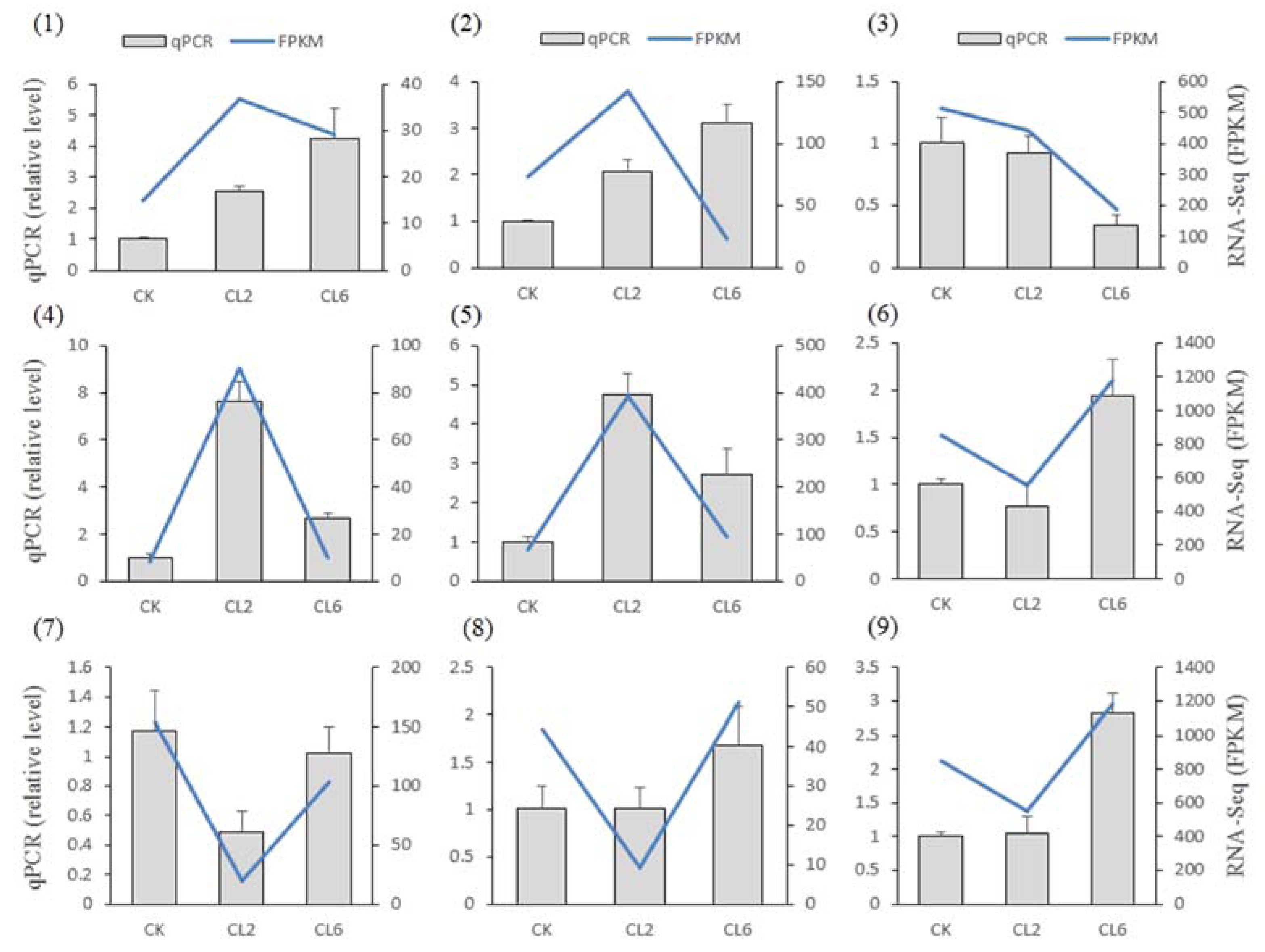
3.4. Verification of the RNA-Seq Results by qPCR
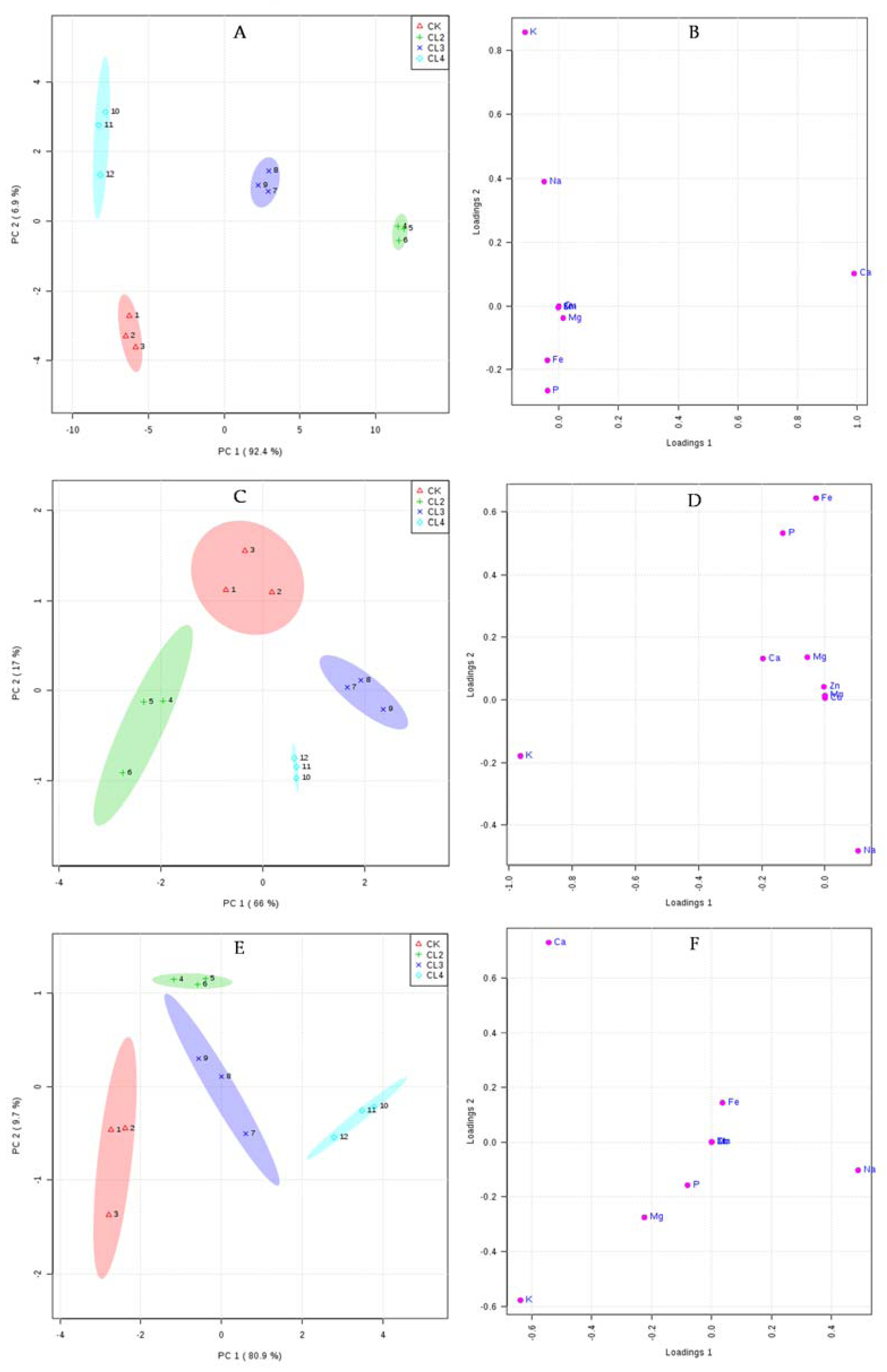
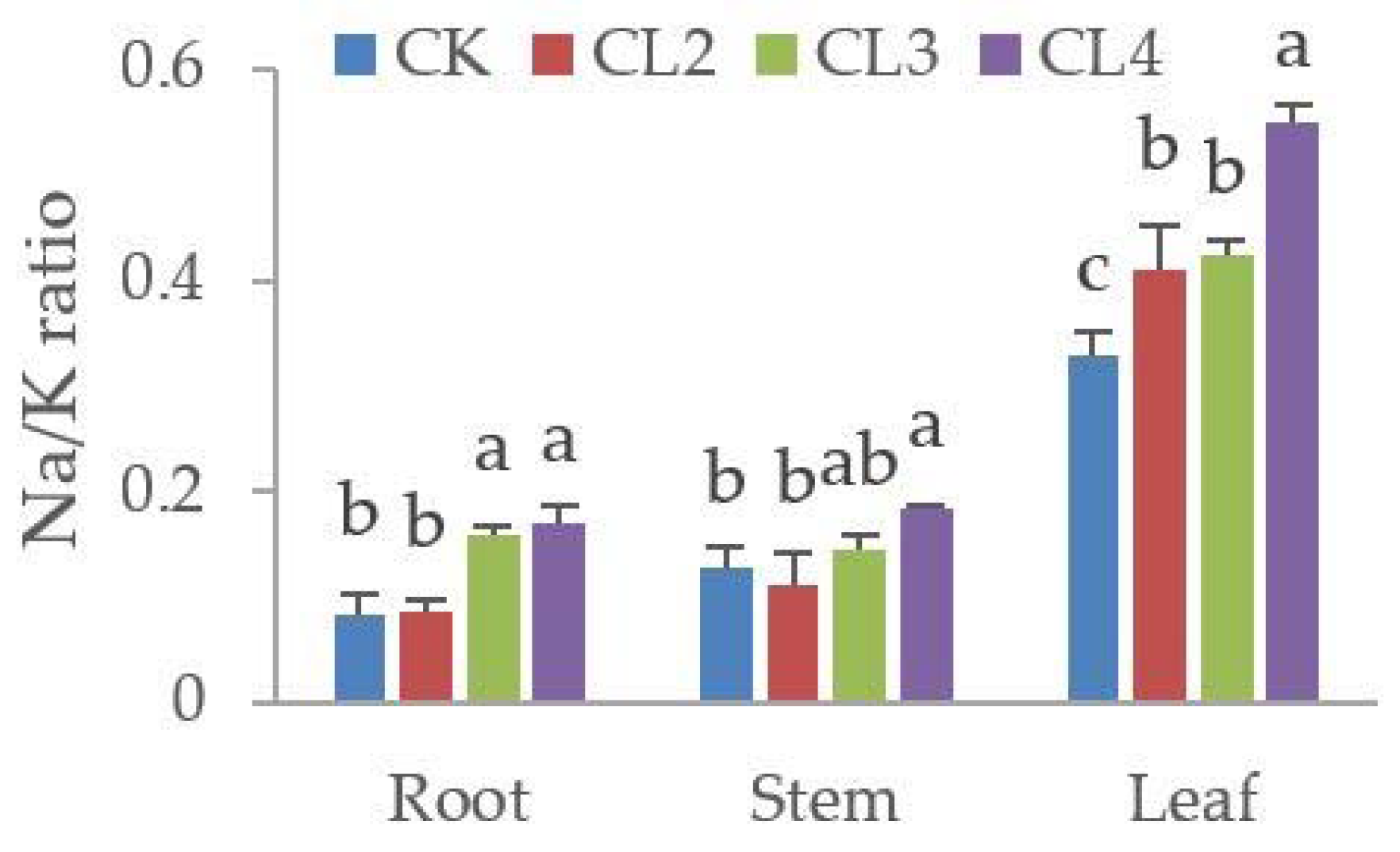
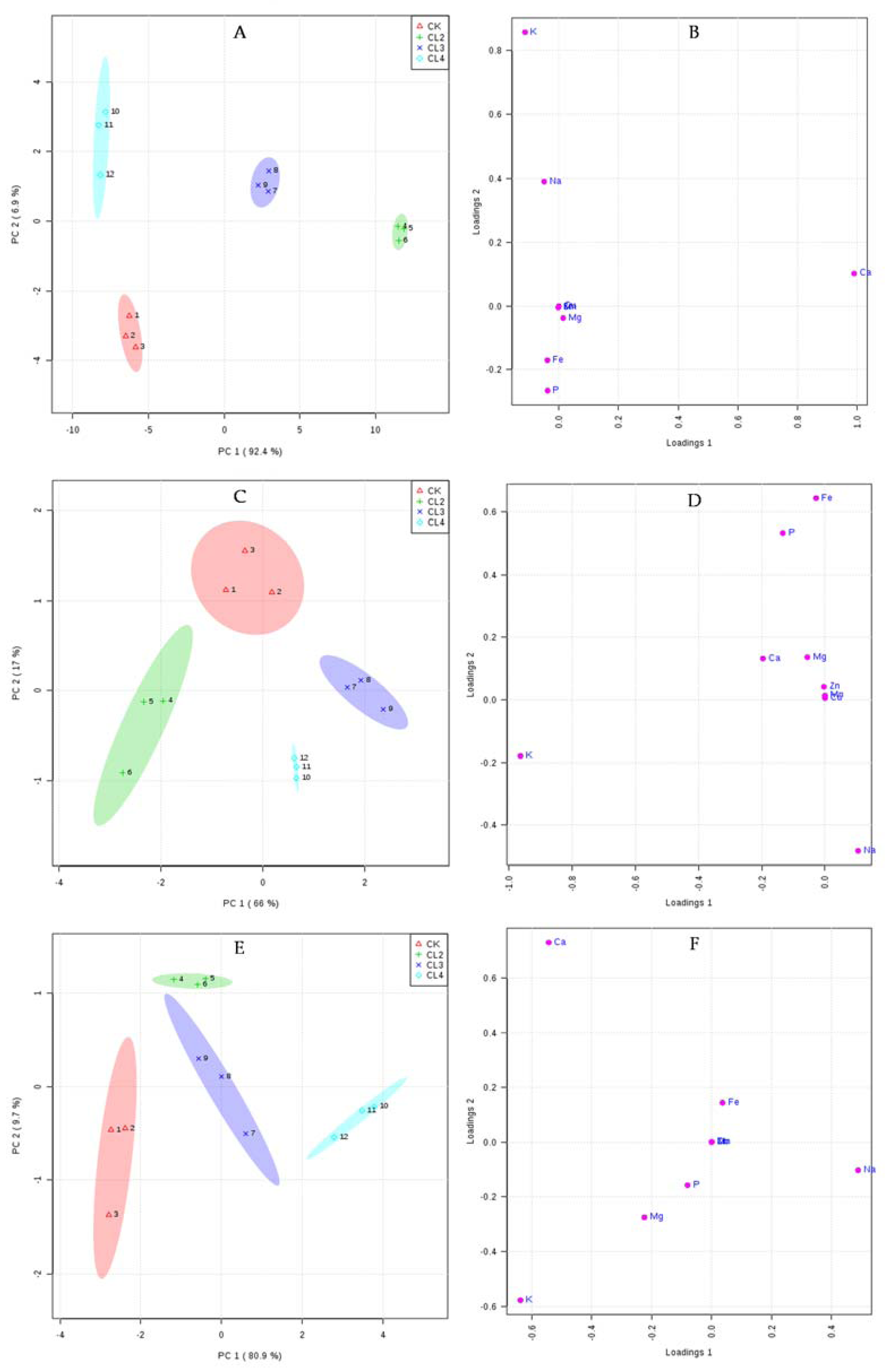
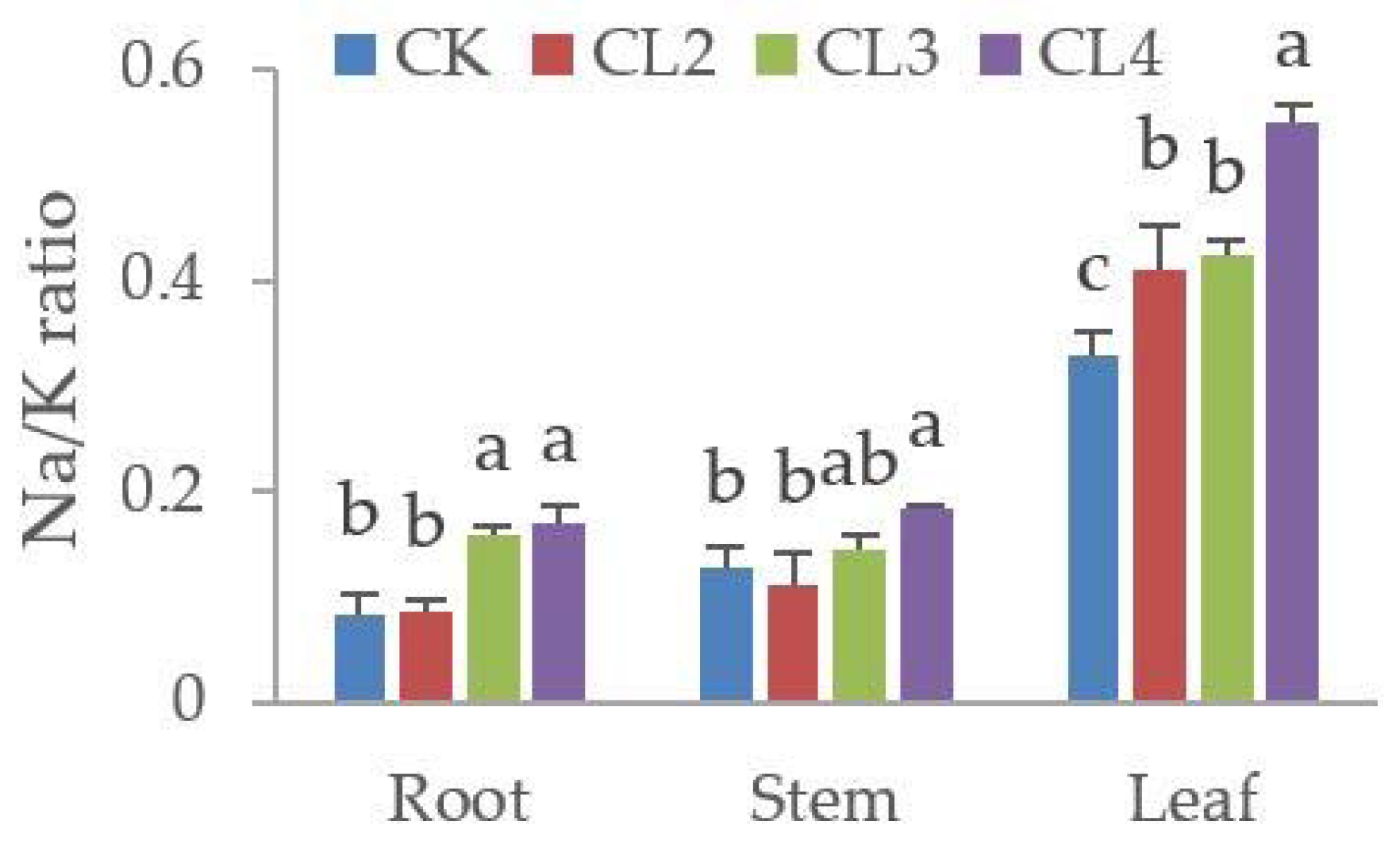
3.5. The Influence of Salt Stress on the Ionome of N. sibirica Pall.
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhu, J.-K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-K. Salt and drought stress signal transduction in plants. Ann. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed]
- Nawar, S.; Buddenbaum, H.; Hill, J. Estimation of soil salinity using three quantitative methods based on visible and near-infrared reflectance spectroscopy: A case study from Egypt. Arab. J. Geosci. 2015, 8, 5127–5140. [Google Scholar] [CrossRef]
- FAO. Global Network on Integrated Soil Management for Sustainable Use of Salt Affected Soils; FAO Land and Plant Nutricion Management Services: Rome, Italy, 2005. [Google Scholar]
- Munns, R. Genes and salt tolerance: Bringing them together. New Phytol. 2005, 167, 645–663. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Shao, H.; Shao, C.; Chen, P.; Zhao, S.; Brestic, M.; Chen, X. Physiological adaptive mechanisms of plants grown in saline soil and implications for sustainable saline agriculture in coastal zone. Acta Physiol. Plant. 2013, 35, 2867–2878. [Google Scholar] [CrossRef]
- Suzuki, N.; Koussevitzky, S.; Mittler, R.; Miller, G. ROS and redox signalling in the response of plants to abiotic stress. Plant Cell Environ. 2012, 35, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Shi, D.; Wang, D. Comparative effects of salt and alkali stresses on growth, osmotic adjustment and ionic balance of an alkali-resistant halophyte Suaeda glauca (Bge.). Plant Growth Regul. 2008, 56, 179. [Google Scholar] [CrossRef]
- Li, H.; Li, D.; Chen, A.; Tang, H.; Li, J.; Huang, S. RNA-seq for comparative transcript profiling of kenaf under salinity stress. J. Plant Res. 2017, 130, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, S.; Horie, T.; Hauser, F.; Deinlein, U.; Schroeder, J.I.; Uozumi, N. HKT transporters mediate salt stress resistance in plants: From structure and function to the field. Curr. Opin. Biotechnol. 2015, 32, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Polle, A. Salinity tolerance of populus. Plant Biol. 2010, 12, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Xu, X.; Zhu, H.; Liu, A.; Liu, L.; Li, J.; Hua, X. Comparative transcriptomic profiling of a salt-tolerant wild tomato species and a salt-sensitive tomato cultivar. Plant Cell Physiol. 2010, 51, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Zhang, D.; Li, X.; Yang, H.; Zhang, Y.; Wood, A.J. De novo transcriptome characterization and gene expression profiling of the desiccation tolerant moss Bryum argenteum following rehydration. BMC Genom. 2015, 16, 416. [Google Scholar] [CrossRef] [PubMed]
- Bahieldin, A.; Atef, A.; Sabir, J.S.; Gadalla, N.O.; Edris, S.; Alzohairy, A.M.; Radhwan, N.A.; Baeshen, M.N.; Ramadan, A.M.; Eissa, H.F.; et al. RNA-seq analysis of the wild barley (H. Spontaneum) leaf transcriptome under salt stress. C. R. Biol. 2015, 338, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.-D.; Wang, J.-Q.; Yang, N.; Dong, Y.-Y.; Liu, L.; Wang, F.-W.; Wang, N.; Chen, H.; Liu, W.-C.; Sun, Y.-P.; et al. Gene expression profiling of soybean leaves and roots under salt, saline–alkali and drought stress by high-throughput Illumina sequencing. Gene 2013, 512, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Postnikova, O.A.; Shao, J.; Nemchinov, L.G. Analysis of the alfalfa root transcriptome in response to salinity stress. Plant Cell Physiol. 2013, 54, 1041–1055. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Zhang, X.; Zhao, X.; Liu, C.; Wang, C.; Zhang, Z.; Zhang, C.; Wei, Q.; Wang, Q.; Yan, H.; et al. Transcriptome analysis reveals salt-stress-regulated biological processes and key pathways in roots of cotton (Gossypium hirsutum L.). Genomics 2011, 98, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yang, P.; Cui, F.; Zhang, F.; Luo, X.; Xie, J. Transcriptome analysis of salt stress responsiveness in the seedlings of dongxiang wild rice (Oryza rufipogon griff.). PLoS ONE 2016, 11, e0146242. [Google Scholar] [CrossRef] [PubMed]
- Atienza, S.G.; Faccioli, P.; Perrotta, G.; Dalfino, G.; Zschiesche, W.; Humbeck, K.; Stanca, A.M.; Cattivelli, L. Large scale analysis of transcripts abundance in barley subjected to several single and combined abiotic stress conditions. Plant Sci. 2004, 167, 1359–1365. [Google Scholar] [CrossRef]
- Barragán, V.; Leidi, E.O.; Andrés, Z.; Rubio, L.; De Luca, A.; Fernández, J.A.; Cubero, B.; Pardo, J.M. Ion exchangers NHX1 and NHX2 mediate active potassium uptake into vacuoles to regulate cell turgor and stomatal function in arabidopsis. Plant Cell. 2012, 24, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tang, R.J.; Jiang, C.M.; Li, B.; Kang, T.; Liu, H.; Zhao, N.; Ma, X.J.; Yang, L.; Chen, S.L.; et al. Overexpression of the PtSOS2 gene improves tolerance to salt stress in transgenic poplar plants. Plant Biotechnol. J. 2015, 13, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Taji, T.; Seki, M.; Satou, M.; Sakurai, T.; Kobayashi, M.; Ishiyama, K.; Narusaka, Y.; Narusaka, M.; Zhu, J.-K.; Shinozaki, K. Comparative genomics in salt tolerance between arabidopsis and arabidopsis-related halophyte salt cress using arabidopsis microarray. Plant Physiol. 2004, 135, 1697–1709. [Google Scholar] [CrossRef]
- Singh, U.M.; Sareen, P.; Sengar, R.S.; Kumar, A. Plant ionomics: A newer approach to study mineral transport and its regulation. Acta Physiol. Plant. 2013, 35, 2641–2653. [Google Scholar] [CrossRef]
- Zhu, J.-K. Regulation of ion homeostasis under salt stress. Curr. Opin. Plant Biol. 2003, 6, 441–445. [Google Scholar] [CrossRef]
- Munns, R. Approaches to identifying genes for salinity tolerance and the importance of timescale. Methods Mol. Biol. 2010, 639, 25–38. [Google Scholar]
- Tavakkoli, E.; Fatehi, F.; Coventry, S.; Rengasamy, P.; McDonald, G.K. Additive effects of Na+ and Cl–ions on barley growth under salinity stress. J. Exp. Bot. 2011, 62, 2189–2203. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Newman, I.; Zhou, M.; Mendham, N.; Zhang, G.; Shabala, S. Screening plants for salt tolerance by measuring K+ flux: A case study for barley. Plant Cell Environ. 2005, 28, 1230–1246. [Google Scholar] [CrossRef]
- Nazar, R.; Iqbal, N.; Masood, A.; Syeed, S.; Khan, N.A. Understanding the significance of sulfur in improving salinity tolerance in plants. Environ. Exp. Bot. 2011, 70, 80–87. [Google Scholar] [CrossRef]
- Yousfi, S.; Mahmoudi, H.; Abdelly, C.; Gharsalli, M. Effect of salt on physiological responses of barley to iron deficiency. Plant Physiol. Biochem. 2007, 45, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Lotfi, N.; Vahdati, K.; Kholdebarin, B.; Hassani, D.; Amiri, R. Mineral composition of some walnut cultivars (Juglans regia L.) for evaluation of ionome and ionomics under salt stress condition. In I International Symposium on Biotechnology of Fruit Species: BIOTECHFRUIT2008; ISHS Acta Horticulturae 839: Dresden, Germany, 2008; pp. 293–300. [Google Scholar]
- Hill, C.B.; Jha, D.; Bacic, A.; Tester, M.; Roessner, U. Characterization of ion contents and metabolic responses to salt stress of different arabidopsis ATHKT1; 1 genotypes and their parental strains. Mol. Plant. 2013, 6, 350–368. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Shen, Q.; Cai, S.; Chen, Z.-H.; Dai, F.; Zhang, G. Ionomic responses and correlations between elements and metabolites under salt stress in wild and cultivated barley. Plant Cell Physiol. 2013, 54, 1976–1988. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Fu, L.; Qiu, L.; Xue, F.; Zhang, G.; Wu, D. Time-course of ionic responses and proteomic analysis of a tibetan wild barley at early stage under salt stress. Plant Growth Regul. 2017, 81, 11–21. [Google Scholar] [CrossRef]
- Ni, J.W.; Wu, X.; Zhang, H.X.; Liu, T.; Zhang, L. Comparative analysis of salt tolerance of three Nitraria species. For. Res. 2012, 25, 48–53. [Google Scholar]
- Li, Q.H.; Wu, S.X.; Xu, J.; Ren, W.J.; Zhao, Y.M. Comprehensive evaluation on salt tolerance of different desert shrubs in Ulan Buh desert regions. Pratacult. Sci. 2012, 29, 1132–1136. [Google Scholar]
- Yang, S.; Zhang, H.-X.; Liu, T. Effect of salt stress on osmotic adjustment substances in plants. For. Res. 2012, 25, 269–277. [Google Scholar]
- Zhang, G.L. Effects of iso-osmotic salt and water stresses on growth and ionic absorption and distribution in Nitraria sibirica seedlings. Agric. Res. Arid Areas 2013, 31, 114–118. [Google Scholar]
- Rina, S.; Guilin, C. Effect of exogenous spermidine on antioxidant enzyme system in leaves of Nitraria sibirica pall. Seedlings under salt stress. Acta Bot. Boreali Occident. Sin. 2013, 33, 352–356. [Google Scholar]
- Cheng, T.L.; Li, H.Y.; Wu, H.W.; Liu, X.Z.; Wu, X.; Yang, S.; Zhang, H.X.; Yang, X.Y. Comparison on osmotica accumulation of different salt-tolerant plants under salt stress. For. Res. 2015, 28, 826–832. [Google Scholar]
- Wu, X.; Ni, J.; Zhang, H.; Liu, T.; Zhang, L. Effects of salt stress on osmotic adjustment substances in three species of Nitraria. J. Northeast Univ. 2012, 40, 44–47. [Google Scholar]
- Wang, L.; Li, F.-F.; Zhang, W.-B.; Chen, G.-L.; Lin, X.-F. Isolation and characterization of Nitraria sibirica actin gene. Acta Pratacult. Sin. 2012, 21, 151–158. [Google Scholar]
- Wang, L.; Ma, Y.; Li, N.; Zhang, W.; Mao, H.; Lin, X. Isolation and characterization of a tonoplast Na+/H+ antiporter from the halophyte Nitraria sibirica. Biol. Plant. 2016, 60, 113–122. [Google Scholar] [CrossRef]
- Tang, X.; Wang, R.H.; Yan, X.Y.; Zhu, J.F.; Liu, Z.X.; Ni, J.W.; Zhang, H.X. Isolation and expression analysis of a vacuolar membrane Na+/H+ antiporter gene ntnhx1 from Nitraria tangutorum. Sci. Silvae Sin. 2014, 50, 38–44. [Google Scholar]
- Zhu, J.; Yang, X.; Liu, Z.; Zhang, H. Identification and target prediction of micrornas in Ulmus pumila L. Seedling roots under salt stress by high-throughput sequencing. Forests 2016, 7, 318. [Google Scholar] [CrossRef]
- Shavrukov, Y. Salt stress or salt shock: Which genes are we studying? J. Exp. Bot. 2013, 64, 119–127. [Google Scholar] [CrossRef]
- Sanchez, D.H.; Lippold, F.; Redestig, H.; Hannah, M.A.; Erban, A.; Krämer, U.; Kopka, J.; Udvardi, M.K. Integrative functional genomics of salt acclimatization in the model legume Lotus japonicus. Plant J. 2008, 53, 973–987. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Parra, G.; Bradnam, K.; Ning, Z.; Keane, T.; Korf, I. Assessing the gene space in draft genomes. Nucleic Acids Res. 2009, 37, 289–297. [Google Scholar] [CrossRef]
- Parra, G.; Bradnam, K.; Korf, I. Cegma: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Bedell, J.; Korf, I.; Yandell, M. Basic Local Alignment Search Tool; O’Reilly: Sebastopol, CA, USA, 2003. [Google Scholar]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011, 40, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. Bingo: A cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.-L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Hong, S.-Y.; Seo, P.J.; Yang, M.-S.; Xiang, F.; Park, C.-M. Exploring valid reference genes for gene expression studies in Brachypodium distachyon by real-time PCR. BMC Plant Biol. 2008, 8, 112. [Google Scholar] [CrossRef]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting go annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.O. The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef]
- Shen, Q.; Fu, L.; Dai, F.; Jiang, L.; Zhang, G.; Wu, D. Multi-omics analysis reveals molecular mechanisms of shoot adaption to salt stress in Tibetan wild barley. BMC Genom. 2016, 17, 889. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.-Q.; Pan, X.-Y.; Yu, Y.-M.; Wang, W.-Y.; Zhang, F.; Ge, Y.-Y.; Shen, X.-L.; Shen, F.-Q.; Liu, X.-J. De novo characterization of the Anthurium transcriptome and analysis of its digital gene expression under cold stress. BMC Genom. 2013, 14, 827. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Bao, A.-K.; Chai, W.-W.; Wang, W.-Y.; Zhang, J.-L.; Li, Y.-X.; Wang, S.-M. Transcriptomic analysis of the succulent xerophyte Zygophyllum xanthoxylum in response to salt treatment and osmotic stress. Plant Soil 2016, 402, 343–361. [Google Scholar] [CrossRef]
- Xu, P.; Liu, Z.; Fan, X.; Gao, J.; Zhang, X.; Zhang, X.; Shen, X. De novo transcriptome sequencing and comparative analysis of differentially expressed genes in Gossypium aridum under salt stress. Gene 2013, 525, 26–34. [Google Scholar] [CrossRef]
- Long, W.; Zou, X.; Zhang, X. Transcriptome analysis of canola (Brassica napus) under salt stress at the germination stage. PLoS ONE 2015, 10, e0116217. [Google Scholar] [CrossRef]
- Wang, G.; Zhu, Q.; Meng, Q.; Wu, C. Transcript profiling during salt stress of young cotton (Gossypium hirsutum) seedlings via solexa sequencing. Acta Physiol. Plant. 2012, 34, 107–115. [Google Scholar] [CrossRef]
- Gharat, S.A.; Parmar, S.; Tambat, S.; Vasudevan, M.; Shaw, B.P. Transcriptome analysis of the response to nacl in Suaeda maritima provides an insight into salt tolerance mechanisms in halophytes. PLoS ONE 2016, 11, e0163485. [Google Scholar] [CrossRef]
- Shabala, S.; Cuin, T.A. Potassium transport and plant salt tolerance. Physiol. Plant. 2008, 133, 651–669. [Google Scholar] [CrossRef]
- Tester, M.; Davenport, R. Na+ tolerance and Na+ transport in higher plants. Ann. Bot. 2003, 91, 503–527. [Google Scholar] [CrossRef]
- Kırmızı, S.; Bell, R.W. Responses of barley to hypoxia and salinity during seed germination, nutrient uptake, and early plant growth in solution culture. J. Plant Nutr. Soil Sci. 2012, 175, 630–640. [Google Scholar] [CrossRef]
- Kopittke, P.M. Interactions between Ca, Mg, Na and K: Alleviation of toxicity in saline solutions. Plant Soil 2012, 352, 353–362. [Google Scholar] [CrossRef]
- Shabala, S.; Shabala, S.; Cuin, T.A.; Pang, J.; Percey, W.; Chen, Z.; Conn, S.; Eing, C.; Wegner, L.H. Xylem ionic relations and salinity tolerance in barley. Plant J. 2010, 61, 839–853. [Google Scholar] [CrossRef]
- Amtmann, A.; Armengaud, P. Effects of N, P, K and S on metabolism: New knowledge gained from multi-level analysis. Curr. Opin. Plant Biol. 2009, 12, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Cai, S.; Chen, M.; Ye, L.; Chen, Z.; Zhang, H.; Dai, F.; Wu, F.; Zhang, G. Tissue metabolic responses to salt stress in wild and cultivated barley. PLoS ONE 2013, 8, e55431. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Names | The Time Points of Salt Treatment and Sample Collection/day | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| −14.25 | −13 | −12 | −11 | −10 | −9 | −8.25 | −7 | −6.25 | −6 | −5 | −4.25 | −4 | −3 | −2.25 | −2 | −1 | −0.25 | 0 | |
| CK | sample collection | ||||||||||||||||||
| CL1 | √ | ||||||||||||||||||
| CL2 | √ | √ | |||||||||||||||||
| CL3 | √ | √ | √ | √ | |||||||||||||||
| CL4 | √ | √ | √ | √ | √ | √ | |||||||||||||
| CL5 | √ | √ | √ | √ | √ | √ | |||||||||||||
| CL6 | √ | √ | √ | √ | √ | √ | |||||||||||||
| Item | Number |
|---|---|
| Total number of raw reads | 109,605,726 |
| Total number of clean reads | 103,965,286 |
| Total clean bases (G) | 13 |
| Q20 percentage (%) | 95.59 |
| GC percentage (%) | 45.71 |
| Total number of unigenes | 137,421 |
| Min length of unigenes (bp) | 201 |
| Max length of unigenes (bp) | 15,714 |
| Mean length of unigenes (bp) | 555 |
| Length of N50 (bp) | 846 |
| Item | CK | CL2 | CL6 |
|---|---|---|---|
| Raw reads | 17,939,409 | 14,784,837 | 16,231,582 |
| Clean reads | 17,866,852 | 14,742,618 | 16,158,118 |
| Clean bases (G) | 0.89 | 0.74 | 0.81 |
| Total mapped | 16,622,090 93.03% | 13,714,511 93.03% | 15,023,640 92.98% |
| Q20 (%) | 98.11 | 98.2 | 98.11 |
| GC (%) | 45.68 | 45.48 | 45.81 |
| Tissue | Treatment | Na | Ca | K | Fe | Mg | P | Mn | Cu | Zn |
|---|---|---|---|---|---|---|---|---|---|---|
| Roots | CK | 1.33 c | 18.75 c | 15.92 c | 2.40 a | 1.81 ab | 8.71 a | 0.1221 a | 0.0189 a | 0.1923 a |
| CL2 | 1.43 c | 36.67 a | 16.34 c | 0.92 c | 1.72 ab | 7.10 b | 0.0594 d | 0.0091 d | 0.1388 d | |
| CL3 | 2.97 b | 28.04 b | 18.58 b | 1.65 b | 2.19 a | 7.40 b | 0.0959 b | 0.0132 c | 0.1887 b | |
| CL4 | 3.53 a | 17.41 d | 20.86 a | 1.19 c | 1.39 c | 7.11 b | 0.0890 c | 0.0139 b | 0.1576 c | |
| Stems | CK | 3.04 b | 12.84 a | 23.46 b | 1.72 a | 2.27 a | 4.75 a | 0.0531 a | 0.0156 a | 0.1133 a |
| CL2 | 2.86 b | 13.34 a | 25.64 a | 0.39 b | 2.02 a | 3.81 b | 0.0230 d | 0.0060 c | 0.0279 b | |
| CL3 | 3.13 b | 12.51 a | 21.40 c | 0.48 b | 1.83 a | 3.46 b | 0.0265 b | 0.0094 b | 0.0247 c | |
| CL4 | 4.21 a | 12.42 a | 22.92 b | 0.34 b | 1.84 a | 3.57 b | 0.0251 c | 0.0047 d | 0.0243 c | |
| Leaves | CK | 7.88 c | 20.31 a | 23.81 a | 0.16 b | 7.87 a | 5.06 a | 0.0532 a | 0.0047 c | 0.0307 bc |
| CL2 | 8.86 bc | 20.76 a | 21.54 b | 0.58 a | 7.03 b | 4.70 a | 0.0513 b | 0.0067 a | 0.0319 a | |
| CL3 | 9.32 b | 19.43 b | 22.04 b | 0.67 a | 6.63 b | 4.52 a | 0.0481 c | 0.0057 b | 0.0303 c | |
| CL4 | 10.85 a | 17.39 c | 19.78 c | 0.50 a | 6.44 b | 4.54 a | 0.0477 c | 0.0059 ab | 0.0311 b |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Tang, X.; Zhu, J.; Yang, X.; Zhang, H. De Novo Transcriptome Characterization, Gene Expression Profiling and Ionic Responses of Nitraria sibirica Pall. under Salt Stress. Forests 2017, 8, 211. https://doi.org/10.3390/f8060211
Li H, Tang X, Zhu J, Yang X, Zhang H. De Novo Transcriptome Characterization, Gene Expression Profiling and Ionic Responses of Nitraria sibirica Pall. under Salt Stress. Forests. 2017; 8(6):211. https://doi.org/10.3390/f8060211
Chicago/Turabian StyleLi, Huanyong, Xiaoqian Tang, Jianfeng Zhu, Xiuyan Yang, and Huaxin Zhang. 2017. "De Novo Transcriptome Characterization, Gene Expression Profiling and Ionic Responses of Nitraria sibirica Pall. under Salt Stress" Forests 8, no. 6: 211. https://doi.org/10.3390/f8060211
APA StyleLi, H., Tang, X., Zhu, J., Yang, X., & Zhang, H. (2017). De Novo Transcriptome Characterization, Gene Expression Profiling and Ionic Responses of Nitraria sibirica Pall. under Salt Stress. Forests, 8(6), 211. https://doi.org/10.3390/f8060211