
Molecular Phylogenetics of the Orchid Genus Spathoglottis (Orchidaceae: Collabieae) in Peninsular Malaysia and Borneo

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Taxon Sampling
2.2. Morphological Observations
2.3. DNA Extraction
2.4. Amplification
2.5. Sequence Editing and Alignment
2.6. Database Search—BLAST
2.7. Maximum Likelihood Analysis
2.8. Maximum Parsimony Analysis
2.9. Bayesian Inference Analysis
2.10. Test of Incongruence
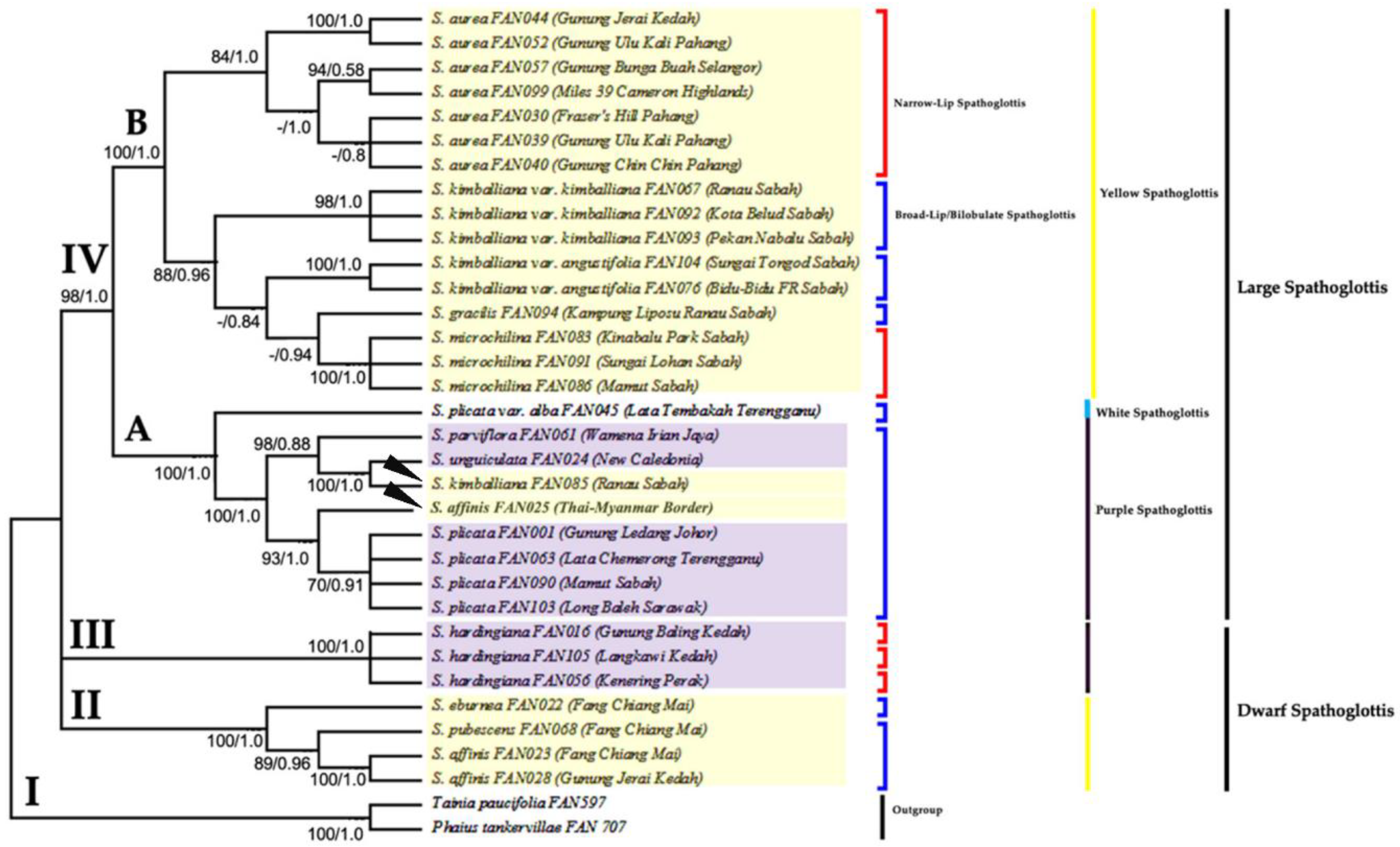
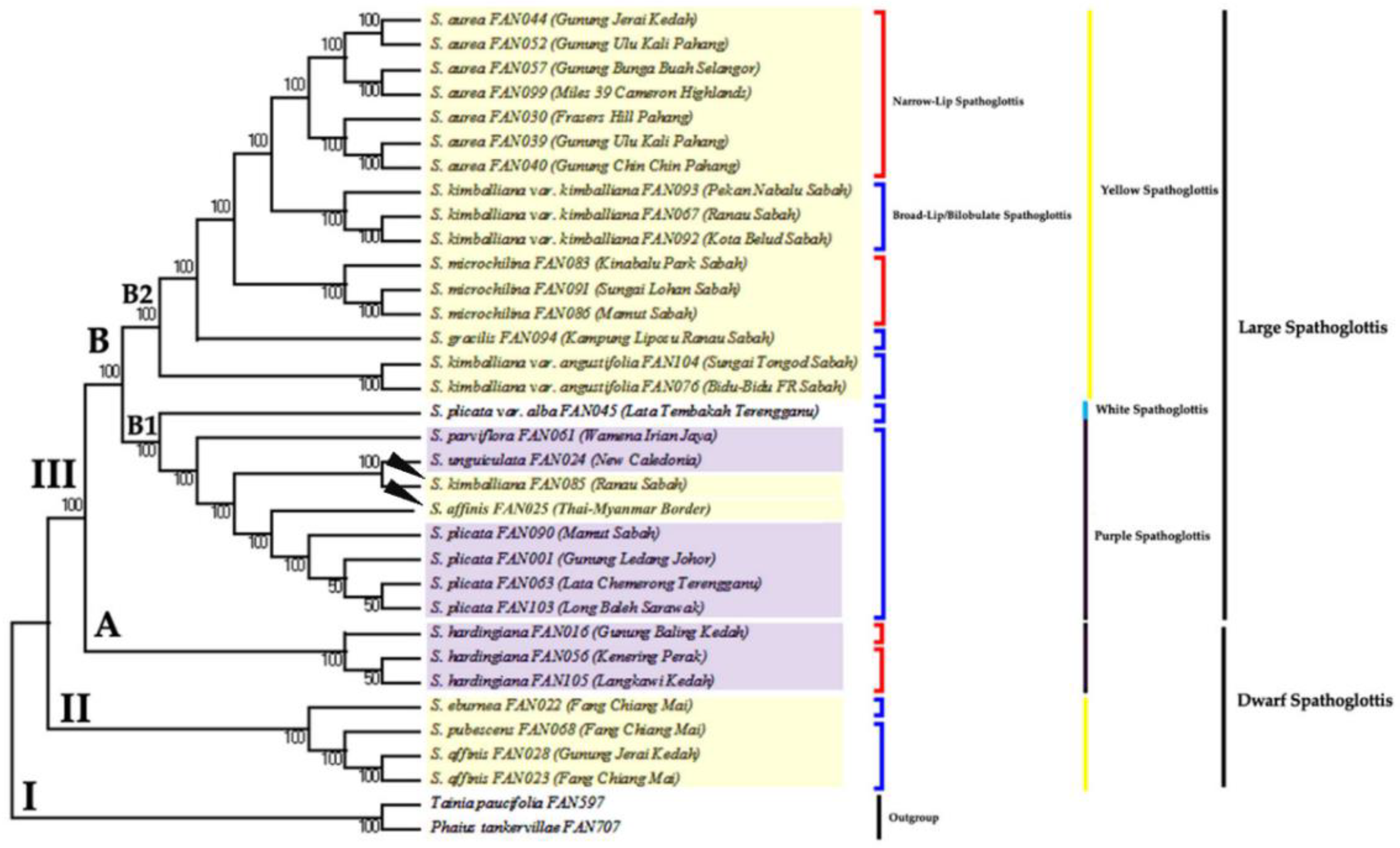
3. Results
3.1. Groupings of Taxa Based on Selected Morphological Characters
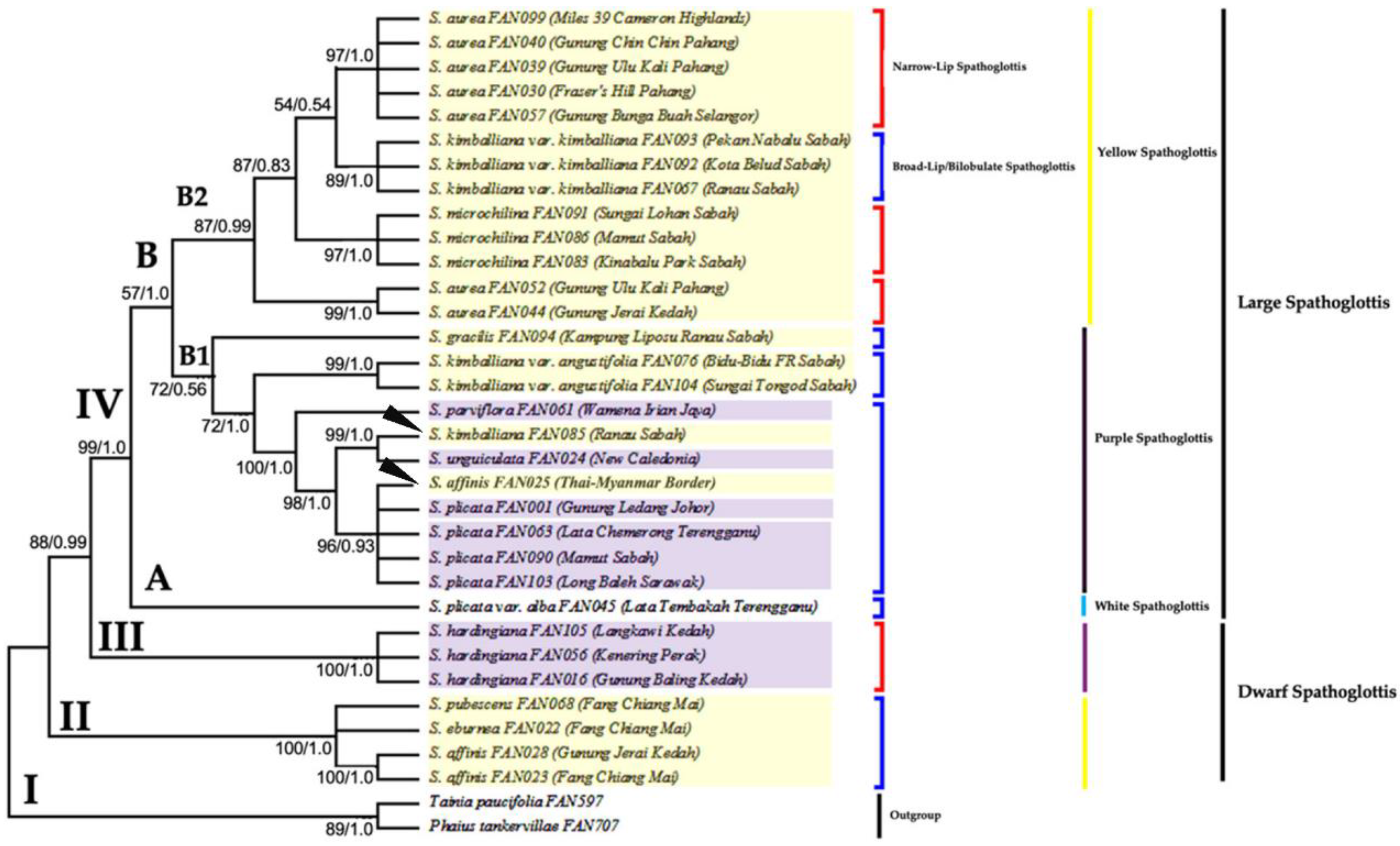
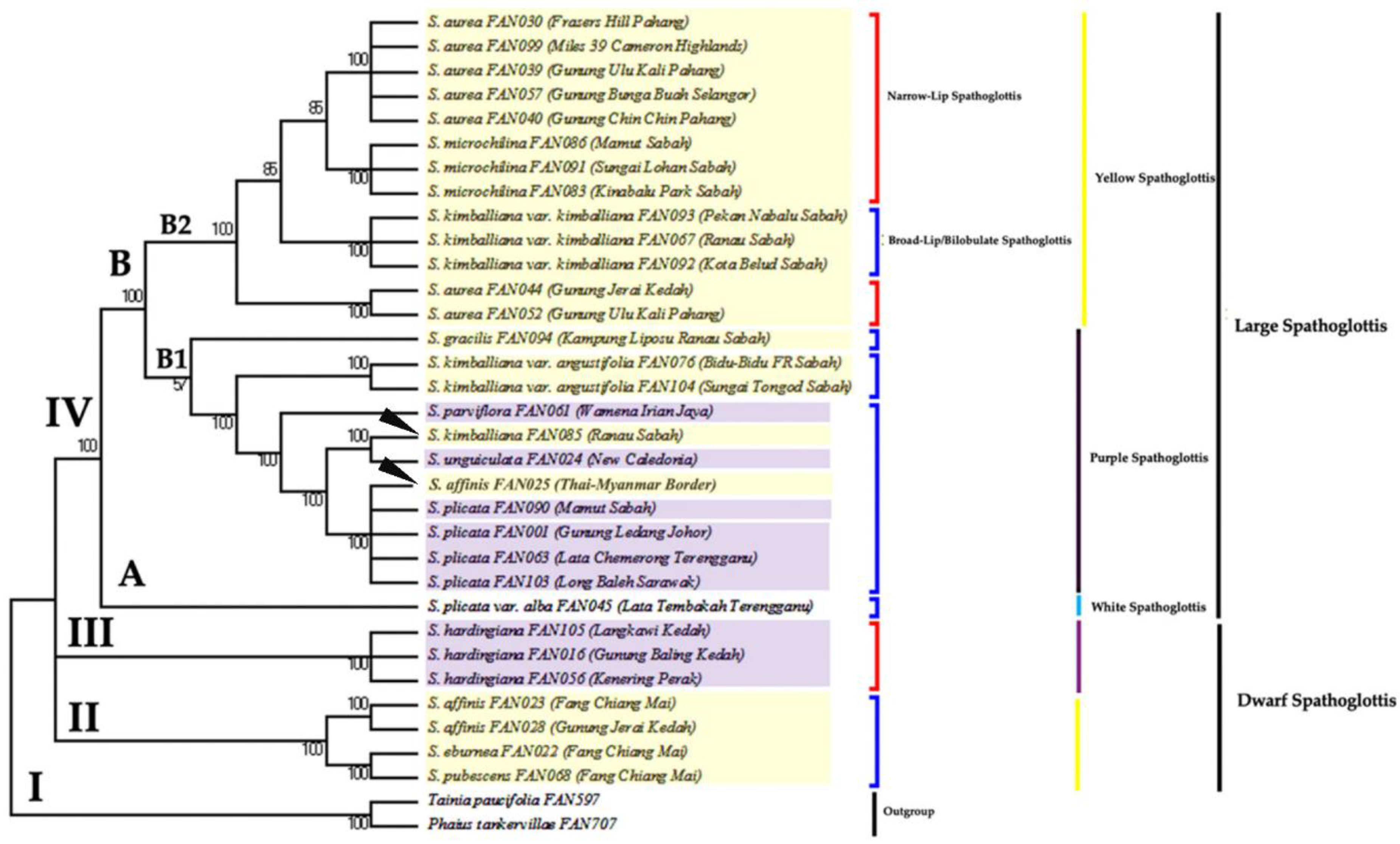
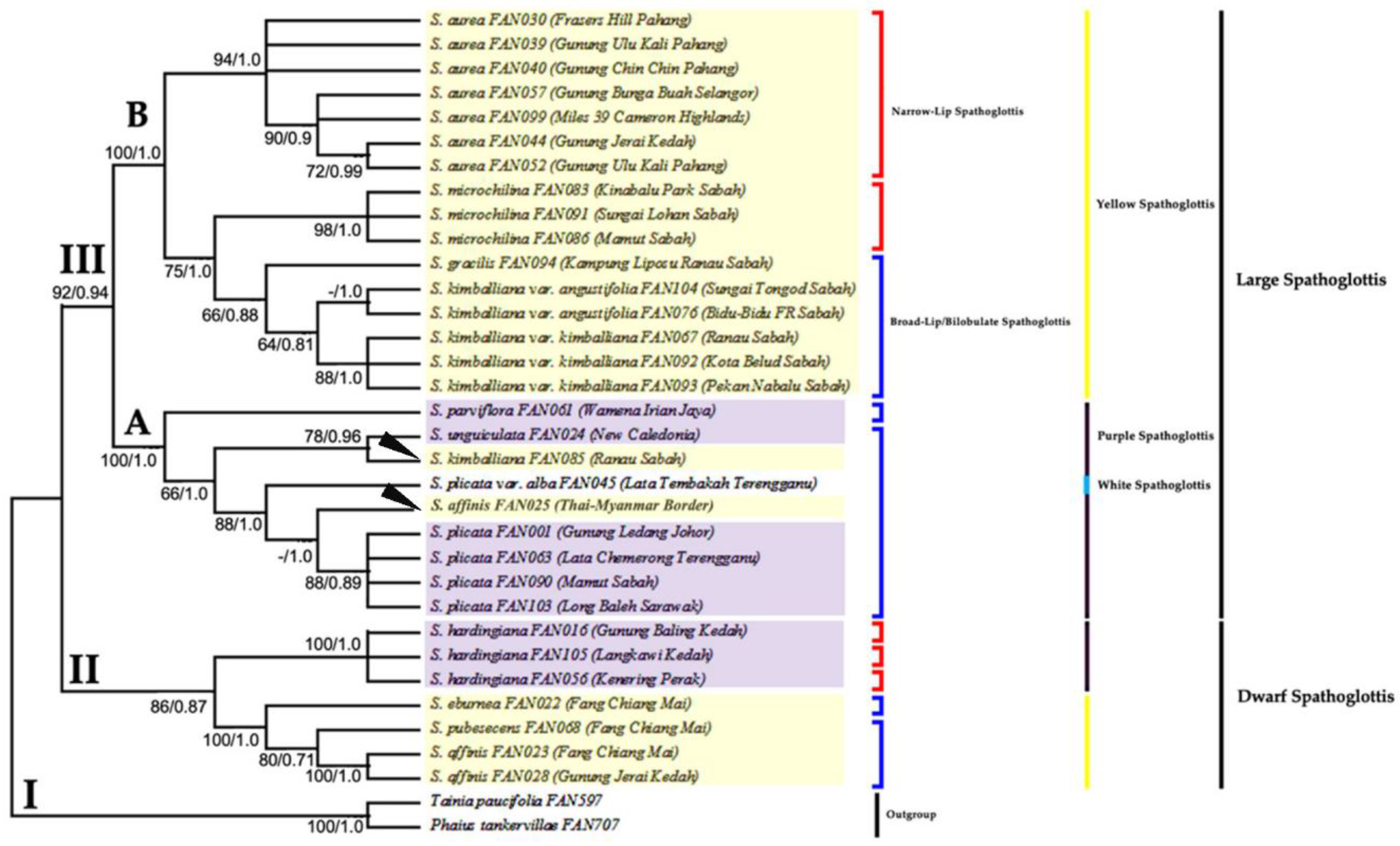
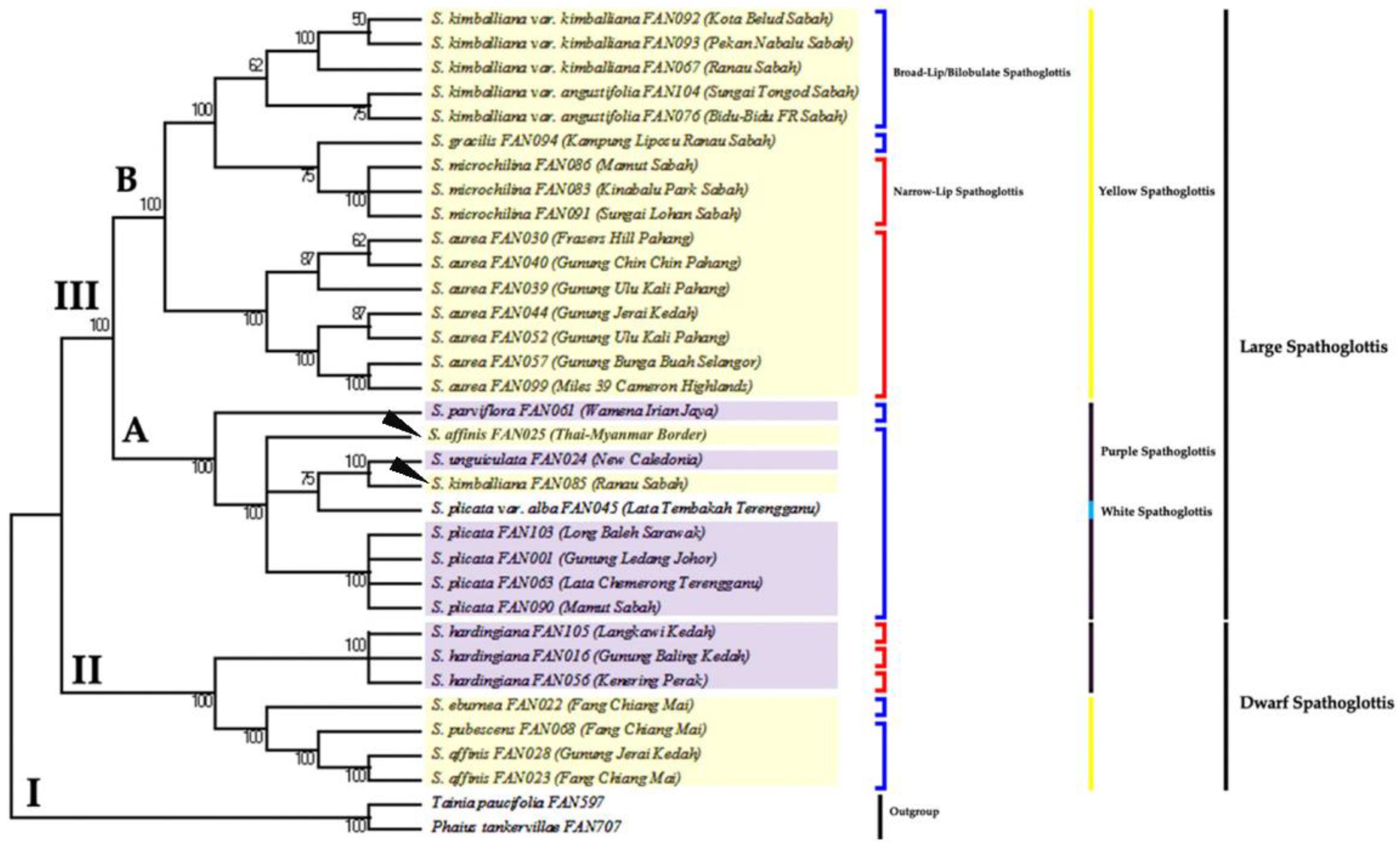
3.2. Phylogenetic Analysis Based on ITS Data Matrix
3.3. Phylogenetic Analysis Based on Combined Plastid Sequence Data
3.4. Phylogenetic Analysis Based on Combined Plastid and nrITS Data
4. Discussion
4.1. The Efficacy of Nuclear and Plastid Gene Regions in Inferring Phylogenetic Relationships
4.2. Monophyly of Genus Spathoglottis
4.3. Proposed Taxonomic and Nomenclatural Changes for Spathoglottis plicata var. alba
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- POWO, Plants of the World Online (Facilitated by the Royal Botanic Gardens, Kew). Available online: https://powo.science.kew.org/taxon/urn:lsid:ipni.org:names:325895- (accessed on 15 October 2022).
- Cribb, P.J.; Tang, C.Z. Spathoglottis (Orchidaceae) in Australia and the Pacific Islands. Kew Bull. 1981, 36, 721–729. [Google Scholar] [CrossRef]
- Sosa, V. A molecular and morphological phylogenetic study of subtribe Bletiinae (Epidendreae, Orchidaceae). Syst. Bot. 2007, 32, 34–42. [Google Scholar] [CrossRef]
- Xiang, X.G.; Jin, W.T.; Li, D.Z.; Schuiteman, A.; Huang, W.C.; Li, J.W.; Jin, X.; Li, Z.Y. Phylogenetics of tribe Collabieae (Orchidaceae, Epidendroideae) based on four chloroplast genes with morphological appraisal. PLoS ONE 2014, 9, e98721. [Google Scholar] [CrossRef]
- Chase, M.W.; Cameron, K.M.; Freudenstein, J.V.; Pridgeon, A.M.; Salazar, G.; Van Den Berg, C.; Schuiteman, A. An updated classification of Orchidaceae. Bot. J. Linn. Soc. 2015, 177, 151–174. [Google Scholar] [CrossRef]
- Dockrill, A. Australian Indigenous Orchids Vol. 1. In: Cribb, P.J.; Tang, C.Z. Spathoglottis (Orchidaceae) in Australia and the Pacific Islands (Eds.). Kew Bull. 1969, 36, 721–729. [Google Scholar]
- Hallê, N. Flore de la Nouvelle-Calêdonie et Dêpendances and Orchidacées. In: Cribb, P.J.; Tang, C.Z. Spathoglottis (Orchidaceae) in Australia and the Pacific Islands (Eds.). Kew Bull. 1977, 36, 721–729. [Google Scholar]
- Holttum, R.E. A Revised Flora of Malaya: Orchids of Malaya, 3rd ed.; Government Printing Office: Singapore, 1964; Volume 1.
- Seidenfaden, G.; Wood, J.J. The Orchids of Peninsular Malaysia and Singapore: A Revision of R.E. Holttum: Orchids of Malaya; Olsen and Olsen in Association with The Kew Royal Botanic Gardens, and Singapore Botanic Gardens: Fredensborg, Denmark, 1992. [Google Scholar]
- Solereder, H.; Meyer, F.J. Systematic anatomy of the monocotyledons. In: Freudenstein, J.V.; Rasmussen, F.N. (Eds.) What does morphology tell us about orchid relationships?—A cladistic analysis. Am. J. Bot. 1930, 86, 225–248. [Google Scholar]
- Williams, N.H. Subsidiary cells in the Orchidaceae: Their general distribution with special reference to development in the Oncidieae. Bot. J. Linn. Soc. 1979, 78, 41–66. [Google Scholar] [CrossRef]
- Pridgeon, A.M.; Stern, W.L.; Benzing, D.H. Tilosomes in roots of Orchidaceae: Morphology and systematic occurrence. Am. J. Bot. 1983, 70, 1365–1377. [Google Scholar] [CrossRef]
- Porembski, S.; Barthlottt, W. Velamen radicum micromorphology and classification of Orchidaceae. Nord. J. Bot. 1988, 8, 117–137. [Google Scholar] [CrossRef]
- Teoh, S.B. Polyploid spore formation in diploid orchid species. Genetica 1984, 63, 53–59. [Google Scholar] [CrossRef]
- Brandham, P. Cytogenetics. In Genera Orchidacearum Vol. 1: General Introduction, Apostasioideae, Cypripedioideae; Pridgeon, A.M., Cribb, P.J., Chase, M.W., Rasmussen, F.N., Eds.; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Ginibun, F.C.; Saad, M.R.M.; Hong, T.L.; Othman, R.Y.; Khalid, N.; Bhassu, S. Chloroplast DNA barcoding of Spathoglottis species for genetic conservation. Acta Holticulturae 2010, 878, 453–459. [Google Scholar] [CrossRef]
- Seidenfaden, G.; Smitinand, T. The Orchids of Thailand: A Preliminary List; The Siam Society: Bangkok, Thailand, 1959. [Google Scholar]
- Wood, J.J. Orchids of Borneo Vol. 3: Dendrobium, Dendrochilum and Others; The Sabah Society in Association with The Bentham-Moxon Trust: Kota Kinabalu, Malaysia, 1997. [Google Scholar]
- Chan, C.L.; Lamb, A.; Shim, P.S.; Wood, J.J. Orchids of Borneo Vol. 1 Introduction and a Selection of Species; The Sabah Society in Association with The Bentham-Moxon Trust, England: Kota Kinabalu, Malaysia, 1994. [Google Scholar]
- Comber, J.B. Orchids of Sumatra; Natural History Publications in Association with The Royal Botanic Gardens, Kew and Botanic Gardens, Singapore: Kota Kinabalu, Malaysia, 2001. [Google Scholar]
- Thiers, B. Index Herbariorum: A Global Directory of Public Herbaria and Associated Staff. Available online: https://sweetgumnybg.org/science/ih (accessed on 27 September 2022).
- Doyle, J.J.; Doyle, J.L. A rapid isolation procedure for small amounts of leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Sun, Y.; Skinner, D.Z.; Liang, G.H.; Hulbert, S.H. Phylogenetic analysis of Sorghum and related taxa using internal transcribed spacers of nuclear ribosomal DNA. Theor. Appl. Genet. 1994, 89, 26–32. [Google Scholar] [CrossRef]
- Cuénoud, P.; Savolainen, V.; Chatrou, L.W.; Powel, M.; Grayer, R.J.; Chase, M.W. Molecular phylogenetics of Caryophyllales based on nuclear 18S rDNA and plastid rbcL, atpB, and matK DNA sequences. Am. J. Bot. 2002, 89, 132–144. [Google Scholar] [CrossRef]
- Taberlet, P.; Gielly, L.; Pautou, G.; Bouvet, J. Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol. Biol. 1991, 17, 1105–1109. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user friendly biological sequence alignment editor and analysis performer windows 95/98/NT. Nucleic Acid Symp. Ser. Oxf. Acad. 1999, 41, 95–98. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA 6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Fitch, W.M. Towards defining the course of evolution: Minimum change for a specific tree topology. Syst. Zool. 1971, 20, 406–416. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Milne, I.; Wright, F.; Rowe, G.; Marshal, D.F.; Husmeier, D.; McGuire, G. TOPALi: Software for Automatic Identification of Recombinant Sequences within DNA Multiple Alignments. Bioinformatics 2004, 20, 1806–1807. [Google Scholar] [CrossRef]
- Felsenstein, J. Distance methods for inferring phylogenies: A justification. Evolution 1984, 38, 16–24. [Google Scholar]
- Erixon, P.; Svennblad, B.; Bitton, T.; Oxelman, B. Reliability of Bayesian posterior probabilities and bootstrap frequencies in phylogenetics. Syst. Biol. 2003, 52, 665–673. [Google Scholar] [CrossRef]
- Farris, J.S.; Kallersjo, M.; Kluge, A.G.; Bult, C. Constructing Significance Test for Incongruence. Syst. Biol. 1995, 44, 570–572. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (* and Other Methods), Version 4b10; Sinauer: Sunderland, MA, USA, 2002.
- Hecht, A.; Glasgow, J.; Jaschke, P.R.; Bawazer, L.A.; Munson, M.S.; Cochran, J.R.; Endy, D.; Salit, M. Measurements of translation initiation from all 64 codons in E. coli. Nucleic Acids Res. 2017, 45, 3615–3626. [Google Scholar] [CrossRef]
- Soltis, D.E.; Soltis, P.S. Phylogenetic relationships among Saxifragaceae sensu lato: A comparison of topologies based on 18S and rbcL sequences. Am. J. Bot. 1997, 84, 504–522. [Google Scholar] [CrossRef]
- Baldwin, B.G.; Sanderson, M.J.; Porter, J.M.; Wojciechowski, M.F.; Campbell, C.S.; Donoghue, M.J. The ITS region of nuclear ribosomal DNA: A valuable source of evidence on angiosperm phylogeny. Ann. Mol. Bot. Gard. 1995, 82, 247–277. [Google Scholar] [CrossRef]
- Aceto, S.; Caputo, P.; Cozzolino, S.; Gaudio, L.; Moretti, A. Phylogeny and evolution of Orchis and allied genera based on ITS DNA variation: Morphological gaps and molecular continuity. Mol. Phylogenetics Evol. 1999, 13, 67–76. [Google Scholar] [CrossRef]
- Van Den Berg, C.; Goldman, D.H.; Freudenstein, J.V.; Pridgeon, A.M.; Cameron, K.M.; Chase, M.W. An overview of the phylogenetic relationships within Eidendroideae inferred from multiple DNA regions and recircumscription of Epidendreae and Arethuseae (Orchidaceae). Am. J. Bot. 2005, 92, 613–624. [Google Scholar] [CrossRef]
- Kocyan, A.; Qiu, Y.L.; Endress, P.K.; Conti, E. A phylogenetic analysis of Apostasioideae (Orchidaceae) based on ITS, trnL–F and matK sequences. Plant Syst. Evol. 2004, 247, 203–213. [Google Scholar] [CrossRef]
- Chochai, A.; Leitch, I.J.; Ingrouille, M.J.; Fay, M.F. Molecular phylogenetics of Paphiopedilum (Cypripedioideae; Orchidaceae) based on nuclear ribosomal ITS and plastid sequences. Bot. J. Linn. Soc. 2012, 170, 176–196. [Google Scholar] [CrossRef][Green Version]
- Li, L.; Yan, H. Remarkable new species of Liparis (Orchidaceae) from China and its phylogenetic implications. PLoS ONE 2013, 8, e78112. [Google Scholar] [CrossRef]
- Xiang, X.-G.; Schuiteman, A.; Li, D.-Z.; Huang, W.-C.; Chung, S.-W.; Li, J.-W.; Zhou, H.-L.; Jin, W.-T.; Lai, Y.-J.; Li, Z.-Y.; et al. Molecular systematics of Dendrobium (Orchidaceae, Dendrobieae) from mainland Asia based on plastid and nuclear sequences. Mol. Phylogenetics Evol. 2013, 69, 950–960. [Google Scholar] [CrossRef]
- Barthet, M.M.; Hilu, K.W. Expression of matK: Functional and evolutionary implications. Am. J. Bot. 2007, 94, 1402–1412. [Google Scholar] [CrossRef]
- Bytebier, B.; Bellstedt, D.U.; Linder, H.P. A molecular phylogeny for the large African orchid genus Disa. Mol. Phylogenetics Evol. 2007, 4, 75–90. [Google Scholar] [CrossRef]
- Khew, G.S.W.; Chia, T.F. Parentage determination of Vanda Miss Joaquim (Orchidaceae) through two chloroplast genes rbcL and matK. AoB Plants 2011, 2011, plr018. [Google Scholar] [CrossRef]
- Yu, J.; Xue, J.H.; Zhou, S.L. New universal matK primers for DNA barcoding angiosperms. J. Syst. Evol. 2011, 49, 176–181. [Google Scholar] [CrossRef]
- Tsai, C.C.; Chou, C.H.; Wang, H.V.; Ko, Y.Z.; Chiang, T.Y.; Chiang, Y.C. Biogeography of the Phalaenopsis amabilis complex inferred from nuclear and plastid DNAs. BioMed. Cent. Plant Biol. 2015, 15, 202. [Google Scholar] [CrossRef]
- Bellstedt, D.U.; Linder, H.P.; Harley, E.H. Phylogenetic relationship in Disa based on non-coding TrnL-TrnF chloroplast sequences: Evidence of numerous repeat regions. Am. J. Bot. 2001, 88, 2088–2100. [Google Scholar] [CrossRef]
- Bellusci, F.; Pellegrino, G.; Palermo, A.M.; Musacchio, A. Phylogenetic relationships in the orchid genus Serapias L. based on non-coding regions of the chloroplast genome. Mol. Phylogenetics Evol. 2008, 47, 986–991. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Voucher Number | Locality | GenBank Accession Number | |||
|---|---|---|---|---|---|---|
| ITS | matK | trnL-F | ||||
| 1. | Spathoglottis affinis de Vriese | FAN023 | Fang District, Chiang Mai, Thailand | MG868982 | MG869016 | MG869050 |
| 2. | Spathoglottis affinis de Vriese | FAN025 | Thailand-Myanmar Border | MG869002 | MG869036 | MG869070 |
| 3. | Spathoglottis affinis de Vriese | FAN028 | Gunung Jerai, Kedah, Malaysia | MG868983 | MG869017 | MG869051 |
| 4. | Spathoglottis aurea Lindl. | FAN030 | Fraser’s Hill, Pahang, Malaysia | MG868984 | MG869018 | MG869052 |
| 5. | Spathoglottis aurea Lindl. | FAN039 | Gunung Ulu Kali, Pahang, Malaysia | MG868985 | MG869019 | MG869053 |
| 6. | Spathoglottis aurea Lindl. | FAN040 | Gunung Chin Chin, Pahang, Malaysia | MG868986 | MG869020 | MG869054 |
| 7. | Spathoglottis aurea Lindl. | FAN044 | Gunung Jerai, Kedah, Malaysia | MG868987 | MG869021 | MG869055 |
| 8. | Spathoglottis aurea Lindl. | FAN052 | Gunung Ulu Kali, Pahang, Malaysia | MG868988 | MG869022 | MG869056 |
| 9. | Spathoglottis aurea Lindl. | FAN057 | Gunung Bunga Buah, Selangor, Malaysia | MG868989 | MG869023 | MG869057 |
| 10. | Spathoglottis aurea Lindl. | FAN099 | Tanah Rata, Cameron Highlands, Pahang, Malaysia | MG868990 | MG869024 | MG869058 |
| 11. | Spathoglottis eburnea Gagnep. | FAN022 | Fang District, Chiang Mai, Thailand | MG868991 | MG869025 | MG869059 |
| 12. | Spathoglottis gracilis Rolfe ex Hook.f. | FAN094 | Kg. Liposu, Ranau, Sabah, Malaysia | MG868992 | MG869026 | MG869060 |
| 13. | Spathoglottis hardingiana C.S.P.Parish & Rchb.f. | FAN016 | Gunung Baling, Kedah, Malaysia | MG868993 | MG869027 | MG869061 |
| 14. | Spathoglottis hardingiana C.S.P.Parish & Rchb.f. | FAN056 | G. Pong, Kenering, Perak, Malaysia | MG868994 | MG869028 | MG869062 |
| 15. | Spathoglottis hardingiana C.S.P.Parish & Rchb.f. | FAN105/ K20160013 | Pulau Timun, Langkawi, Kedah, Malaysia | MG868995 | MG869029 | MG869063 |
| 16. | Spathoglottis kimballiana Hook.f. | FAN085 | Ranau, Sabah, Malaysia | MG868996 | MG869030 | MG869064 |
| 17. | Spathoglottis kimballiana var. angustifolia Ames | FAN076 | Bidu-Bidu FR, Telupid, Sabah, Malaysia | MG868997 | MG869031 | MG869065 |
| 18. | Spathoglottis kimballiana var. angustifolia Ames | FAN104 | Sungai Tongod, Telupid, Sabah, Malaysia | MG868998 | MG869032 | MG869066 |
| 19. | Spathoglottis kimballiana var. kimballiana | FAN067 | Gunung Kinabalu, Ranau, Sabah, Malaysia | MG868999 | MG869033 | MG869067 |
| 20. | Spathoglottis kimballiana var. kimballiana | FAN092 | Kota Belud, Sabah, Malaysia | MG869000 | MG869034 | MG869068 |
| 21. | Spathoglottis kimballiana var. kimballiana | FAN093 | Pekan Nabalu, Ranau, Sabah, Malaysia | MG869001 | MG869035 | MG869069 |
| 22. | Spathoglottis microchilina Kraenzl. | FAN083 | Kinabalu Park Research Centre, Sabah, Malaysia | MG869003 | MG869037 | MG869071 |
| 23. | Spathoglottis microchilina Kraenzl. | FAN086 | Mamut Copper Mine, Sabah, Malaysia | MG869004 | MG869038 | MG869072 |
| 24. | Spathoglottis microchilina Kraenzl. | FAN091 | Sg. Lohan, Ranau, Sabah, Malaysia | MG869005 | MG869039 | MG869073 |
| 25. | Spathoglottis parviflora Kraenzl. | FAN061 | Wamena, Irian Jaya | MG869006 | MG869040 | MG869074 |
| 26. | Spathoglottis plicata Blume | FAN001 | Gunung Ledang, Johor, Malaysia | MG869007 | MG869041 | MG869075 |
| 27. | Spathoglottis plicata Blume | FAN063 | Lata Chemerong, Dungun, Terengganu, Malaysia | MG869008 | MG869042 | MG869076 |
| 28. | Spathoglottis plicata Blume | FAN090 | Puncak Post, Mamut Copper Mine, Sabah, Malaysia | MG869009 | MG869043 | MG869077 |
| 29. | Spathoglottis plicata Blume | FAN103 | Long Baleh, Sarawak, Malaysia | MG869010 | MG869044 | MG869078 |
| 30. | Spathoglottis plicata var. alba | FAN045 | Lata Tembakah, Terengganu, Malaysia | MG869011 | MG869045 | MG869079 |
| 31. | Spathoglottis pubescens Lindl. | FAN068 | Fang District, Chiang Mai, Thailand | MG869012 | MG869046 | MG869080 |
| 32. | Spathoglottis unguiculata (Labill.) Rcbh.f. | FAN024 | Isle of Pines, New Caledonia | MG869013 | MG869047 | MG869081 |
| 33. | Tainia paucifolia (Breda) J.J.Sm. | FAN597 | Taman Rimba Kenong, Jerantut, Pahang, Malaysia | MG869014 | MG869048 | MG869082 |
| 34. | Calanthe tankervilleae (Banks) M.W.Chase, Christenh. & Schuit. | FAN707 | Kundasang, Sabah, Malaysia | MG869015 | MG869049 | MG869083 |
| Region | Primers Sequences (5′ to 3′) | Primer Name | Primer Length | PCR Product Size |
|---|---|---|---|---|
| ITS | F: ACGAATTCATGGTCCGGTGAAGTGTTCG R: TAGAATTCCCCGGTTCGCTCGCCGTTAC | 17SE 26SE | 28 bp 28 bp | 800–1000 bp |
| matK (partial sequence) | F: CGATCTATTCATTCAATATTTC R: TCTAGCACACGAAAGTCGAAGT | 360F 1326R | 22 bp 22 bp | 900 bp |
| trnL-F | F: CGAAATCGGTAGACGCTACG R: ATTTGAACTGGTGACACGAG | c f | 20 bp 20 bp | 1000 bp |
| Taxa | Plant Size | Flower Size | Shape of Labellum |
|---|---|---|---|
| Dwarf | Small | Broad/Bilobulate |
| Dwarf | Small | Broad/Bilobulate |
| Dwarf | Small | Broad/Bilobulate |
| Dwarf | Small | Broad/Bilobulate |
| Large | Medium | Narrow |
| Large | Large | Broad/Bilobulate |
| Large | Medium | Broad/Bilobulate |
| Large | Large | Broad/Bilobulate |
| Large | Medium | Broad/Bilobulate |
| Large | Medium | Narrow |
| Large | Medium | Broad/Bilobulate |
| Large | Medium | Broad/Bilobulate |
| Large | Medium | Broad/Bilobulate |
| Large | Medium | Broad/Bilobulate |
| Region | Aligned Nucleotide Length (bp) | Conserved Site (%) | Variable Site(%) | Parsimony Informative Site (%) | G+C Content (%) |
|---|---|---|---|---|---|
| ITS1 | 244–260 | 137 (53.7) | 116 (45.5) | 85 (33.3) | 57.6 |
| 5.8S | 157 | 140 (89.2) | 17 (11) | 10 (6.4) | 56.8 |
| ITS2 | 318-345 | 208 (60.3) | 125 (36.2) | 88 (25.5) | 64.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nordin, F.A.; Saibeh, K.; Go, R.; Mangsor, K.N.A.; Othman, A.S. Molecular Phylogenetics of the Orchid Genus Spathoglottis (Orchidaceae: Collabieae) in Peninsular Malaysia and Borneo. Forests 2022, 13, 2079. https://doi.org/10.3390/f13122079
Nordin FA, Saibeh K, Go R, Mangsor KNA, Othman AS. Molecular Phylogenetics of the Orchid Genus Spathoglottis (Orchidaceae: Collabieae) in Peninsular Malaysia and Borneo. Forests. 2022; 13(12):2079. https://doi.org/10.3390/f13122079
Chicago/Turabian StyleNordin, Farah Alia, Kartini Saibeh, Rusea Go, Khairul Nasirudin Abu Mangsor, and Ahmad Sofiman Othman. 2022. "Molecular Phylogenetics of the Orchid Genus Spathoglottis (Orchidaceae: Collabieae) in Peninsular Malaysia and Borneo" Forests 13, no. 12: 2079. https://doi.org/10.3390/f13122079
APA StyleNordin, F. A., Saibeh, K., Go, R., Mangsor, K. N. A., & Othman, A. S. (2022). Molecular Phylogenetics of the Orchid Genus Spathoglottis (Orchidaceae: Collabieae) in Peninsular Malaysia and Borneo. Forests, 13(12), 2079. https://doi.org/10.3390/f13122079