Transcriptome Analysis of a Multiple-Branches Mutant Terminal Buds in Betula platyphylla × B. pendula

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Methods
2.2.1. Observation of Biomass and Growth Traits
2.2.2. Observation of Apical Buds Microstructure
2.2.3. RNA Extraction, Library Construction, and Sequencing
2.2.4. Data Filtering, Assembly, Gene Annotation, and DEG Analysis
2.2.5. Verification of qRT-PCR
2.2.6. Statistical Analysis
3. Results
3.1. Identification of br Mutation Characteristics
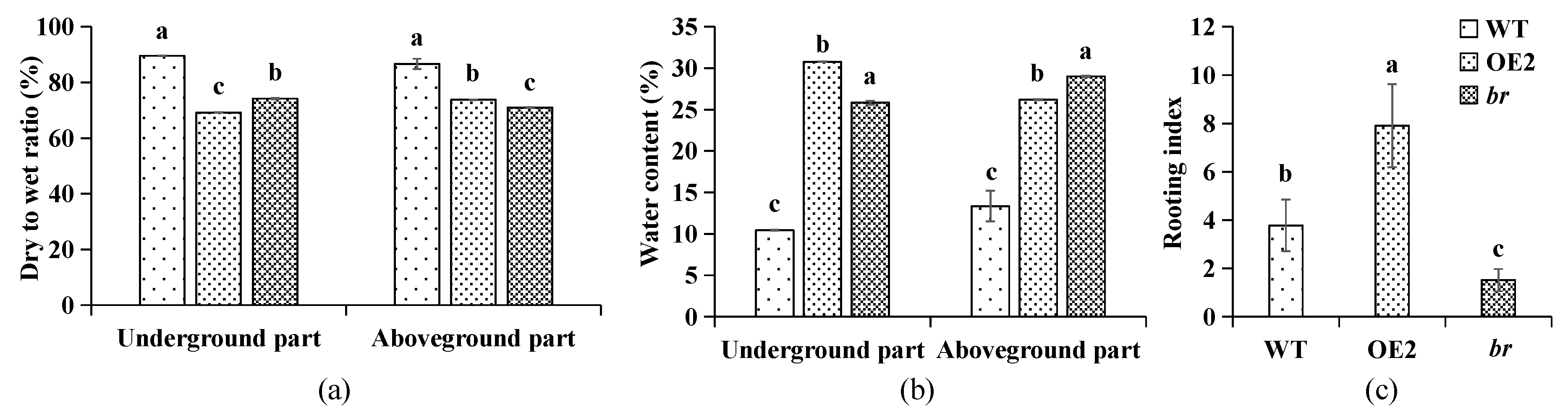
3.1.1. Variations in Biomass
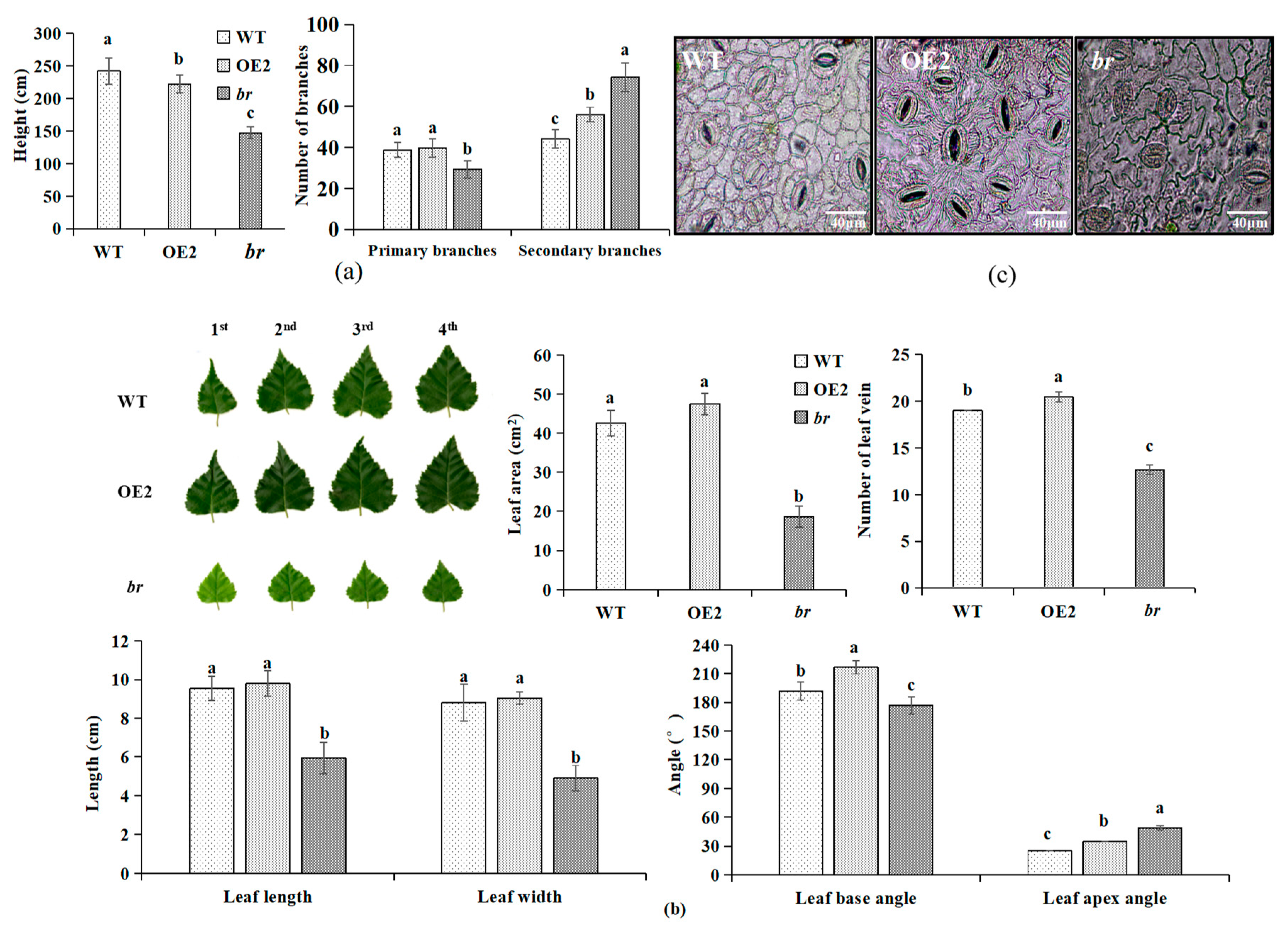
3.1.2. Variations in Growth
3.1.3. Observation of Morphology and the Microstructures of Apical Buds
3.2. Transcriptomic Analyses
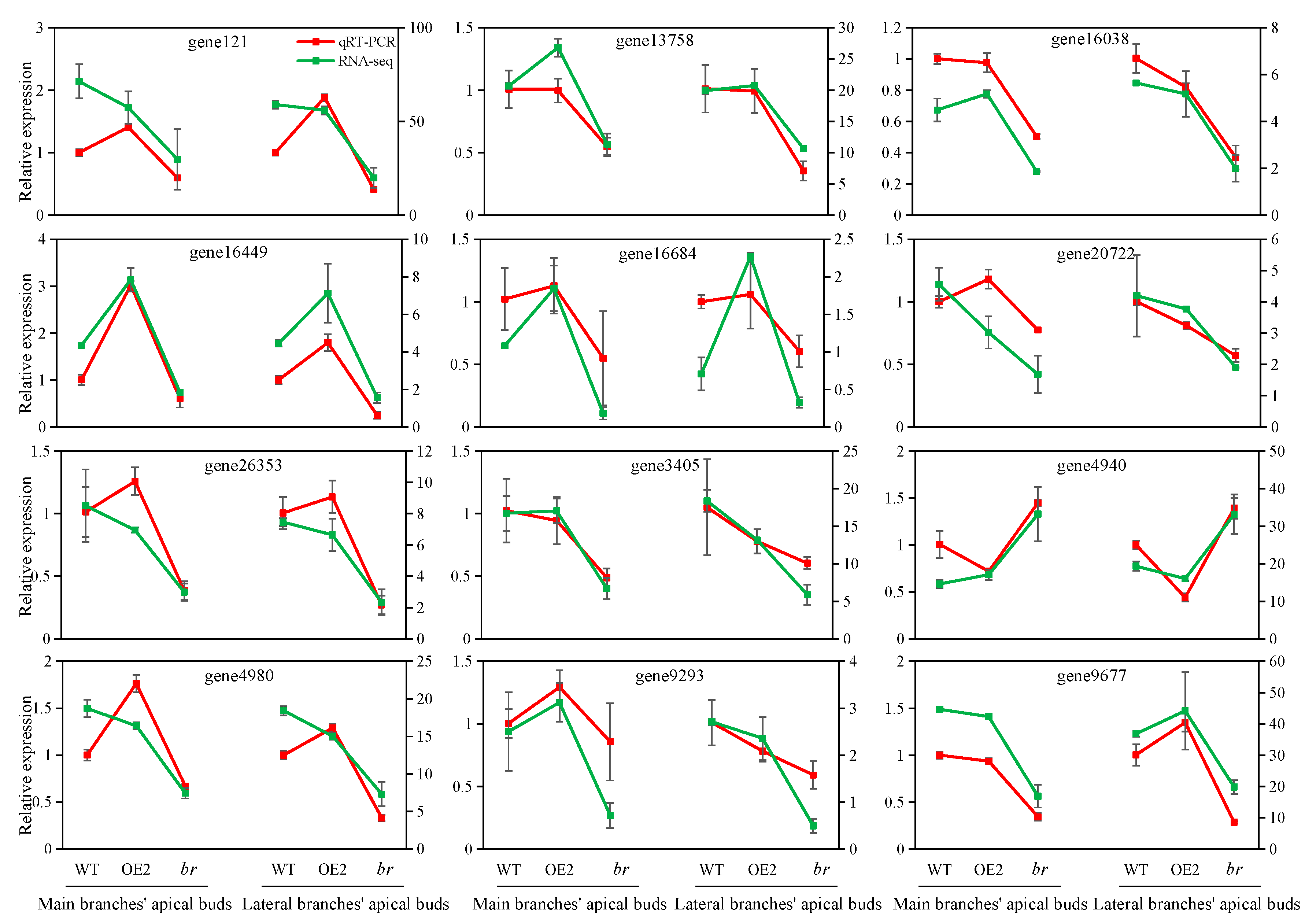
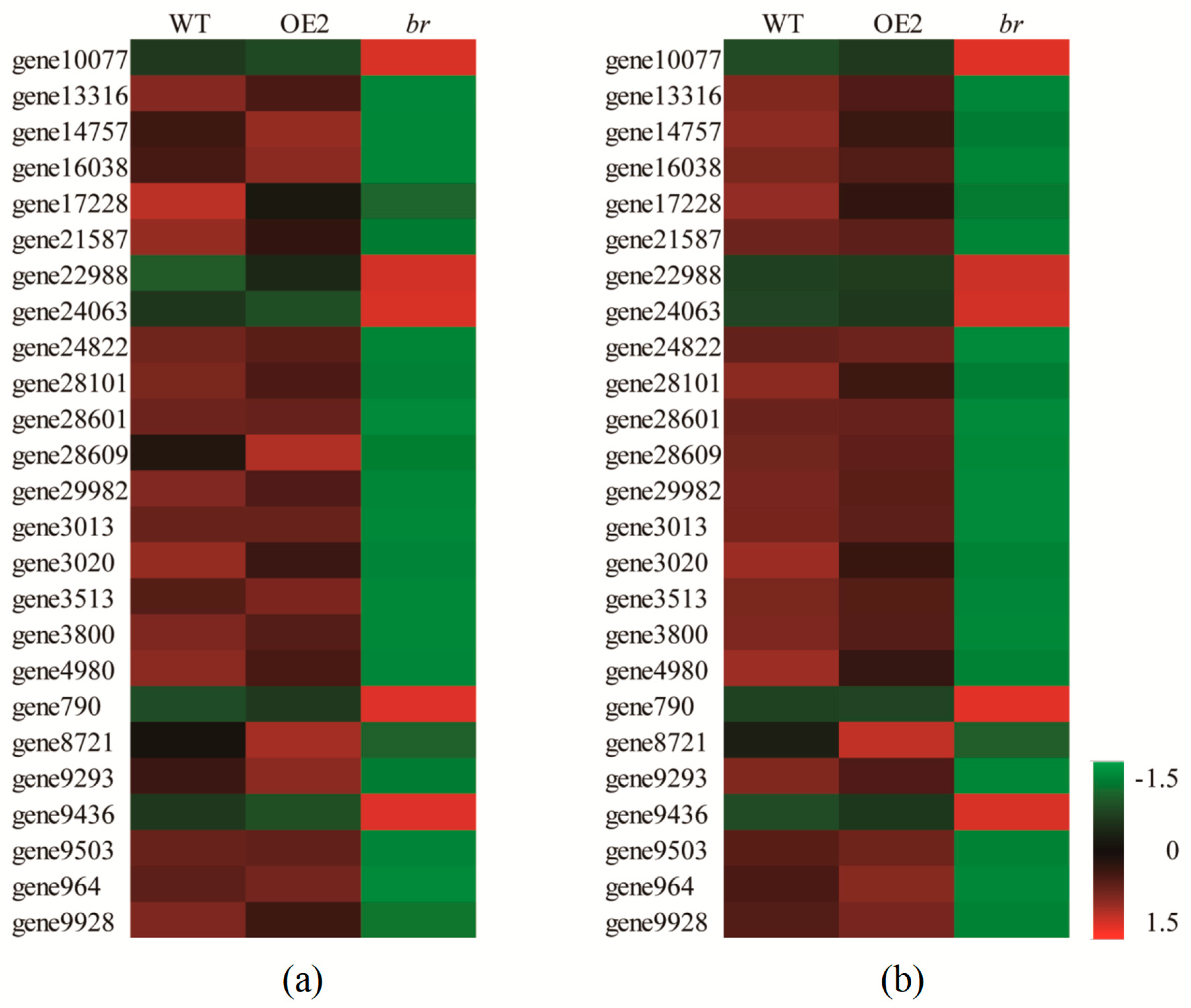
3.2.1. Sequencing Quality Evaluation and qRT-PCR Verification
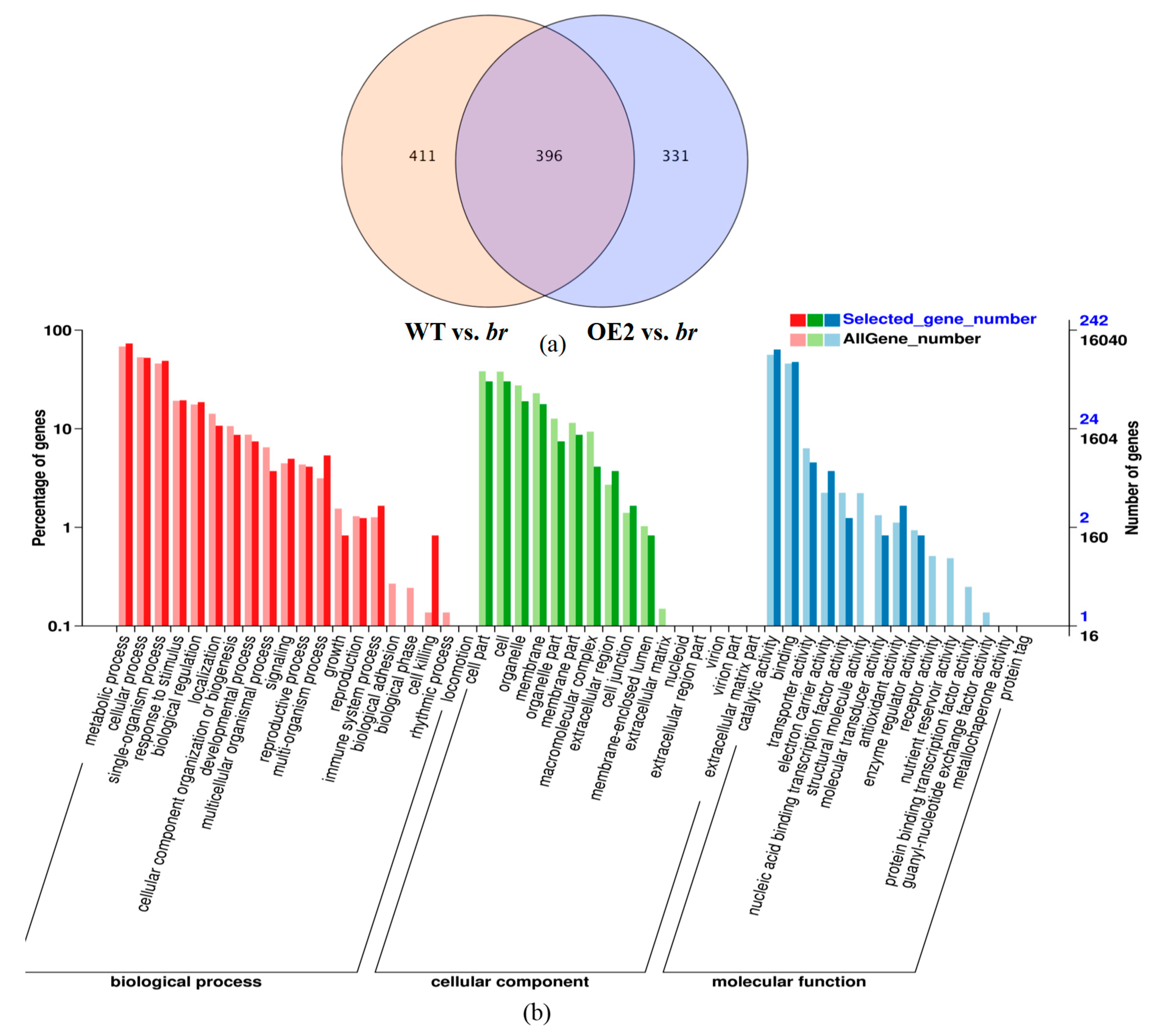
3.2.2. Transcriptomic Analyses of the Apical Buds of the Main Branches
3.2.3. Transcriptomic Analyses of the Lateral Branches’ Apical Buds
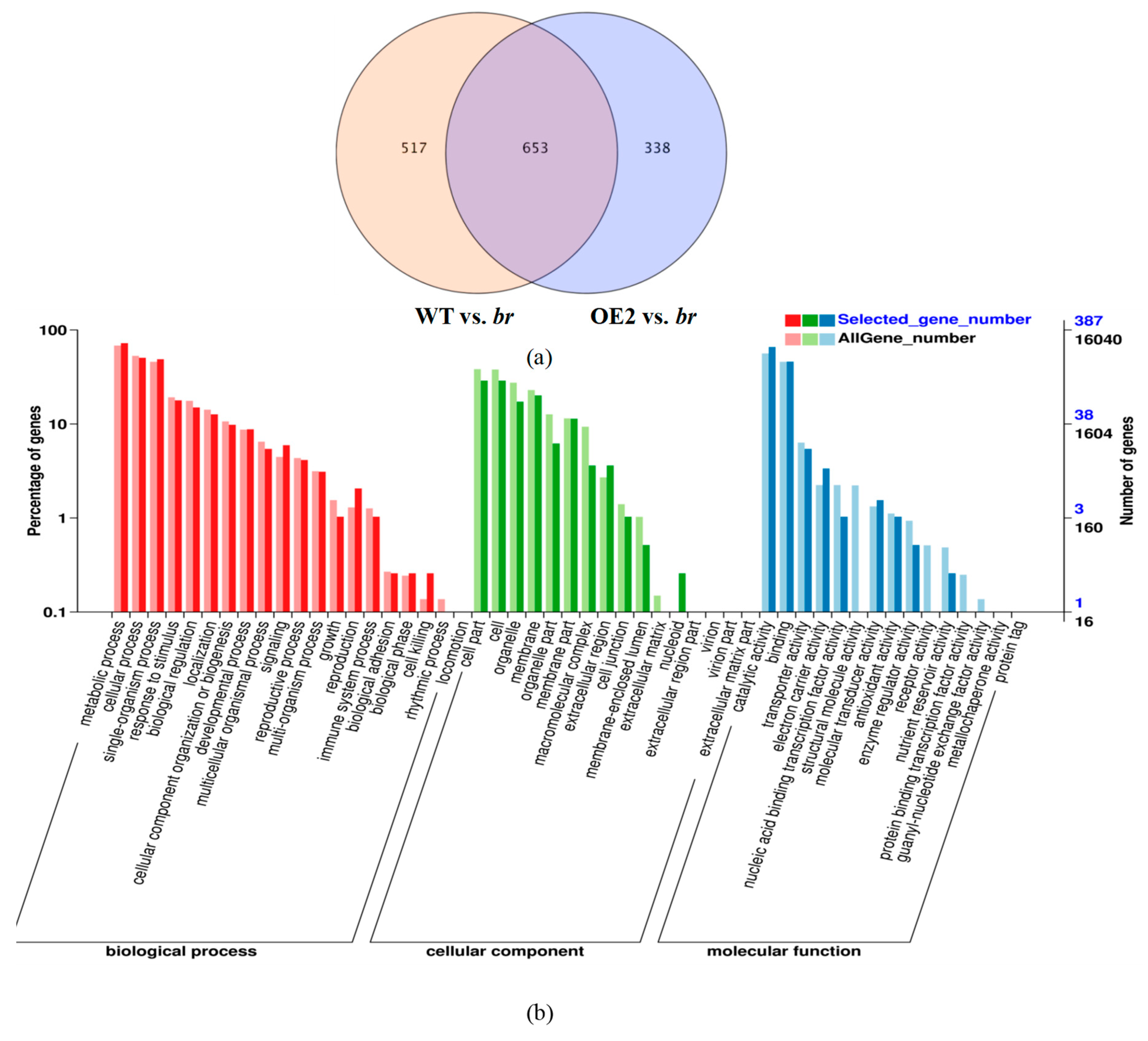
3.2.4. Transcriptomic Analyses of the Apical Buds
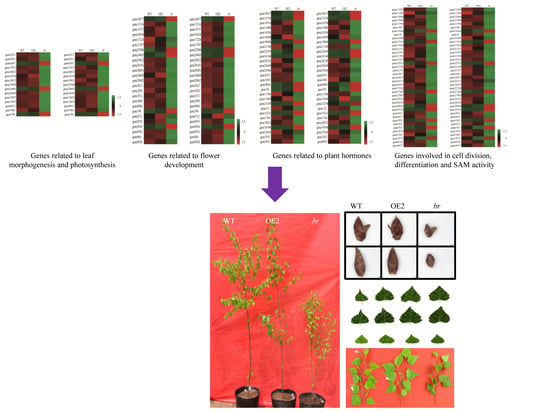
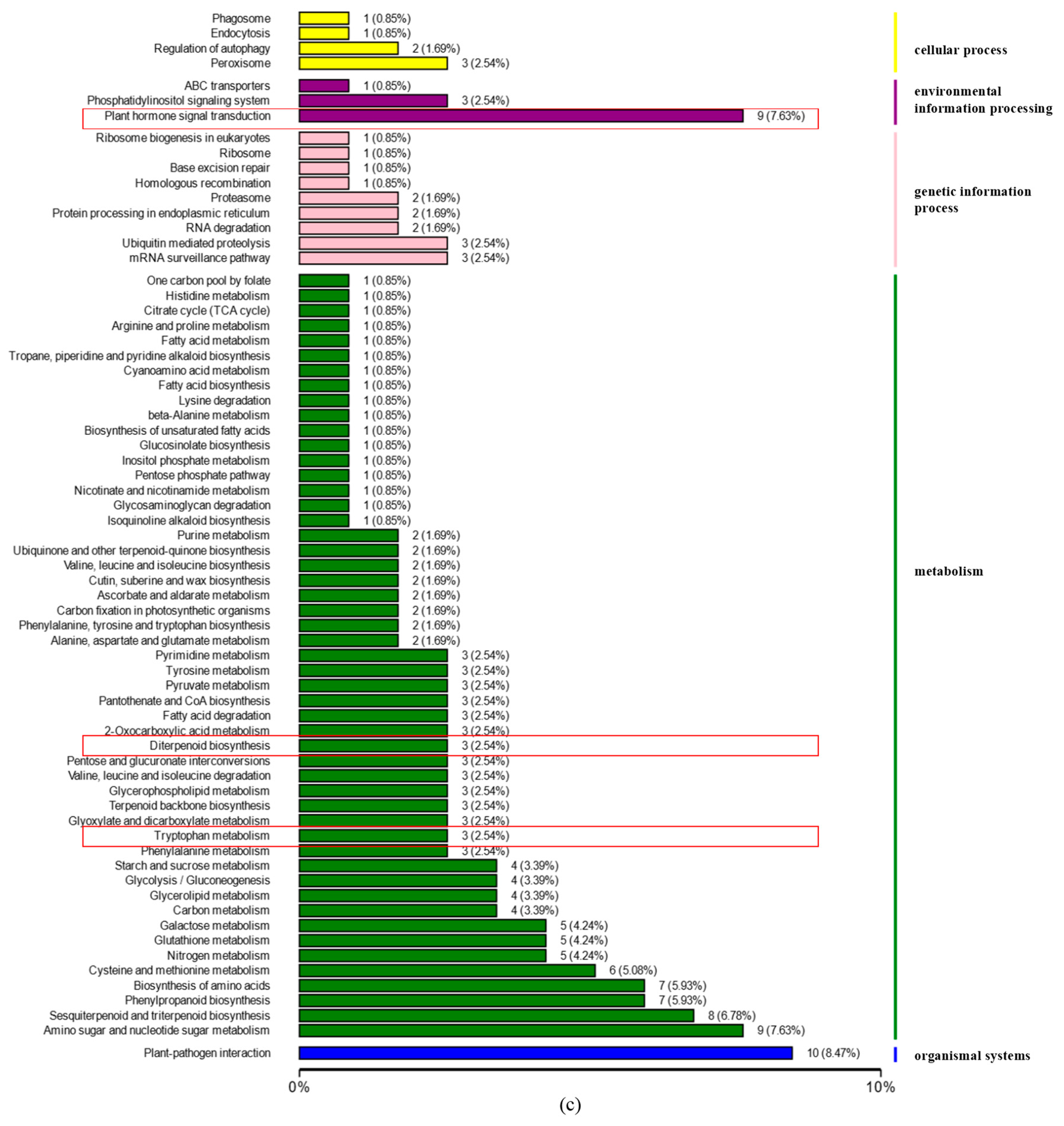
3.3. DEGs Analyses
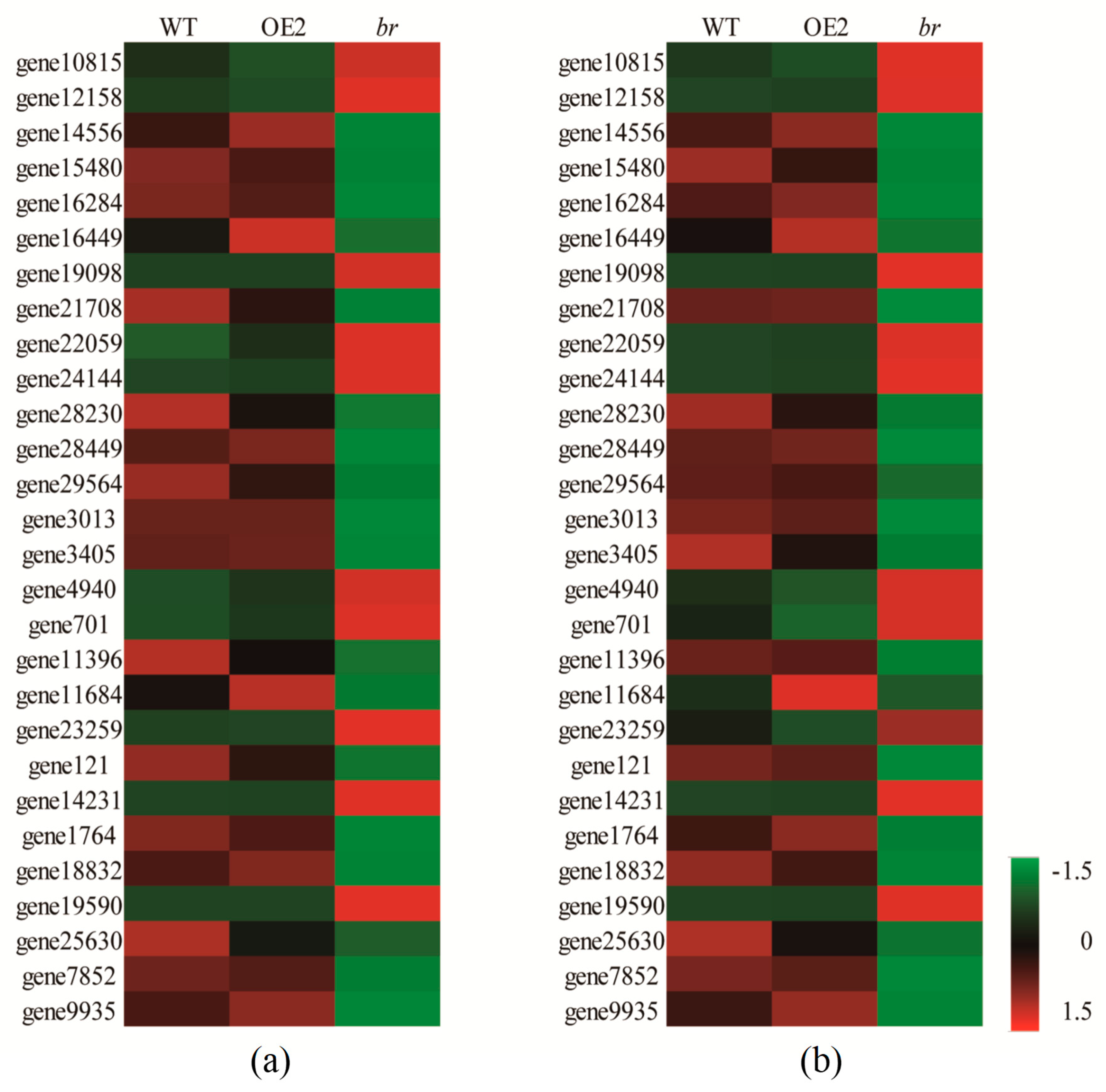
3.3.1. DEGs Related to Flower Development
3.3.2. DEGs Related to Cell Division, Differentiation, and SAM Activity
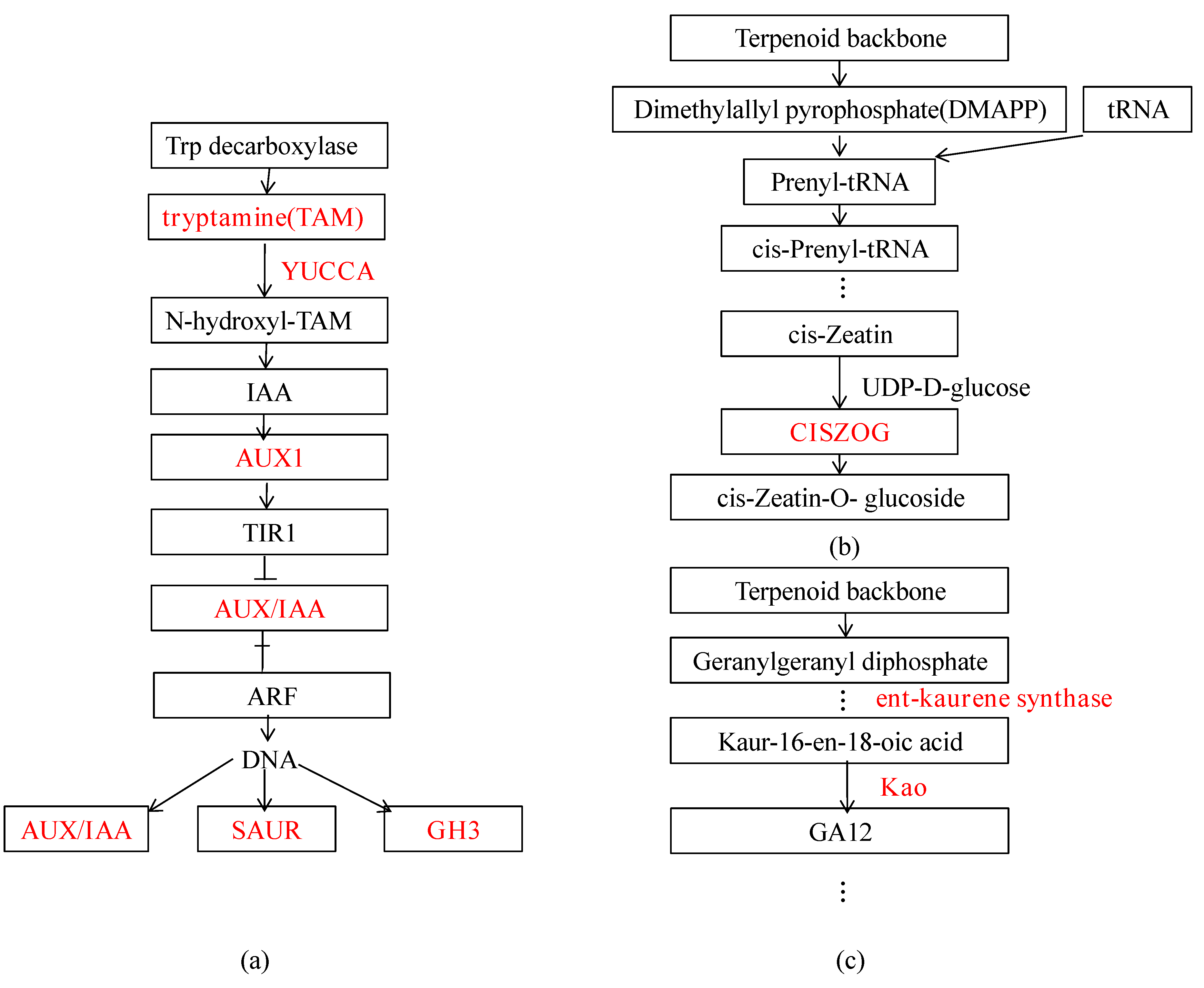
3.3.3. DEGs Related to Plant Hormones
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tanaka, W.; Pautler, M.; Jackson, D.; Hirano, H.-Y. Grass Meristems II: Inflorescence Architecture, Flower Development and Meristem Fate. Plant Cell Physiol. 2013, 54, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Bleecker, A.B.; Grbić, V. Axillary meristem development in Arabidopsis thaliana. Plant J. 2000, 21, 215–223. [Google Scholar]
- Welling, A.; Rinne, P.; Viherä-Aarnio, A.; Kontunen-Soppela, S.; Heino, P.; Palva, E.T. Photoperiod and temperature differentially regulate the expression of two dehydrin genes during overwintering of birch (Betula pubescens Ehrh.). J. Exp. Bot. 2004, 55, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Girault, T.; Bergougnoux, V.; Combes, D.; Viemont, J.D.; Leduc, N. Light controls shoot meristem organogenic activity and leaf primordia growth during bud burst in Rosa sp. Plant Cell Environ. 2008, 31, 1534–1544. [Google Scholar] [CrossRef]
- Evers, J.B.; Vos, J.; Andrieu, B.; Struik, P.C. Cessation of tillering in spring wheat in relation to light interception and red: Far-red ratio. Annu. Bot. 2006, 97, 649–658. [Google Scholar] [CrossRef]
- Vernoux, T.; Besnard, F.; Traas, J. Auxin at the shoot apical meristem. Cold Spring Harb. Perspect. Biol. 2010, 2, a001487. [Google Scholar] [CrossRef]
- Reinhardt, D.; Mandel, T.; Kuhlemeier, C. Auxin regulates the initiation and radial position of plant lateral organs. Plant Cell 2000, 12, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Carraro, N.; Peaucelle, A.; Laufs, P.; Traas, J. Cell differentiation and organ initiation at the shoot apical meristem. Plant Mol. Biol. 2006, 60, 811–826. [Google Scholar] [CrossRef]
- Werner, T.; Motyka, V.; Strnad, M.; Schmülling, T. Regulation of plant growth by cytokinin. Proc. Nat. Acad. Sci. USA 2001, 98, 10487–10492. [Google Scholar] [CrossRef] [PubMed]
- Werner, T.; Motyka, V.; Laucou, V.; Smets, R.; Onckelen, H.V. Cytokinin-deficient transgenic Arabidopsis plants show multiple developmental alterations indicating opposite functions of cytokinins in the regulation of shoot and root meristem activity. Plant Cell 2003, 15, 2532–2550. [Google Scholar] [CrossRef]
- Zhuang, W.B.; Gao, Z.H.; Wang, L.J.; Zhong, W.J.; Ni, Z.J.; Zhang, Z. Comparative proteomic and transcriptomic approaches to address the active role of GA4 in Japanese apricot flower bud dormancy release. J. Exp. Bot. 2013, 64, 4953–4966. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.B.; Gao, Z.H.; Wen, L.H.; Huo, X.M.; Cai, B.H.; Zhang, Z. Metabolic changes upon flower bud break in Japanese apricot are enhanced by exogenous GA4. Hortic Res. 2015, 2, 15046. [Google Scholar] [CrossRef] [PubMed]
- Perales, M.; Reddy, G.V. Stem cell maintenance in shoot apical meristems. Curr. Opin. Plant Biol. 2012, 15, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Tataw, M.; Reddy, G.V. Towards a functional understanding of cell growth dynamics in shoot meristem stem cell niche. Semin. Cell Dev. Biol. 2009, 20, 1126–1133. [Google Scholar] [CrossRef]
- Laufs, P.; Grandjean, O.; Jonak, C.; Kieu, K.; Traas, J. Cellular parameters of the shoot apical meristem in Arabidopsis. Plant. Cell. 1998, 10, 1375–1390. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.K.; Perales, M.; Gruel, J.; Girke, T.; Jönsson, H.; Reddy, G.V. WUSCHEL protein movement mediates stem cell homeostasis in the Arabidopsis shoot apex. Genes Dev. 2011, 25, 2025–2030. [Google Scholar] [CrossRef]
- Brand, U.; Grünewald, M.; Hobe, M.; Simon, R. Regulation of CLV3 expression by two homeobox genes in Arabidopsis. Plant Physiol. 2002, 129, 565–575. [Google Scholar] [CrossRef]
- Schoof, H.; Lenhard, M.; Haecker, A.; Mayer, K.F.; Jürgens, G.; Laux, T. The stem cell population of Arabidopsis shoot meristems is maintained by a regulatory loop betweenthe CLAVATA and WUSCHEL genes. Cell 2000, 100, 635–644. [Google Scholar] [CrossRef]
- Gordon, S.P.; Chickarmane, V.S.; Ohno, C.; Meyerowitz, E.M. Multiple feedback loops through cytokinin signaling control stem cell number within the Arabidopsis shoot meristem. Proc. Nat. Acad. Sci. 2009, 106, 16529–16534. [Google Scholar] [CrossRef]
- Jasinski, S.; Piazza, P.; Craft, J.; Hay, A.; Woolley, L.; Rieu, I.; Phillips, A.; Hedden, P.; Tsiantis, M. KNOX action in Arabidopsis is mediated by coordinate regulation of cytokinin and gibberellin activities. Curr. Biol. 2005, 15, 1560–1565. [Google Scholar] [CrossRef]
- Hay, A.; Tsiantis, M. KNOX genes: Versatile regulators of plant development and diversity. Development 2010, 137, 3153–3165. [Google Scholar] [CrossRef]
- Mudunkothge, J.S.; Krizek, B.A. Three Arabidopsis AIL/PLT genes act in combination to regulate shoot apical meristem function. Plant J. 2012, 71, 108–121. [Google Scholar] [CrossRef]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef]
- Jung, J.H.; Seo, Y.H.; Seo, P.J.; Reyes, J.L.; Yun, J.; Chua, N.H.; Park, C.M. The GIGANTEA-regulated microRNA172 mediates photoperiodic flowering independent of CONSTANS in Arabidopsis. Plant Cell 2007, 19, 2736–2748. [Google Scholar] [CrossRef]
- Raman, S.; Greb, T.; Peaucelle, A.; Blein, T.; Laufs, P.; Theres, K. Interplay of miR164, CUP-SHAPED COTYLEDON genes and LATERAL SUPPRESSOR controls axillary meristem formation in Arabidopsis. Plant J. 2008, 55, 65–76. [Google Scholar] [CrossRef]
- Laufs, P.; Peaucelle, A.; Morin, H.; Traas, J. microRNA regulation of the CUC genes is required for boundary size control in Arabidopsis meristems. Development 2004, 131, 4311–4322. [Google Scholar] [CrossRef]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Ai, X.Y.; Sun, L.M.; Zhang, D.L.; Guo, W.W.; Deng, X.X.; Hu, G.G. Transcriptome profile analysis of flowering molecular processes of early flowering trifoliate orange mutant and the wild-type [Poncirus trifoliata (L.) Raf.] by massively parallel signature sequencing. BMC Genom. 2011, 12, 63. [Google Scholar] [CrossRef]
- Hoshika, Y.; Watanabe, M.; Carrari, E.; Paoletti, E.; Koike, T. Ozone-induced stomatal sluggishness changes stomatal parameters of Jarvis-type model in white birch and deciduous oak. Plant Biol. 2018, 20, 20–28. [Google Scholar] [CrossRef]
- Gang, H.X.; Liu, G.F.; Chen, S.; Jiang, J. Physiological and transcriptome analysisof a yellow-green leaf mutant in birch (Betula platyphylla × B. Pendula). Forests 2019, 10, 120. [Google Scholar] [CrossRef]
- Guo, H.Y.; Wang, Y.C.; Hu, P.; Wang, Y.M.; Jiang, Y.; Yang, C.P.; Wang, C. Gene expression profiles in different stem internodes reveal the genetic regulation of primary and secondary stem development in Betula platyphylla. Tree Genet. Genomes 2016, 12, 113. [Google Scholar] [CrossRef]
- Zhang, W.B.; Wei, R.; Chen, S.; Jiang, J.; Li, H.Y.; Wang, S.; Wei, H.R.; Liu, G.F. Functional Characterization of Cinnamoyl-CoA Reductase (CCR) in birch (B. platyphylla × B. pendula) through overexpression and suppression analysis. Physiol. Plant. 2015, 154, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.J.; Puryear, J.; Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 1993, 11, 113–116. [Google Scholar] [CrossRef]
- Dobin, A.; Gingeras, T.R. Comment on “TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions” by Kim et al. Bioinformatics 2013, 14, 000851. [Google Scholar]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.Z.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.Z.; Huang, J.J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef]
- Madera, M. Profile Comparer: A program for scoring and aligning profile hidden Markov models. Bioinformatics 2008, 24, 2630–2631. [Google Scholar] [CrossRef]
- Wang, L.K.; Feng, Z.X.; Wang, X.; Wang, X.W.; Zhang, X.G. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Huang, H.J.; Wang, S.; Jiang, J.; Liu, G.F.; Li, H.Y.; Chen, S.; Xu, H.W. Overexpression of BpAP1 induces early flowering and produces dwarfism in Betula platyphylla × Betula pendula. Physiol. Plant. 2013, 151, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Mu, H.Z.; Lin, L.; Liu, G.F.; Jiang, J. Transcriptomic analysis of incised leaf-shape determination in birch. Gene 2013, 531, 263–269. [Google Scholar] [CrossRef]
- Lin, L.; Mu, H.Z.; Jiang, J.; Liu, G.F. Transcriptomic analysis of purple leaf determination in birch. Gene 2013, 526, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Uhlenhaut, N.H.; Godard, F.; Demar, M.; Bressan, R.; Weigel, D.; Lohmann, J.U. Dissection of floral induction pathways using global expression analysis. Development 2003, 130, 6001–6012. [Google Scholar] [CrossRef]
- Silva, G.F.F.E.; Silva, E.M.; da Silva Azevedo, M.; Guivin, M.A.; Ramiro, D.A.; Fiqueiredo, C.R.; Carrer, H.; Peres, L.E.; Nogueira, F.T. microRNA156-targeted SPL/SBP box transcription factors regulate tomato ovary and fruit development. Plant J. 2014, 78, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Salinas, M.; Garcia-Molina, A.; Höhmann, S.; Berndtgen, R.; Huijser, P. SPL8 and miR156-targeted SPL genes redundantly regulate Arabidopsis gynoecium differential patterning. Plant J. 2013, 75, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Ju, Y.; Seo, P.J.; Lee, J.H.; Park, C.M. The SOC1-SPL module integrates photoperiod and gibberellic acid signals to control flowering time in Arabidopsis. Plant J. 2012, 69, 577–588. [Google Scholar] [CrossRef]
- Smaczniak, C.; Immink, R.G.; Angenent, G.C.; Kaufmann, K. Developmental and evolutionary diversity of plant MADS-domain factors: Insights from recent studies. Development 2012, 139, 3081–3098. [Google Scholar] [CrossRef]
- Adamczyk, B.J.; Fernandez, D.E. MIKC*MADS domain heterodimers are required for pollen maturation and tube growth in Arabidopsis. Plant Physiol. 2009, 149, 1713–1723. [Google Scholar] [CrossRef]
- Del Pozo, J.C.; Boniotti, M.B.; Gutierrez, C. Arabidopsis E2Fc functions in cell division and is degraded by the ubiquitin-SCF(AtSKP2) pathway in response to light. Plant Cell 2002, 14, 3057–3071. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.M.; Lin, W.C.; Husbangds, A.Y.; Jaganatha, V.; Mangeon, A.; Neff, M.M.; Girke, T.; Springer, P.S. Arabidopsis lateral organ boundaries negatively regulates brassinosteroid accumulation to limit growth in organ boundaries. Proc. Natl. Acad. Sci. USA 2012, 109, 21146–21151. [Google Scholar] [CrossRef] [PubMed]
- Efroni, I.; Blum, E.; Golshmid, A.; Eshd, Y. A protracted and dynamic maturation schedule underlies Arabidopsis leaf development. Plant Cell 2008, 20, 2293–2306. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Irish, V.F. Temporal control of plant organ growth by TCP transcription factors. Curr. Biol. Rep. 2015, 25, 1765–1770. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.J. The plant hormones: Their nature, occurrence, and functions. Plant Horm. 2010, 1–15. [Google Scholar] [CrossRef]
- Qin, G.J.; Gu, H.Y.; Zhao, Y.D.; Ma, Z.Q.; Shi, G.L.; Yang, Y.; Pichersky, E.; Chen, H.D.; Liu, M.H.; Chen, Z.L.; Qu, L.J. An indole-3-acetic acid carboxyl methyltransferase regulates Arabidopsis leaf development. Plant Cell 2005, 17, 2693–2704. [Google Scholar] [CrossRef] [PubMed]
- Korasick, D.A.; Enders, T.A.; Strader, L.C. Auxin biosynthesis and storage forms. J. Exp. Bot. 2013, 64, 2541–2555. [Google Scholar] [CrossRef]
- Mano, Y.; Nemoto, K. The pathway of auxin biosynthesis in plants. J. Exp. Bot. 2012, 63, 2853–2872. [Google Scholar] [CrossRef]
- Mashiguchi, K.; Tanaka, K.; Sakai, T.; Sugawara, S.; Kawaide, H.; Natsume, M.; Hanada, A.; Yaeno, T.; Shiasu, K.; Yao, H.; et al. The main auxin biosynthesis pathway in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 18512–18517. [Google Scholar] [CrossRef]
- Zhao, Y.; Christensen, S.K.; Fankhauser, C.; Cashman, J.R.; Cohen, J.D.; Weigel, D.; Chory, J. A role for flavin monooxygenase-like enzymes in auxin biosynthesis. Science 2001, 291, 306–309. [Google Scholar] [CrossRef]
- Sakakibara, H. Cytokinins: Activity, biosynthesis, and translocation. Annu. Rev. Plant Biol. 2006, 57, 431–449. [Google Scholar] [CrossRef]
- Tanaka, M.; Takei, K.; Kijima, M.; Sakakibara, H.; Mori, H. Auxin controls local cytokinin biosynthesis in the nodal stem in apical dominance. Plant J. 2006, 45, 1028–1036. [Google Scholar] [CrossRef]
- Müller, D.; Waldie, T.; Miyawaki, K.; To, J.P.; Melnyk, C.W.; Kieber, J.J.; Kakimoto, T.; Leyser, O. Cytokinin is required for escape but not release from auxin mediated apical dominance. Plant J. 2015, 82, 874–886. [Google Scholar] [CrossRef]
- Haberer, G.; Kieber, J.J. Cytokinins. New insights into a classic phytohormone. Plant Physiol. 2002, 128, 354–362. [Google Scholar] [CrossRef]
- Schmitz, R.Y.; Skoog, F. Cytokinins: Synthesis and biological activity of geometric and position isomers of zeatin. Plant Physiol. 1972, 50, 702–705. [Google Scholar] [CrossRef]
- Emery, R.J.; Leport, L.; Barton, J.E.; Turner, N.C.; Atkins, C.A. cis-isomers of cytokinins predominate in chickpea seeds throughout their development. Plant Physiol. 1998, 117, 1515–1523. [Google Scholar] [CrossRef]
- Louis, J.P.; Augur, C.; Teller, G. Cytokinins and differentiation processes in mercurialis annua. Plant Physiol. 1990, 94, 1535–1541. [Google Scholar] [CrossRef]
- Martin, R.C.; Mok, M.C.; Habben, J.E.; Mok, D.W. A maize cytokinin gene encoding an O-glucosyltransferase specific to cis-zeatin. Proc. Natl. Acad. Sci. USA 2001, 98, 5922–5926. [Google Scholar] [CrossRef]
- Hedden, P. The genes of the green revolution. Trends Genet. 2003, 19, 5–9. [Google Scholar] [CrossRef]
- Yamaguchi, S. Gibberellin metabolism and its regulation. Annu. Rev. Plant Biol. 2008, 59, 225–251. [Google Scholar] [CrossRef]
- García-Martínez, J.L.; López-Diaz, I.; Sánchez-Beltrán, M.J.; Phillips, A.L.; Ward, D.A.; Gaskin, P.; Hedden, P. Isolation and transcript analysis of gibberellin 20-oxidase genes in pea and bean in relation to fruit development. Plant Mol. Biol. 1997, 33, 1073–1084. [Google Scholar] [CrossRef]
- Willige, B.C.; Isono, E.; Richter, R.; Zourelidou, M.; Schwechheimer, C. Gibberellin regulates PIN-FORMED abundance and is required for auxin transport-dependent growth and development in Arabidopsis thaliana. Plant Cell 2011, 23, 2184–2195. [Google Scholar] [CrossRef] [PubMed]
- Fleishon, S.; Shani, E.; Ori, N.; Weiss, D. Negative reciprocal interactions between gibberellin and cytokinin in tomato. New Phytol. 2011, 190, 609–617. [Google Scholar] [CrossRef]
- Yaish, M.W.; El-Kereamy, A.; Zhu, T.; Beatty, P.H.; Good, A.G.; Bi, Y.M.; Rothstein, S.J. The APETALA-2-like transcription factor OsAP2-39 controls key interactions between abscisic acid and gibberellin in rice. PLoS Genet. 2011, 6, e1001098. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Cadenas, A.; Zentella, R.; Walker-Simmons, M.K.; Ho, T.-H.D. Gibberellin/Abscisic acid antagonism in barley aleurone cells: Site of action of the protein kinase PKABA1 in relation to gibberellin signaling molecules. Plant Cell 2001, 13, 667–679. [Google Scholar] [CrossRef]
- Dubois, V.; Moritz, T.; García-Martínez, J.L. Examination of two lowland rice cultivars reveals that gibberellin-dependent early response to submergence is not necessarily mediated by ethylene. Plant Signal Behav. 2011, 6, 134–136. [Google Scholar] [CrossRef][Green Version]
- Ferguson, B.J.; Foo, E.; Ross, J.J.; Reid, J.B. Relationship between gibberellin, ethylene and nodulation in Pisum sativum. New Phytol. 2011, 189, 829–842. [Google Scholar] [CrossRef]
- Navarro, L.; Bari, R.; Achard, P.; Lisón, P.; Nemri, A.; Harberd, N.P.; Jones, J.D. DELLAs control plant immune responses by modulating the balance of jasmonic acid and salicylic acid signaling. Curr. Biol. 2008, 18, 650–655. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Line | Stem Length (cm) | The Average Root Length (cm) | Root Number | Aboveground Part | Underground Part | ||
|---|---|---|---|---|---|---|---|
| Fresh Weight (g) | Dry Weight (g) | Fresh Weight (g) | Dry Weight (g) | ||||
| WT | 1.47 ± 0.35b | 1.49 ± 0.61a | 2.67 ± 0.90a | 0.0894 ± 0.0035a | 0.0119 ± 0.0012b | 0.0294 ± 0.0041a | 0.0031 ± 0.0004b |
| OE2 | 1.80 ± 0.36a | 1.38 ± 0.45a | 6.15 ± 1.28b | 0.0772 ± 0.0013b | 0.0202 ± 0.0004a | 0.0186 ± 0.0031b | 0.0057 ± 0.0010a |
| br | 1.22 ± 0.30b | 0.87 ± 0.23b | 1.73 ± 0.70c | 0.0361 ± 0.0005c | 0.0105 ± 0.0001b | 0.0049 ± 0.0012c | 0.0013 ± 0.0003c |
| Samples | Clean Reads | Clean Bases | GC Content | % ≥Q30 | Total Reads | Mapped Reads |
|---|---|---|---|---|---|---|
| T1 | 15,781,488 | 4,734,446,400 | 46.66% | 94.51% | 31,562,976 | 29,638,044 (93.90%) |
| T2 | 14,560,716 | 4,368,214,800 | 46.73% | 93.71% | 29,121,432 | 27,269,226 (93.64%) |
| T3 | 15,447,705 | 4,634,311,500 | 46.78% | 94.23% | 30,895,410 | 29,080,761 (94.13%) |
| T4 | 15,587,054 | 4,676,116,200 | 46.72% | 94.11% | 31,174,108 | 29,278,066 (93.92%) |
| T5 | 15,785,947 | 4,735,784,100 | 46.54% | 94.24% | 31,571,894 | 29,547,843 (93.59%) |
| T6 | 15,605,416 | 4,681,624,800 | 46.83% | 94.25% | 31,210,832 | 29,297,703 (93.87%) |
| T7 | 15,346,068 | 4,603,820,400 | 46.82% | 94.15% | 30,692,136 | 28,791,530 (93.81%) |
| T8 | 15,205,628 | 4,561,688,400 | 47.25% | 94.24% | 30,411,256 | 28,635,215 (94.16%) |
| T9 | 15,521,452 | 4,656,435,600 | 47.01% | 94.52% | 31,042,904 | 29,292,901 (94.36%) |
| T10 | 15,223,613 | 4,567,083,900 | 47.64% | 94.46% | 30,447,226 | 28,809,352 (94.62%) |
| T11 | 15,137,989 | 4,541,396,700 | 46.93% | 94.64% | 30,275,978 | 28,600,140 (94.46%) |
| T12 | 14,686,313 | 4,405,893,900 | 47.19% | 94.22% | 29,372,626 | 27,687,905 (94.26%) |
| Annotated Databases | New Gene Number |
|---|---|
| COG | 329 |
| GO | 853 |
| KEGG | 492 |
| KOG | 799 |
| Pfam | 890 |
| Swiss-Prot | 986 |
| eggNOG | 1365 |
| Nr | 1532 |
| All | 1550 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, R.; Wang, S.; Liu, C.; Xu, W.; Bian, X.; Liu, G.; Jiang, J. Transcriptome Analysis of a Multiple-Branches Mutant Terminal Buds in Betula platyphylla × B. pendula. Forests 2019, 10, 374. https://doi.org/10.3390/f10050374
Han R, Wang S, Liu C, Xu W, Bian X, Liu G, Jiang J. Transcriptome Analysis of a Multiple-Branches Mutant Terminal Buds in Betula platyphylla × B. pendula. Forests. 2019; 10(5):374. https://doi.org/10.3390/f10050374
Chicago/Turabian StyleHan, Rui, Shuo Wang, Chaoyi Liu, Wendi Xu, Xiuyan Bian, Guifeng Liu, and Jing Jiang. 2019. "Transcriptome Analysis of a Multiple-Branches Mutant Terminal Buds in Betula platyphylla × B. pendula" Forests 10, no. 5: 374. https://doi.org/10.3390/f10050374
APA StyleHan, R., Wang, S., Liu, C., Xu, W., Bian, X., Liu, G., & Jiang, J. (2019). Transcriptome Analysis of a Multiple-Branches Mutant Terminal Buds in Betula platyphylla × B. pendula. Forests, 10(5), 374. https://doi.org/10.3390/f10050374