Chemical Structure and Mechanical Properties of Wood Cell Walls Treated with Acid and Alkali Solution


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Acid and Alkali Treatment
2.3. Confocal Raman Microscopy (CRM)
2.4. Nanoindentation
2.5. X-ray Diffraction
2.6. Data Statistical Analysis
3. Results
3.1. Changes in Chemical Structure after Acid and Alkali Treatments
3.1.1. Acid Treatment
3.1.2. Alkali Treatment
3.1.3. Different Changes in Chemical Structure after Acid and Alkali Treatment
3.2. Changes in Mechanical Properties after Acid and Alkali Treatments
3.2.1. Hot Water and B/E Extraction Treatment
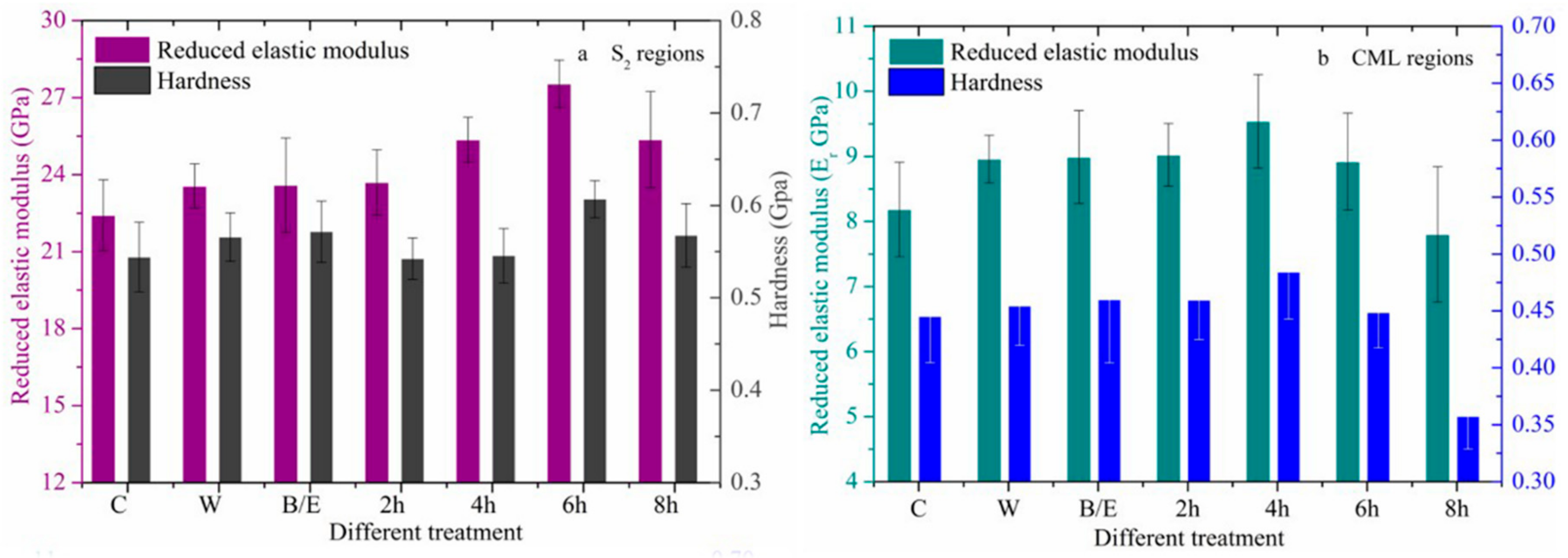
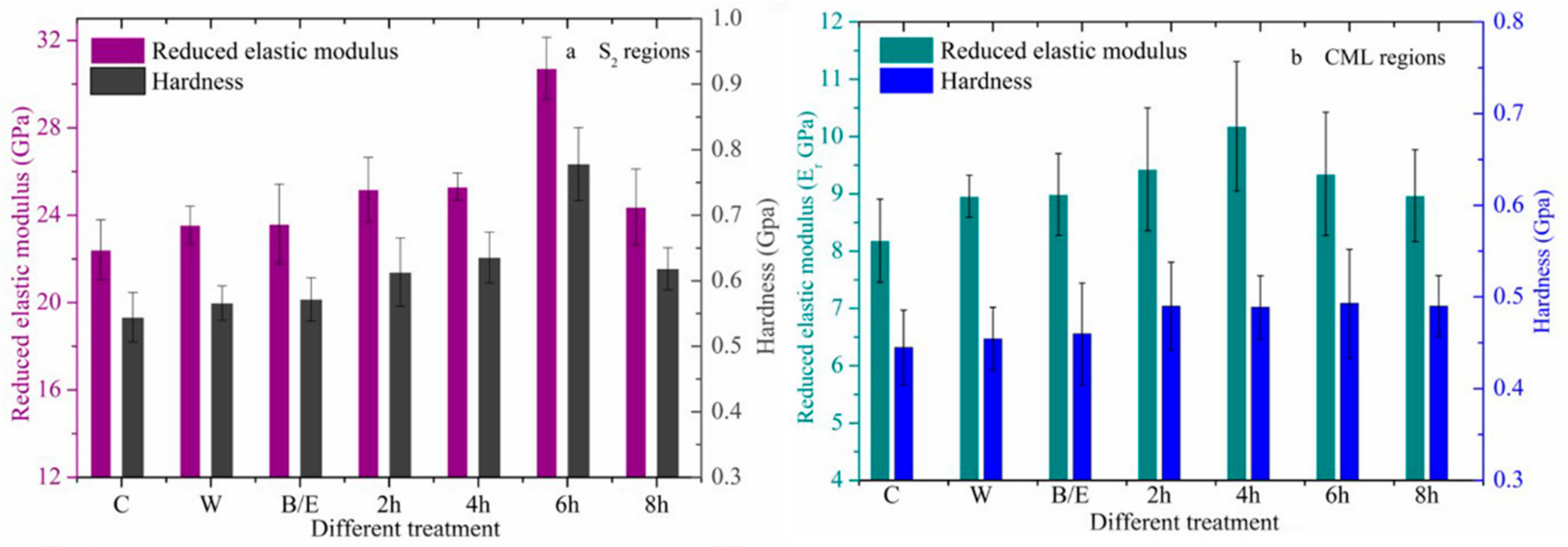
3.2.2. Changes in Mechanical Properties after Acid Treatment
3.2.3. Changes in Mechanical Properties after Alkali Treatment
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Magistris, F.D.; Salmén, L. Mechanical behaviour of wet wood in sequences of compression and combined compression and shear. Nord. Pulp Pap. Res. J. 2006, 21, 231–236. [Google Scholar] [CrossRef]
- Heitner, C.; Karnis, A.; Atack, D. Ultra-high yield pulping. IV. High strength pulp from mechanical pulp rejects [Sulphonation of wood chips]. Proc. Tech. Assoc. Pulp Pap. Ind. (USA) 1983, 85, 133–139. [Google Scholar]
- Sixta, H.; Potthast, A.; Krotschek, A.W. Chemical Pulping Processe. In Handbook of Pulp, 1st ed.; Sixta, H., Ed.; Wiley-VCH: Weinheim, Germany, 2006; Volume 1, pp. 109–229. ISBN 9783527309993. [Google Scholar]
- Germer, E.I. Production of bleachable pulp through catalytic oxygen-alkaline delignification of high-yield mechanical pulp. Tappi J. 1995, 78, 121–124. [Google Scholar]
- Zhang, H.; Hou, Q.; Liu, W.; Yue, Z.; Jiang, X.; Ma, X. Improved diffusivity of NaOH solution in autohydrolyzed poplar sapwood chips for chemi-mechanical pulp production. Bioresour. Technol. 2018, 259, 61–66. [Google Scholar] [CrossRef]
- Sundberg, K.E.; Holmbom, B.R.; Pranovich, A.V. Chemical changes in thermomechanical pulp at alkaline conditions. J. Wood Chem. Technol. 2003, 23, 89–112. [Google Scholar] [CrossRef]
- Zanuttini, M.; Marzocchi, V.; Mocchiutti, P.; Inalbon, M. Deacetylation consequences in pulping processes. Holz Als Roh-Und Werkstoff. 2005, 63, 149–153. [Google Scholar] [CrossRef]
- Fahlén, J. The Cell Wall Ultrastructure of Wood Fibres: Effects of the Chemical Pulp Fibre Line. Ph.D. Thesis, Kungliga Tekniska Högskolan, Stockholm, Sweden, January 2005. [Google Scholar]
- Vinod, A.; Vijay, R.; Singaravelu, D.L.; Sanjay, M.R.; Siengchin, S.; Moure, M.M. Characterization of untreated and alkali treated natural fibers extracted from the stem of Catharanthus roseus. Mater. Res. Express 2019, 6, 085460. [Google Scholar] [CrossRef]
- Kathirselvam, M.; Kumaravel, A.; Arthanarieswaran, V.P.; Saravanakumar, S.S. Characterization of cellulose fibers in Thespesia populnea barks: Influence of alkali treatment. Carbohyd. Polym. 2019, 217, 178–189. [Google Scholar] [CrossRef]
- Reddy, K.O.; Shukla, M.; Maheswari, C.U.; Rajulu, A.V. Mechanical and physical characterization of sodium hydroxide treated Borassus fruit fibers. J. For. Res. 2012, 23, 667–674. [Google Scholar] [CrossRef]
- Chen, H.; Yu, Y.; Zhong, T.; Wu, Y.; Li, Y.; Wu, Z.; Fei, B. Effect of alkali treatment on microstructure and mechanical properties of individual bamboo fibers. Cellulose 2016, 24, 333–347. [Google Scholar] [CrossRef]
- Evans, R.; Newman, R.H.; Roick, U.C.; Suckling, I.D.; Wallis, A.F. Changes in cellulose crystallinity during kraft pulping. Comparison of infrared, x-ray diffraction and solid state NMR results. Holzforschung 1995, 49, 498–504. [Google Scholar] [CrossRef]
- Hult, E.L.; Larsson, P.T.; Iversen, T. A comparative CP/MAS 13C-NMR study of cellulose structure in spruce wood and kraft pulp. Cellulose 2000, 7, 35–55. [Google Scholar] [CrossRef]
- Hult, E.L.; Larsson, P.T.; Iversen, T. Cellulose fibril aggregation—An inherent property of kraft pulps. Polymer 2001, 42, 3309–3314. [Google Scholar] [CrossRef]
- Hult, E.L.; Larsson, P.T.; Iversen, T. A comparative CP/MAS 13C-NMR study of the supermolecular structure of polysaccharides in sulphite and kraft pulps. Holzforschung 2002, 56, 179–184. [Google Scholar] [CrossRef]
- Gassan, J.; Bledzki, A.K. Alkali treatment of jute fibers: Relationship between structure and mechanical properties. J. Appl. Polym. Sci. 1999, 71, 623–629. [Google Scholar] [CrossRef]
- Ahlgren, P.A.; Goring, D.A.I. Removal of wood components during chlorite delignification of black spruce. Can. J. Chem. 1971, 49, 1272–1275. [Google Scholar] [CrossRef]
- Gierer, J. Chemistry of delignification. Wood Sci. Technol. 1985, 19, 289–312. [Google Scholar] [CrossRef]
- Kumar, R.; Mago, G.; Balan, V.; Wyman, C.E. Physical and chemical characterizations of corn stover and poplar solids resulting from leading pretreatment technologies. Bioresour. Technol. 2009, 100, 3948–3962. [Google Scholar] [CrossRef]
- Millet, M.A.; Moore, W.E.; Saeman, J.E. Preparation and properties of hydrocelluloses. Ind. Eng. Chem. 1954, 46, 1493–1497. [Google Scholar] [CrossRef]
- Lewin, M.; Epstein, J.A. Functional groups and degradation of cotton oxidized by hypochlorite. J. Polym. Sci. 1962, 58, 1023–1037. [Google Scholar] [CrossRef]
- Singh, O.P. Kinetics and mechanism of hypochlorite oxidation of cellulose. Text. Dry. Print. 1982, 15, 35–38. [Google Scholar]
- Ji, Z.; Ma, J.; Xu, F. Multi-scale visualization of dynamic changes in poplar cell walls during alkali pretreatment. Microsc. Microanal. 2014, 20, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Deng, Y.; Yu, W.; Xie, X. Contributions of basic chemical components to the mechanical behavior of wood fiber cell walls as evaluated by nanoindentation. Bioresource 2016, 11, 6026–6039. [Google Scholar] [CrossRef]
- Agarwal, U.P. Higher acid-chlorite reactivity of cell corner middle lamella lignin in black spruce. In Proceedings of the 14th International Symposium on Wood Fibre and Pulping Chemistry, Durban, South Africa, 25–28 June 2007. [Google Scholar]
- Hubbell, C.A.; Ragauskas, A.J. Effect of acid-chlorite delignification on cellulose degree of polymerization. Bioresour. Technol. 2010, 101, 7410–7415. [Google Scholar] [CrossRef] [PubMed]
- Kerr, A.J.; Goring, D.A.I. The role of hemicellulose in the delignification of wood. Can. J. Chem. 1975, 53, 952–959. [Google Scholar] [CrossRef]
- Virtanen, T.; Maunu, S.L.; Tamminen, T.; Bo Hortling, B.; Liitia, T. Changes in fiber ultrastructure during various kraft pulping conditions evaluated by 13C CPMAS NMR spectroscopy. Carbohydr. Polym. 2008, 73, 156–163. [Google Scholar] [CrossRef]
- Whiting, P.; Pulp, D.G. The topochemistry of delignification shown by pulping middle lamella and secondary wall tissue from black spruce wood. J. Wood Chem. Technol. 1981, 1, 111–122. [Google Scholar] [CrossRef]
- Hult, E.L.; Liitiä, T.; Maunu, S.L.; Hortling, B.; Iversen, T. A CP/MAS 13C-NMR study of cellulose structure on the surface of refined kraft pulp fibers. Carbohydr. Polym. 2002, 49, 231–234. [Google Scholar] [CrossRef]
- Page, D.H. The origin of the differences between sulfite and kraft pulps. Pulp Pap. Can. 1983, 84, 15–20. [Google Scholar]
- Mantanis, G.I.; Young, R.A.; Rowell, R.M. Swelling of wood. Part II. Swelling in organic liquids. Holzforschung 1994, 48, 480–490. [Google Scholar] [CrossRef]
- Paredes, J.J.; Shaler, S.; Howell, C.; Jakes, J. Influence of hot water extraction on cell wall and OSB strand mechanics. Wood Sci. Technol. 2017, 51, 1307–1319. [Google Scholar] [CrossRef]
- Krogell, J.; Korotkova, E.; Eränen, K.; Pranovich, A.; Salmi, T.; Murzin, D.; Willför, S. Intensification of hemicellulose hot-water extraction from spruce wood in a batch extractor—Effects of wood particle size. Bioresour. Technol. 2013, 143, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Shelat, B.R.; Radhakrishnan, T.; Iyer, B.V. The relation between crystallite orientation and mechanical properties of mercerized cottons. Text. Res. J. 1960, 30, 836–842. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample 1 | CrI 2 (%) | 2θ 3(002) (°) | FWHM 4 (°) | Dhkl 5 (nm) |
|---|---|---|---|---|
| Control | 56.72 ± 0.14 (A) | 22.30 ± 0.01 | 2.81 ± 0.04 | 2.85 ± 0.00 (AB) |
| Hot water | 52.40 ± 0.08 (C) | 22.52 ± 0.37 | 2.91 ± 0.03 | 2.78 ± 0.03 (BC) |
| B/E | 47.54 ± 0.37 (E) | 22.20 ± 0.03 | 3.10 ± 0.14 | 2.61 ± 0.12 (D) |
| 2H | 49.40 ± 0.53 (D) | 22.20 ± 0.02 | 3.02 ± 0.09 | 2.68 ± 0.08 (CD) |
| 4H | 50.02 ± 0.59 (D) | 22.15 ± 0.01 | 2.94 ± 0.16 | 2.75 ± 0.14 (CD) |
| 6H | 53.80 ± 0.25 (B) | 22.26 ± 0.00 | 2.71 ± 0.19 | 2.99 ± 0.11 (A) |
| 8H | 52.36 ± 0.77 (C) | 22.19 ± 0.02 | 2.96 ± 0.05 | 2.74 ± 0.05 (C) |
| Control | 56.72 ± 0.14 (A) | 22.30 ± 0.01 | 2.81 ± 0.04 | 2.85 ± 0.00 (CD) |
| Hot water | 52.40 ± 0.08 (C) | 22.52 ± 0.37 | 2.91 ± 0.03 | 2.78 ± 0.03 (D) |
| B/E | 47.54 ± 0.37 (E) | 22.20 ± 0.03 | 3.10 ± 0.14 | 2.61 ± 0.12 (E) |
| 2OH | 53.54 ± 0.12 (B) | 22.40 ± 0.18 | 2.81 ± 0.06 | 2.88 ± 0.06 (CD) |
| 4OH | 53.71 ± 0.50 (B) | 22.31 ± 0.02 | 2.53 ± 0.11 | 3.34 ± 0.03 (A) |
| 6OH | 52.52 ± 0.18 (C) | 22.28 ± 0.02 | 2.64 ± 0.12 | 3.21 ± 0.07 (B) |
| 8OH | 50.71 ± 0.33 (D) | 22.30 ± 0.01 | 2.75 ± 0.13 | 2.94 ± 0.05 (C) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, E.; Wang, D.; Lin, L. Chemical Structure and Mechanical Properties of Wood Cell Walls Treated with Acid and Alkali Solution. Forests 2020, 11, 87. https://doi.org/10.3390/f11010087
Xu E, Wang D, Lin L. Chemical Structure and Mechanical Properties of Wood Cell Walls Treated with Acid and Alkali Solution. Forests. 2020; 11(1):87. https://doi.org/10.3390/f11010087
Chicago/Turabian StyleXu, Enguang, Dong Wang, and Lanying Lin. 2020. "Chemical Structure and Mechanical Properties of Wood Cell Walls Treated with Acid and Alkali Solution" Forests 11, no. 1: 87. https://doi.org/10.3390/f11010087
APA StyleXu, E., Wang, D., & Lin, L. (2020). Chemical Structure and Mechanical Properties of Wood Cell Walls Treated with Acid and Alkali Solution. Forests, 11(1), 87. https://doi.org/10.3390/f11010087