


Effects of Thinning of the Infected Trees and Cultivating of the Resistant Pines on Soil Microbial Diversity and Function


Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Protocol and Sample Collection
2.2. Soil Sampling
2.3. Determination of Enzymatic Activity and Physicochemical Properties of the Soil
2.4. DNA Extraction, PCR Amplification, and Illumina MiSeq Sequencing
2.5. Bioinformatics Analysis
2.6. Statistical Analyses
3. Results
3.1. Soil Enzyme Activities in Three Pine Plots
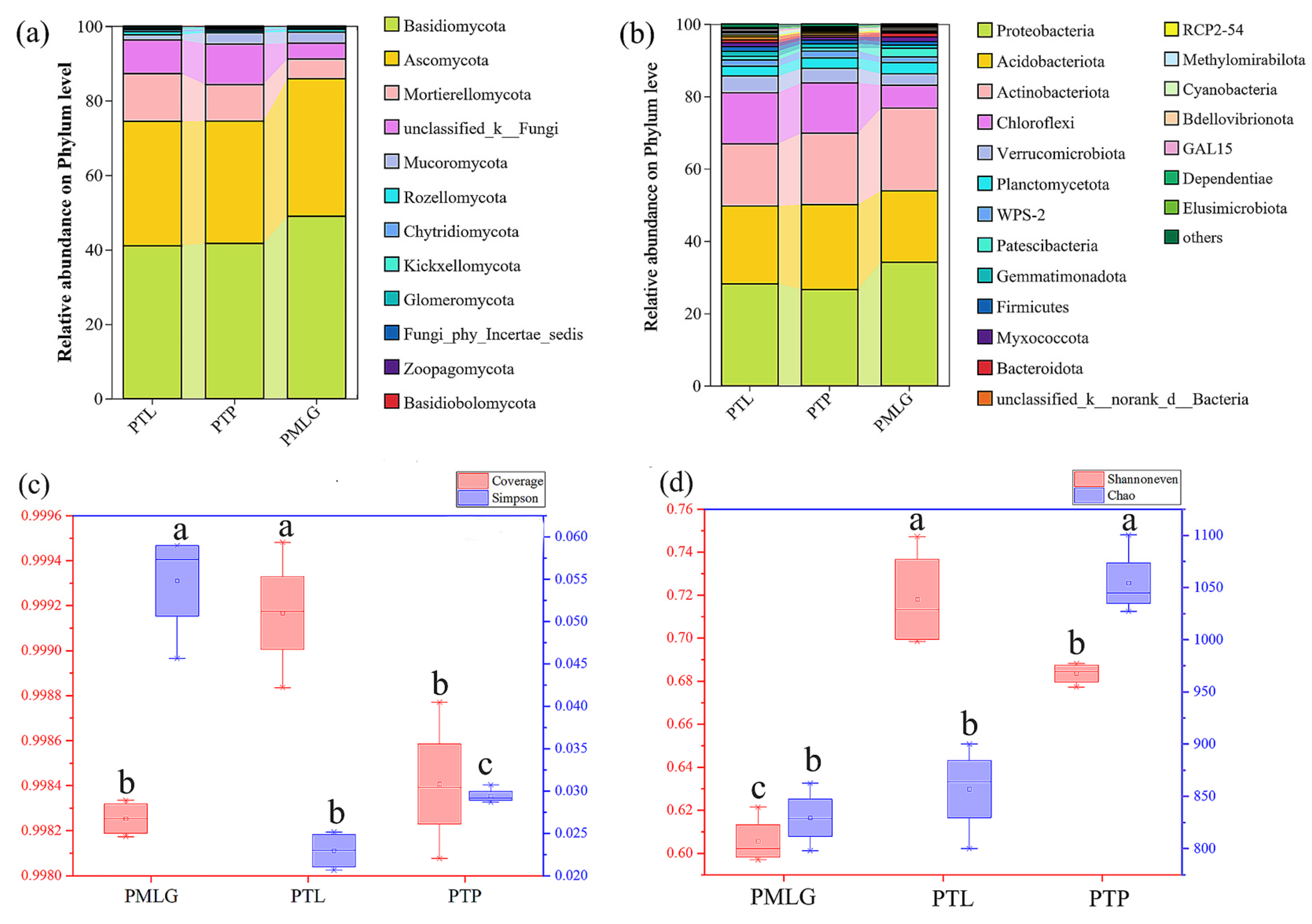
3.2. Bacterial and Fungal Community Composition and Diversity
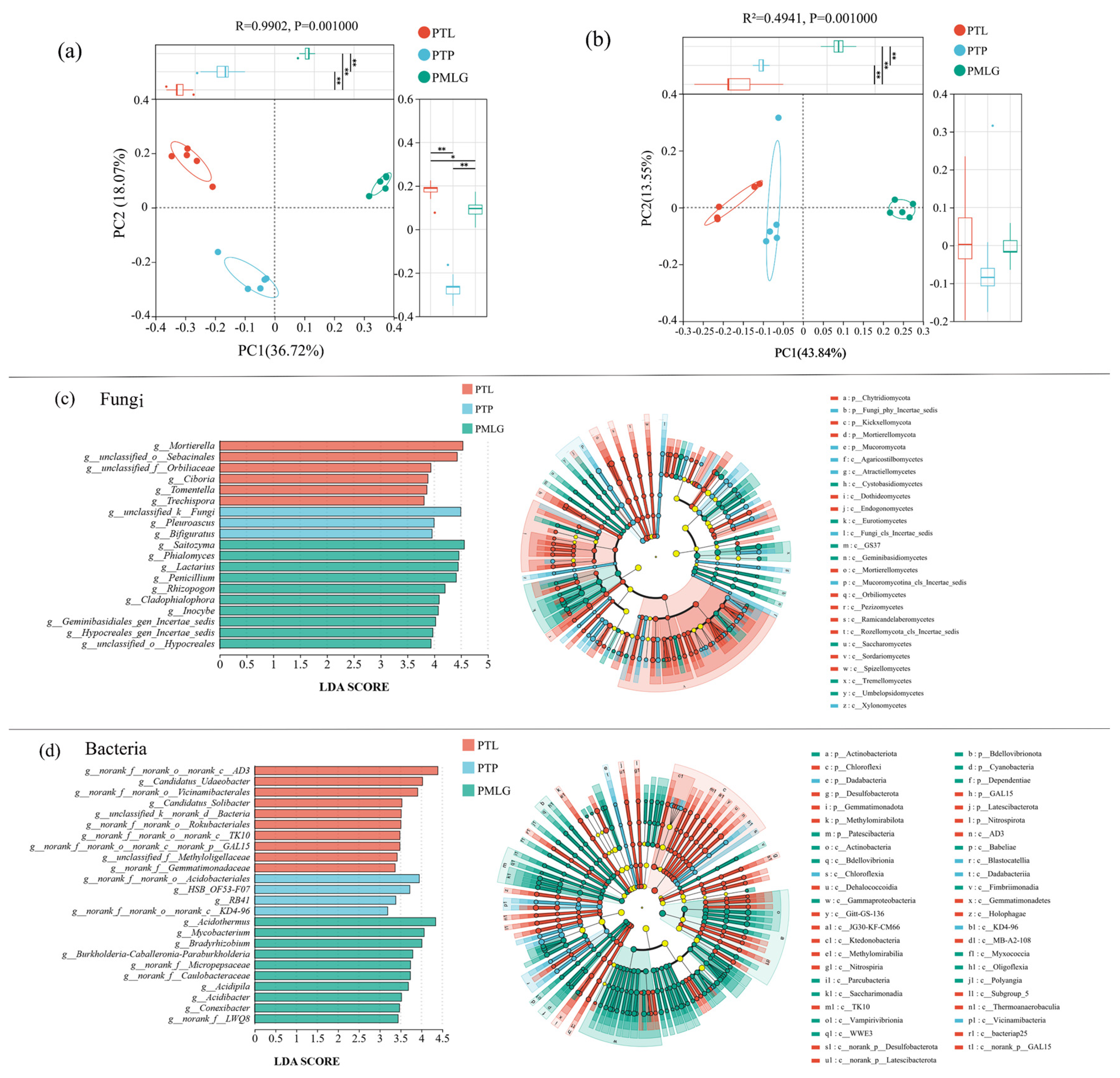
3.3. Beta Diversity
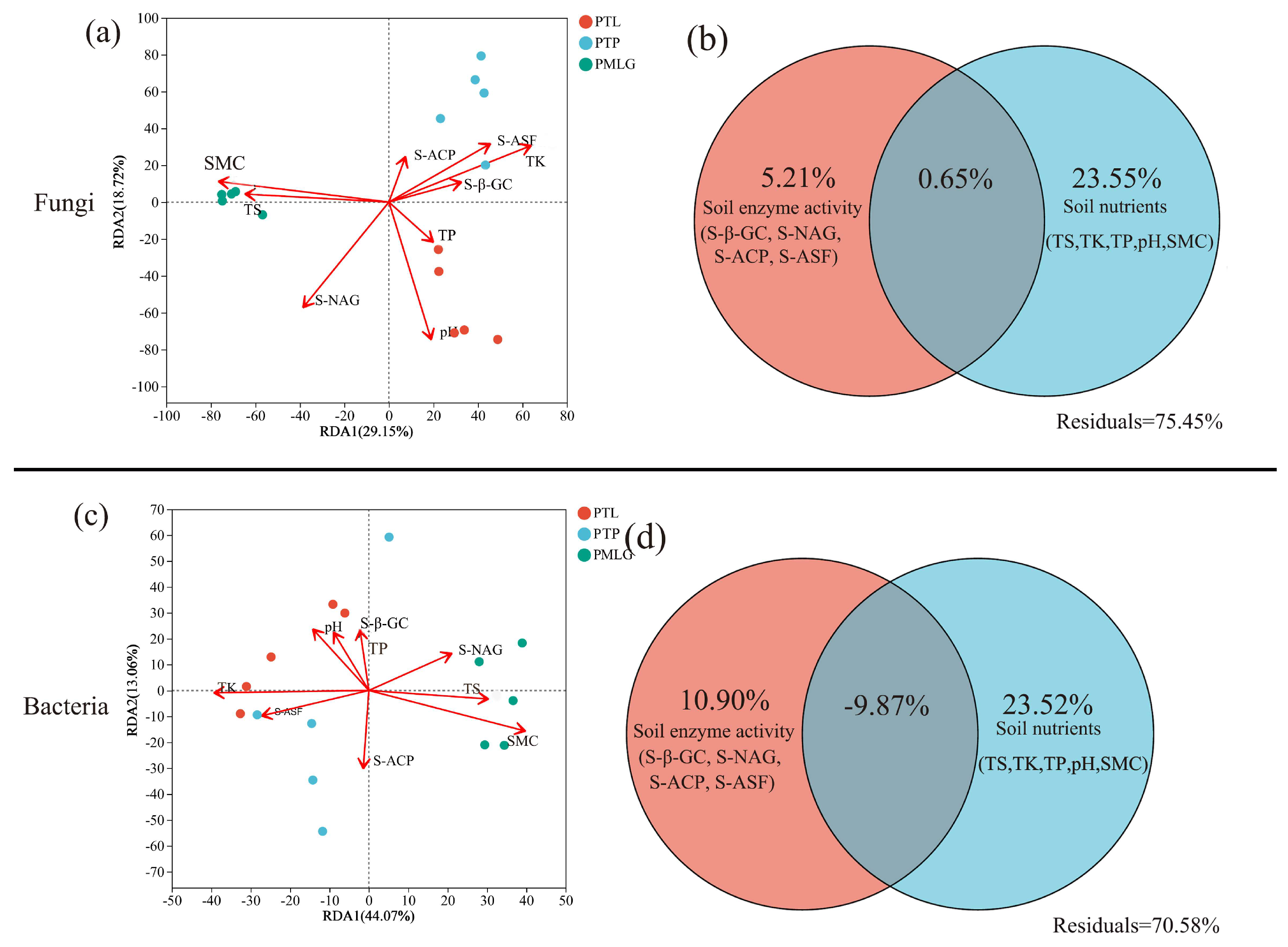
3.4. Correlations of Soil Microbial Communities with Environmental Variables
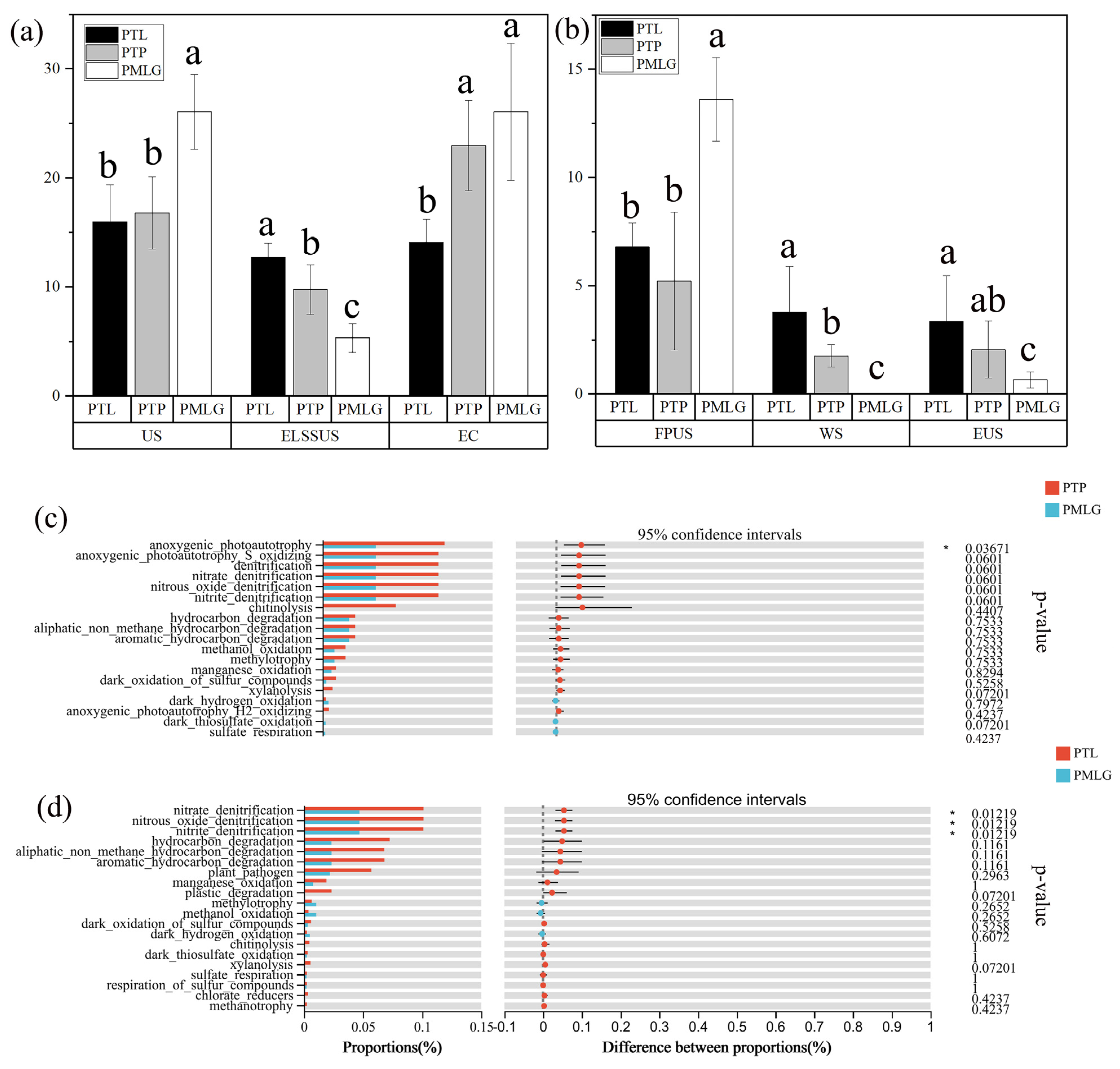
3.5. Potential Functional Structure of Different Microbial Communities
3.6. Deterministic and Stochastic Assembly of Soil Microbial Communities
4. Discussion
4.1. Effects of Infected Wood Harvesting and Planting of Disease-Resistant Pine Trees on Soil Enzyme Activity
4.2. Microbial Communities and Their Diversity
4.3. Relationships Between Soil Properties and Microorganisms
4.4. Functional Changes in Soil Microbial Communities
4.5. Soil Microbial Community Assembly
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Torres, I.; Moreno, J.M.; Morales-Molino, C.; Arianoutsou, M. Ecosystem services provided by pine forests. In Pines and Their Mixed Forest Ecosystems in the Mediterranean Basin; Springer: Berlin/Heidelberg, Germany, 2021; pp. 617–629. [Google Scholar]
- Dering, M.; Baranowska, M.; Beridze, B.; Chybicki, I.J.; Danelia, I.; Iszkuo, G.; Kvartskhava, G.; Kosiński, P.; Rczka, G.; Thomas, P.A. The evolutionary heritage and ecological uniqueness of Scots pine in the Caucasus ecoregion is at risk of climate changes. Sci. Rep. 2021, 11, 22845. [Google Scholar] [CrossRef] [PubMed]
- Aierken, N.; Wang, G.; Chen, M.Y.; Chai, G.Q.; Han, X.Y.; Qian, Z.H.; Zhang, X.L. Assessing global pine wilt disease risk based on ensemble species distribution models. Ecol. Indic. 2024, 167, 112691. [Google Scholar] [CrossRef]
- Qi, J.; Nan, J.; Zhao, X.; Liang, C.; Fan, J.; He, H. Genetic Structure of Monochamus alternatus (Hope) in Qinling-Daba Mountains and Expansion Trend: Implications for Pest Prevention and Management. Ecol. Evol. 2024, 14, e70373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.F.; Chen, S.Y.; Liu, G.; Wang, Y.X. Effects of sanitation cutting pine wilt diseased trees on the stand structure of pure Pinus massoniana plantatio. J. Zhejiang AF Univ. 2020, 37, 745–751. [Google Scholar]
- Huang, Y.A. Control Effects of Pine Wood Nematode Disease Transmitted by Monochamus alternatus Hope by Sanitation Cuttings. Biol. Disaster Sci. 2015, 38, 31–34. [Google Scholar]
- Roberts, M.; Gilligan, C.A.; Kleczkowski, A.; Hanley, N.; Whalley, A.E.; Healey, J.R. The effect of forest management options on forest resilience to pathogens. Front. For. Glob. Change 2020, 3, 7. [Google Scholar] [CrossRef]
- Wang, X.Y.; Wu, X.Q.; Wen, T.Y.; Feng, Y.Q.; Zhang, Y. Transcriptomic analysis reveals differentially expressed genes associated with pine wood nematode resistance in resistant Pinus thunbergii. Tree Physiol. 2023, 43, 995–1008. [Google Scholar] [CrossRef]
- Nose, M.; Shiraishi, S. Breeding for Resistance to Pine Wilt Disease; Springer: Tokyo, Japan, 2008. [Google Scholar]
- Zhu, S.W.; Liu, L.X.; Hu, X.F.; Dai, W.; Wang, Y.R. The effects of different thinning intensities on the understory vegetation characteristics of mixed forests of Larix principis-rupprechtii. For. Eng. 2024, 40, 47–55. [Google Scholar]
- Han, S.; Tan, S.; Wang, A.; Chen, W.; Huang, Q. Bacterial rather than fungal diversity and community assembly drive soil multifunctionality in a subtropical forest ecosystem. Environ. Microbiol. Rep. 2022, 14, 85–95. [Google Scholar] [CrossRef]
- Singh Rawat, V.; Kaur, J.; Bhagwat, S.; Arora Pandit, M.; Dogra Rawat, C. Deploying microbes as drivers and indicators in ecological restoration. Restor. Ecol. 2023, 31, e13688. [Google Scholar] [CrossRef]
- Yang, G.; Ryo, M.; Roy, J.; Lammel, D.R.; Ballhausen, M.B.; Jing, X.; Zhu, X.F.; Rillig, M.C. Multiple anthropogenic pressures eliminate the effects of soil microbial diversity on ecosystem functions in experimental microcosms. Nat. Commun. 2022, 13, 4260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Nunan, N.; Hirsch, P.R.; Sun, B.; Zhou, J.; Liang, Y. Theory of microbial coexistence in promoting soil–plant ecosystem health. Biol. Fertil. Soils 2021, 57, 897–911. [Google Scholar] [CrossRef]
- She, J.R.; Zheng, Q.; Zhang, X.; Yu, N.H.; Li, H. Study on rhizosphere microbial community structure and soil physical and chemical properties of rare trees in Xiangxi Region. J. Cent. South Univ. For. Technol. 2023, 43, 104–115. [Google Scholar]
- Wang, N.; Gao, J.; Wei, J.; Liu, Y.; Zhuang, X.L.; Zhuang, G.Q. Effects of Wetland Reclamation on Soil Microbial Community Structure in the Sanjiang Plain. Environ. Sci. 2019, 40, 2375–2381. [Google Scholar]
- Chen, X.L.; Hu, M.Y.; Zheng, G.C.; Chen, H.Y.H. Persistent soil organic carbon deficits from converting primary forests to plantations and secondary forests in subtropical China. Glob. Ecol. Conserv. 2023, 45, e02530. [Google Scholar] [CrossRef]
- Yang, X.M.; Feng, Q.; Zhu, M.; Zhang, J.T.; Yang, L.S.; Li, R.L. The Impact of Artificial Restoration of Alpine Grasslands in the Qilian Mountains on Vegetation, Soil Bacteria, and Soil Fungal Community Diversity. Microorganisms 2024, 12, 854. [Google Scholar] [CrossRef]
- Meng, W.J.; Li, Y.L.; Qu, Z.L.; Zhang, Y.M.; Liu, B.; Liu, K.; Gao, Z.W.; Dong, L.N.; Sun, H. Fungal community structure shifts in litter degradation along forest succession induced by pine wilt disease. Microbiol. Res. 2024, 280, 127588. [Google Scholar] [CrossRef]
- Qiao, H.; Mo, X.Q.; Luo, Y.H.; Liu, X.Y.; Hu, Y.J.; Chen, X.B.; Su, Y.R. Patterns of soil ecoenzymatic stoichiometry and its influencing factors during stand development in Camellia oleifera plantations. Acta Ecol. Sin. 2019, 39, 1887–1896. [Google Scholar]
- Xun, W.B.; Li, W.; Xiong, W.; Ren, Y.; Liu, Y.P.; Miao, Y.Z.; Xu, Z.H.; Zhang, N.; Shen, Q.R.; Zhang, R.F. Diversity-triggered deterministic bacterial assembly constrains community functions. Nat. Commun. 2019, 10, 3833. [Google Scholar] [CrossRef]
- Yan, Y.; Klinkhamer, P.G.L.; Veen, J.A.V.; Kuramae, E.E. Environmental filtering: A case of bacterial community assembly in soil. Soil Biol. Biochem. 2019, 136, 107531. [Google Scholar] [CrossRef]
- Lan, G.Y.; Wu, Z.X.; Yang, C.; Sun, R.; Chen, B.Q.; Zhang, X.C. Forest conversion alters the structure and functional processes of tropical forest soil microbial communities. Land Degrad. Dev. 2021, 32, 613–627. [Google Scholar] [CrossRef]
- Nemergut, D.R.; Schmidt, S.K.; Fukami, T.; O’Neill, S.P.; Bilinski, T.M.; Stanish, L.F.; Knelman, J.E.; Darcy, J.L.; Lynch, R.C.; Wickey, P.; et al. Patterns and processes of microbial community assembly. Microbiol. Mol. Biol. Rev. 2013, 77, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Liu, J.; Jia, T.; Luo, Z.; Li, C.; Chai, B. Assembly mechanisms of soil bacterial communities in subalpine coniferous forests on the Loess Plateau, China. J. Microbiol. 2019, 57, 461–469. [Google Scholar] [CrossRef]
- Zhou, J.Z.; Liu, W.Z.; Deng, Y.; Jiang, Y.H.; Wang, A.J. Stochastic Assembly Leads to Alternative Communities with Distinct Functions in a Bioreactor Microbial Community. Mbio 2013, 4, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.; Parsons, T.L.; Ijaz, U.Z.; Lahti, L.; Holmes, I.; Quince, C. Linking statistical and ecological theory: Hubbell’s unified neutral theory of biodiversity as a hierarchical Dirichlet process. Proc. IEEE 2015, 105, 516–529. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, H.; Sun, Y.; Yang, Q.; Fan, J. Contrasting the assembly of phytoplankton and zooplankton communities in a polluted semi-closed sea: Effects of marine compartments and environmental selection. Environ. Pollut. (Barking Essex 1987) 2021, 285, 117256. [Google Scholar] [CrossRef]
- Vignola, M.; Werner, D.; Wade, M.J.; Meynet, P.; Davenport, R.J. Medium shapes the microbial community of water filters with implications for effluent quality. Water Res. 2018, 129, 499–508. [Google Scholar] [CrossRef]
- Yu, C.F.; Zhu, Z.Y.; Zhang, M.X. Unveiling the impact and mechanisms of Cd-driven ecological assembly and coexistence of bacterial communities in coastal sediments of Yellow Sea. J. Hazard. Mater. 2023, 460, 132309. [Google Scholar] [CrossRef]
- Goss-Souza, D.; Mendes, L.W.; Rodrigues, J.L.M.; Tsai, S.M. Ecological processes shaping bulk soil and rhizosphere microbiome assembly in a long-term Amazon forest-to-agriculture conversion. Microb. Ecol. 2020, 79, 110–122. [Google Scholar] [CrossRef]
- He, H.R.; Xu, M.Z.; Li, W.T.; Chen, L.; Chen, Y.A.; Moorhead, D.L.; Brangari, A.C.; Liu, J.; Cui, Y.X.; Zeng, Y.; et al. Linking soil depth to aridity effects on soil microbial community composition, diversity and resource limitation. Catena 2023, 232, 107393. [Google Scholar] [CrossRef]
- Ghazouani, S.; Béjaoui, Z.; Michael, P.; Spiers, G.; Beckett, P.; Gtari, M.; Nkongolo, K. Rhizobioaugmentation of Casuarina glauca with N-fixing actinobacteria Frankia decreases enzymatic activities in wastewater irrigated soil: Effects of Frankia on C. glauca growth. Ecotoxicology 2020, 29, 417–428. [Google Scholar] [CrossRef]
- Shang, H.B. Methods of Conventional Analysis of Resources and Environment; Northwest Agriculture and Forestry University Press: Yangling, China, 2010. [Google Scholar]
- Sun, Y.; Shi, Y.; Tang, Y.; Tian, J.; Wu, X. Correlation between plant diversity and the physicochemical properties of soil microbes. Appl. Ecol. Environ. Res. 2019, 17, 10371–10388. [Google Scholar] [CrossRef]
- Afonin, F.V.P. X-ray fluorescence analysis. Z. Vsesoyuznogo Khimicheskogo Obs. Im. DI Mendeleeva 1980, 25, 610–615. [Google Scholar]
- Wang, J.P.; Chen, G.R.; Ji, S.H.; Zhong, Y.Q.; Zhao, Q.; He, Q.Q.; Wu, Y.H.; Bing, H.J. Close relationship between the gene abundance and activity of soil extracellular enzyme: Evidence from a vegetation restoration chronosequence. Soil Biol. Biochem. 2023, 177, 108929. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.T.; Liu, F.; Liang, J.M.; Zhao, P.; Tsui, C.K.M.; Cai, L. Cross-kingdom synthetic microbiota supports tomato suppression of Fusarium wilt disease. Nat. Commun. 2022, 13, 7890. [Google Scholar] [CrossRef] [PubMed]
- Tanunchai, B.; Ji, L.; Schroeter, S.A.; Wahdan, S.F.M.; Hossen, S.; Buscot, Y.F.; Lehnert, A.S.; Alves, E.G.; Hilke, I.; Gleixner, G.; et al. FungalTraits vs. FUNGuild: Comparison of Ecological Functional Assignments of Leaf- and Needle-Associated Fungi Across 12 Temperate Tree Species. Microb. Ecol. 2023, 85, 411–428. [Google Scholar] [CrossRef]
- Yang, Z.C.; Peng, C.Y.; Cao, H.M.; Song, J.J.; Gong, B.; Li, L.; Wang, L.; He, Y.; Liang, M.; Lin, J.C. Microbial functional assemblages predicted by the FAPROTAX analysis are impacted by physicochemical properties, but C, N and S cycling genes are not in mangrove soil in the Beibu Gulf, China. Ecol. Indic. 2022, 139, 108887. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.J.; Fredrickson, J.K.; Chen, X.Y.; Kennedy, D.W.; Murray, C.J.; Rockhold, M.L.; Konopka, A. Quantifying community assembly processes and identifying features that impose them. Isme J. 2013, 7, 2069–2079. [Google Scholar] [CrossRef]
- Hou, Y.P.; Liu, L.; Chu, H.; Ma, S.J.; Zhao, D.; Liang, R.R. Effects of exotic plant Rhus typhina invasion on soil properties in different forest types. Acta Ecol. Sin. 2015, 35, 5324–5330. [Google Scholar]
- Guo, J.Y.; Wang, L.; Yang, J.J.; Ma, F.Y.; Ma, X.S.; Wang, W.B.; Dong, Y.F. Understory vegetation patterns and soil characteristics of a Pinus thunbergii plantation in mountainous land of Shandong Province. J. Zhejiang AF Univ. 2018, 35, 209–218. [Google Scholar]
- Mól, P.C.G.; Júnior, J.C.Q.; Veríssimo, L.A.A.; Boscolo, M.; Gomes, E.; Minim, L.A.; Da Silva, R. β-glucosidase: An overview on immobilization and some aspects of structure, function, applications and cost. Process Biochem. 2023, 130, 26–39. [Google Scholar] [CrossRef]
- Wu, H.W.; Cui, H.L.; Fu, C.X.; Li, R.; Qi, F.Y.; Liu, Z.L.; Yang, G.; Xiao, K.Q.; Qiao, M. Unveiling the crucial role of soil microorganisms in carbon cycling: A review. Sci. Total Environ. 2024, 909, 168627. [Google Scholar] [CrossRef]
- Yang, M.; Yang, D.; Yu, X. Soil microbial communities and enzyme activities in sea-buckthorn (Hippophae rhamnoides) plantation at different ages. PLoS ONE 2018, 13, e0190959. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Wang, C.; Wang, H.; Chen, H.; Tang, M. Pine wilt disease alters soil properties and root-associated fungal communities in Pinus tabulaeformis forest. Plant Soil 2016, 404, 237–249. [Google Scholar] [CrossRef]
- Lin, Y.T.; Whitman, W.B.; Coleman, D.C.; Chiu, C.Y. Effects of reforestation on the structure and diversity of bacterial communities in subtropical low mountain forest soils. Front. Microbiol. 2018, 9, 1968. [Google Scholar] [CrossRef]
- Tedersoo, L.; Mikryukov, V.; Anslan, S.; Bahram, M.; Khalid, A.N.; Corrales, A.; Abarenkov, K. The Global Soil Mycobiome consortium dataset for boosting fungal diversity research. Fungal Divers. 2021, 111, 573–588. [Google Scholar] [CrossRef]
- Tian, H.M.; Li, L.Z.; Zhu, Y.P.; Wang, C.C.; Wu, M.X.; Shen, W.X.; Li, C.R.; Li, K. Soil fungal community and co-occurrence network patterns at different successional stages of black locust coppice stands. Front. Microbiol. 2025, 16, 1528028. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, S.H.; Jo, H.Y.; Finneran, K.T.; Kwon, M.J.; Man, J.K. Diversity and composition of soil Acidobacteria and Proteobacteria communities as a bacterial indicator of past land-use change from forest to farmland. Sci. Total Environ. 2021, 797, 148944. [Google Scholar] [CrossRef]
- Rosling, A.; Eshghi Sahraei, S.; Kalsoom Khan, F.; Desirò, A.; Bryson, A.E.; Mondo, S.J.; Grigoriev, L.V.; Bonito, G.; Sánchez-García, M. Evolutionary history of arbuscular mycorrhizal fungi and genomic signatures of obligate symbiosis. BMC Genom. 2024, 25, 529. [Google Scholar] [CrossRef]
- Li, X.; Liu, Y.; Wu, G.P.; Lie, Z.Y.; Sheng, H.; Aguila, L.C.R.; Khan, M.Q.; Liu, X.J.; Zhou, S.Y.D.; Wu, T. Mixed plantations do not necessarily provide higher ecosystem multifunctionality than monoculture plantations. Sci. Total Environ. 2024, 914, 170156. [Google Scholar] [CrossRef]
- Tan, R.; Yu, S.Q.; LI, Y.; Wang, X.F.; Xu, X.Y.; Li, Y.H.; Wang, W.F. Effect of thinning restoration on enzyme activity and enzyme stoichiometry in the topsoil of oak-pine mixed forest. J. Zhejiang AF Univ. 2024, 41, 1201–1210. [Google Scholar]
- Wei, X.; Liang, W.J. Regulation of stand density alters forest structure and soil moisture during afforestation with Robinia pseudoacacia L. and Pinus tabulaeformis Carr on the Loess Plateau. For. Ecol. Manag. 2021, 491, 119196. [Google Scholar] [CrossRef]
- Guo, L.; Chen, X.H.; Sheng, Y.Z.; Yang, N.; Hou, A.K.; Fang, H.S. Impact of soil fissure status on microbial community in mining-disturbed area, the northern Shaanxi province. Front. Microbiol. 2024, 15, 1463665. [Google Scholar] [CrossRef]
- Jing, J.Y.; Cong, F.W.; Bezemer, T.M. Legacies at work: Plant–soil–microbiome interactions underpinning agricultural sustainability. Trends Plant Sci. 2022, 27, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Q.; Guo, Z.Y.; Chen, C.G.; Jia, Z.J. Soil microbial community structure and diversity are largely influenced by soil pH and nutrient quality in 78-year-old tree plantations. Biogeosciences 2017, 14, 2101–2111. [Google Scholar] [CrossRef]
- Wang, X.J.; Zhang, Z.C.; Yu, Z.Q.; Shen, G.F.; Cheng, H.F.; Tao, S. Composition and diversity of soil microbial communities in the alpine wetland and alpine forest ecosystems on the Tibetan Plateau. Sci. Total Environ. 2020, 747, 141358. [Google Scholar] [CrossRef]
- Lehto, T.; Zwiazek, J.J. Ectomycorrhizas and water relations of trees: A review. Mycorrhiza 2011, 21, 71–90. [Google Scholar] [CrossRef]
- Anthony, M.A. Does ectomycorrhizal fungal biodiversity affect tree growth? Fungal Ecol. 2025, 74, 101413. [Google Scholar] [CrossRef]
- Clemmensen, K.E.; Bahr, A.; Ovaskainen, O.; Dahlberg, A.; Ekblad, A.; Wallander, H.; Stenlid, J.; Finlay, R.D.; Wardle, D.A.; Lindahl, B.D. Roots and associated fungi drive long-term carbon sequestration in boreal forest. Science 2013, 339, 1615–1618. [Google Scholar] [CrossRef]
- Maraón-Jiménez, S.; Radujkovi, D.; Verbruggen, E.; Grau, O.; Cuntz, M.; Peuelas, J.; Richter, A.; Schrumpf, M.; Rebmann, C. Shifts in the abundances of saprotrophic and ectomycorrhizal fungi with altered leaf litter inputs. Front. Plant Sci. 2021, 12, 682142. [Google Scholar]
- Du, L.; Zhong, H.; Guo, X.; Li, H.; Xia, J.; Chen, Q. Nitrogen fertilization and soil nitrogen cycling: Unraveling the links among multiple environmental factors, functional genes, and transformation rates. Sci. Total Environ. 2024, 951, 175561. [Google Scholar] [CrossRef] [PubMed]
- George, D.M.; Vincent, A.S.; Mackey, H.R. An overview of anoxygenic phototrophic bacteria and their applications in environmental biotechnology for sustainable Resource recovery. Biotechnol. Rep. 2020, 28, e00563. [Google Scholar] [CrossRef]
- Ma, J.M.; Ma, K.; Liu, J.L.; Chen, N.N. Rhizosphere soil microbial community under ice in a high-latitude wetland: Different community assembly processes shape patterns of rare and abundant microbes. Front. Microbiol. 2022, 13, 783371. [Google Scholar] [CrossRef]
- Willig, M.R.; Moulton, M.P. The role of stochastic and deterministic processes in structuring Neotropical bat communities. J. Mammal. 1989, 70, 323–329. [Google Scholar] [CrossRef]
- Schmidt, S.K.; Nemergut, D.R.; Darcy, J.L.; Lynch, R. Do Bacterial and Fungal Communities Assemble Differently During Primary Succession? Wiley Online Library: Hoboken, NJ, USA, 2014; pp. 254–258. [Google Scholar]
- Dong, L.; Li, M.X.; Li, S.; Yue, L.Y.; Ali, M.; Han, J.; Lian, W.H.; Hu, C.J.; Lin, Z.L.; Shi, G.Y. Aridity drives the variability of desert soil microbiomes across north-western China. Sci. Total Environ. 2024, 907, 168048. [Google Scholar] [CrossRef]
- Lu, M.; Tian, K.; Sun, X.Y.; Ren, Y.L.; Wang, H.; Peng, S.X. Variation of soil fungal community characteristics of typical wetland in Napahai between dry wet seasons under different waterlogging conditions. Sci. Silvae Sin. 2018, 54, 98–109. [Google Scholar]
- Zhou, J.Z.; Ye, D.; Zhang, P.; Xue, K.; Liang, Y.T.; Nostrand, J.D.V.; Yang, Y.F.; He, Z.L.; Wu, L.Y.; Stahl, D.A. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc. Natl. Acad. Sci. USA 2014, 111, E836–E845. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Foresty-Type | S-β-GC (umol/d/g) | S-NAG (umol/d/g) | S-ACP (umol/d/g) | S-ASF (umol/d/g) |
|---|---|---|---|---|
| Mixed pine forests | 2.71 ± 1.87 b | 7.96 ± 1.23 c | 17.48 ± 1.70 a | 0.66 ± 0.07 b |
| Pinus thunbergii forests | 17.06 ± 3.36 a | 18.83 ± 1.67 b | 21.72 ± 8.99 a | 1.99 ± 0.56 a |
| Pinus tada forests | 12.12 ± 1.08 b | 27.72 ± 7.26 a | 21.52 ± 3.66 a | 1.22 ± 0.166 b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Liu, Z.; Cao, M.; Dai, T. Effects of Thinning of the Infected Trees and Cultivating of the Resistant Pines on Soil Microbial Diversity and Function. Forests 2025, 16, 813. https://doi.org/10.3390/f16050813
Zhang X, Liu Z, Cao M, Dai T. Effects of Thinning of the Infected Trees and Cultivating of the Resistant Pines on Soil Microbial Diversity and Function. Forests. 2025; 16(5):813. https://doi.org/10.3390/f16050813
Chicago/Turabian StyleZhang, Xiaorui, Zhuo Liu, Mu Cao, and Tingting Dai. 2025. "Effects of Thinning of the Infected Trees and Cultivating of the Resistant Pines on Soil Microbial Diversity and Function" Forests 16, no. 5: 813. https://doi.org/10.3390/f16050813
APA StyleZhang, X., Liu, Z., Cao, M., & Dai, T. (2025). Effects of Thinning of the Infected Trees and Cultivating of the Resistant Pines on Soil Microbial Diversity and Function. Forests, 16(5), 813. https://doi.org/10.3390/f16050813