
Reduced Nucleotides, Thiols and O2 in Cellular Redox Balance: A Biochemist’s View


Abstract
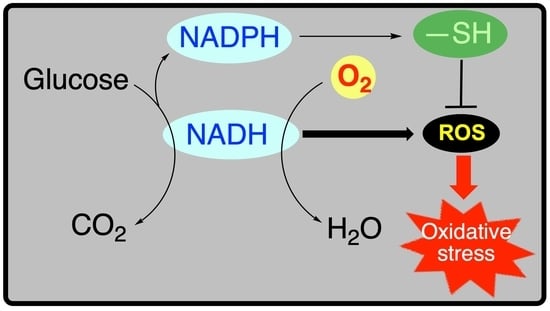
1. Introduction
2. Free Energy Is the Driving Force for Maintaining Cell Dynamics
3. Redox Potential and the Chemical Basis for the Role of O2 as an “Energy-Rich” Molecule
4. The Special Role of Hydrogen in Biochemical Redox Reactions


5. Metabolic Blocks and Metabolic Compartmentation of Redox Couples
- (1)
- Nucleoside triphosphates with a high phosphate group transfer potential—in particular, the ATP/ADP couple.
- (2)
- The redox coupling agents are NAD+/NADH, NADP+/NADPH and FAD/FADH2.
- (3)
- Approximately 10 organic compounds that constitute products of the catabolic block and are used as building blocks for the anabolic block. Among these are two thioesters (acetyl-CoA and succinyl-CoA, with a high acetyl transfer group potential), three 2-oxoacids (pyruvate, oxaloacetate and oxoglutarate), phosphoenolpyruvate and four phosphorylated sugars (from C3 to C6).
6. Extrapolation of the Concept of Redox Potential to Complex Systems: The Redox Environment
7. Cellular Compartmentation of Redox Balance
8. Antioxidant Defense Systems
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CYSS | Cystine |
| ER | Endoplasmic reticulum |
| GSH | Reduced glutathione |
| GSSG | Oxidized glutathione |
| PPS | Pentose phosphate shunt |
| ROS | Reactive Oxygen Species |
| TCA | Tricarboxylic Acid Cycle |
References
- Klotz, I.M. Introduction to Biomolecular Energetics; Academic Press Inc.: Cambridge, MA, USA, 1986. [Google Scholar]
- Chang, A.Y.; Marshall, W.F. Dynamics of living cells in a cytomorphological state space. Proc. Natl. Acad. Sci. USA 2019, 116, 21556–21562. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.; Grãos, M.; Anjo, S.I.; Manadas, B. Modulation of signaling pathways by DJ-1: An updated overview. Redox Biol. 2022, 51, 102283. [Google Scholar] [CrossRef] [PubMed]
- Boas, S.M.; Joyce, K.L.; Cowell, R.M. The NRF2-Dependent Transcriptional Regulation of Antioxidant Defense Pathways: Relevance for Cell Type-Specific Vulnerability to Neurodegeneration and Therapeutic Intervention. Antioxidants 2021, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Tretter, V.; Hochreiter, B.; Zach, M.L.; Krenn, K.; Klein, K.U. Understanding Cellular Redox Homeostasis: A Challenge for Precision Medicine. Int. J. Mol. Sci. 2022, 23, 106. [Google Scholar] [CrossRef]
- Muzio, G.; Barrera, G.; Pizzimenti, S. Peroxisome Proliferator-Activated Receptors (PPARs) and Oxidative Stress in Physiological Conditions and in Cancer. Antioxidants 2021, 10, 1734. [Google Scholar] [CrossRef]
- Buxbaum, E. Fundamentals of Protein Structure and Function, 2nd ed.; Springer: Cham, Switzerland, 2015; ISBN 978-3-319-19919-1. [Google Scholar]
- Fukushi, A.; Kim, H.-D.; Chang, Y.-C.; Kim, C.-H. Revisited Metabolic Control and Reprogramming Cancers by Means of the Warburg Effect in Tumor Cells. Int. J. Mol. Sci. 2022, 23, 10037. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.-D.V.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef]
- Sies, H. Oxidative eustress: On constant alert for redox homeostasis. Redox Biol. 2021, 41, 101867. [Google Scholar] [CrossRef]
- Ge, H.; Qian, H. Mathematical Formalism of Nonequilibrium Thermodynamics for Nonlinear Chemical Reaction Systems with General Rate Law. J. Stat. Phys. 2017, 166, 190–209. [Google Scholar] [CrossRef]
- Beard, D.A.; Qian, H. Relationship between Thermodynamic Driving Force and One-Way Fluxes in Reversible Processes. PLoS ONE 2007, 2, e144. [Google Scholar] [CrossRef]
- Xu, J.; Martien, J.; Gilbertson, C.; Ma, J.; Amador-Noguez, D.; Park, J.O. Metabolic flux analysis and fluxomics-driven determination of reaction free energy using multiple isotopes. Curr. Opin. Biotechnol. 2020, 64, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Ferguson, S.J. Bioenergetics 4; Academic Press: Amsterdam, The Netherlands, 2013; ISBN 978-0-12-388425-1. [Google Scholar]
- Schmidt-Rohr, K. How Batteries Store and Release Energy: Explaining Basic Electrochemistry. J. Chem. Educ. 2018, 95, 1801–1810. [Google Scholar] [CrossRef]
- Schmidt-Rohr, K. Oxygen Is the High-Energy Molecule Powering Complex Multicellular Life: Fundamental Corrections to Traditional Bioenergetics. ACS Omega 2020, 5, 2221–2233. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, N.W.H. Introducing Biological Energetics; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Grattieri, M. Purple bacteria photo-bioelectrochemistry: Enthralling challenges and opportunities. Photochem. Photobiol. Sci. 2020, 19, 424–435. [Google Scholar] [CrossRef]
- Brown, T.L.; LeMAy, H.E.; Bursten, B.E. Chemistry: The Central Science, 6th ed.; Prentice Hall: Englewood Cliffs, NJ, USA, 1994. [Google Scholar]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Alberty, R.A. Standard apparent reduction potentials of biochemical half reactions and thermodynamic data on the species involved. Biophys. Chem. 2004, 111, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, S.; Shimada, A. Reaction mechanism of cytochrome c oxidase. Chem. Rev. 2015, 115, 1936–1989. [Google Scholar] [CrossRef]
- Voet, D.; Voet, J.G.; Pratt, C.W. Principles of Biochemistry; John Wiley & Sons: Singapore, 2013. [Google Scholar]
- Schmidt-Rohr, K. Why combustions are always exothermic, yielding about 418 kJ per mole of O2. J. Chem. Educ. 2015, 92, 2094–2099. [Google Scholar] [CrossRef]
- Weiss, H.M. Appreciating Oxygen. J. Chem. Educ. 2008, 85, 1218. [Google Scholar] [CrossRef]
- Merckel, R.D.; Labuschagne, F.J.W.J.; Heydenrych, M.D. Oxygen consumption as the definitive factor in predicting heat of combustion. Appl. Energy 2019, 235, 1041–1047. [Google Scholar] [CrossRef]
- Borden, W.T.; Hoffmann, R.; Stuyver, T.; Chen, B. Dioxygen: What Makes This Triplet Diradical Kinetically Persistent? J. Am. Chem. Soc. 2017, 139, 9010–9018. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Reinhard, C.T.; Planavsky, N.J. The rise of oxygen in Earth’s early ocean and atmosphere. Nature 2014, 506, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Shang, H.; Rothman, D.H.; Fournier, G.P. Oxidative metabolisms catalyzed Earth’s oxygenation. Nat. Commun. 2022, 13, 1328. [Google Scholar] [CrossRef] [PubMed]
- Hodgskiss, M.S.W.; Crockford, P.W.; Peng, Y.; Wing, B.A.; Horner, T.J. A productivity collapse to end Earth’s Great Oxidation. Proc. Natl. Acad. Sci. USA 2019, 116, 17207–17212. [Google Scholar] [CrossRef] [PubMed]
- Park, B.-K.; Doh, S.-T.; Son, G.-S.; Kim, J.-M.; Lee, G.-Y. MO study of Hydride Transfer between NADH and Flavin Nucleotides. Bull. Korean Chem. Soc. 1994, 15, 291–293. [Google Scholar]
- Pullman, B.; Pullman, A. The Electronic Structure of the Respiratory Coenzymes. Proc. Natl. Acad. Sci. USA 1959, 45, 136–144. [Google Scholar] [CrossRef]
- Archipowa, N.; Kutta, R.J.; Heyes, D.J.; Scrutton, N.S. Stepwise Hydride Transfer in a Biological System: Insights into the Reaction Mechanism of the Light-Dependent Protochlorophyllide Oxidoreductase. Angew. Chem. Int. Ed. Engl. 2018, 57, 2682–2686. [Google Scholar] [CrossRef]
- Garrett, R.H.; Grisham, C.M. Biochemistry, 2nd ed.; Saunders College Publishing: Rochester, NY, USA, 1999; ISBN 0-03-022318-0. [Google Scholar]
- Xiao, W.; Loscalzo, J. Metabolic Responses to Reductive Stress. Antioxid. Redox Signal. 2020, 32, 1330–1347. [Google Scholar] [CrossRef]
- Mosconi, L. Glucose metabolism in normal aging and Alzheimer’s disease: Methodological and physiological considerations for PET studies. Clin. Transl. Imaging 2013, 1, 217–233. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Weiss, M.C.; Preiner, M.; Xavier, J.C.; Zimorski, V.; Martin, W.F. The last universal common ancestor between ancient Earth chemistry and the onset of genetics. PLoS Genet. 2018, 14, e1007518. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.F.; Matschinsky, F.M. Metabolic Homeostasis in Life as We Know It: Its Origin and Thermodynamic Basis. Front. Physiol. 2021, 12, 658997. [Google Scholar] [CrossRef]
- Moriarty-Craige, S.E.; Jones, D.P. Extracellular thiols and thiol/disulfide redox in metabolism. Ann. Rev. Nutr. 2004, 24, 481–509. [Google Scholar] [CrossRef] [PubMed]
- Baba, S.P.; Bhatnagar, A. Role of Thiols in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Piste, P. Cysteine—Master Antioxidant. IJPCBS 2013, 3, 143–149. [Google Scholar]
- Hensley, K.; Danekas, A.; Farrell, W.; Garcia, T.; Mehboob, W.; White, M. At the intersection of sulfur redox chemistry, cellular signal transduction and proteostasis: A useful perspective from which to understand and treat neurodegeneration. Free Radic. Biol. Med. 2022, 178, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Rai, A.; Checker, R.; Patwardhan, R.S.; Singh, B.; Sharma, D.; Sandur, S.K. Role of protein S-Glutathionylation in cancer progression and development of resistance to anti-cancer drugs. Arch. Biochem. Biophys. 2021, 704, 108890. [Google Scholar] [CrossRef]
- Kalinina, E.; Novichkova, M. Glutathione in Protein Redox Modulation through S-Glutathionylation and S-Nitrosylation. Molecules 2021, 26, 435. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, O.; Stepanenko, S.; Chehivska, L.; Sambon, M.; Bettendorff, L.; Yu, P. Thiamine deficiency in rats affects thiamine metabolism possibly through the formation of oxidized thiamine pyrophosphate. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129980. [Google Scholar] [CrossRef]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Watson, W.H.; Jones, D.P. Oxidation of nuclear thioredoxin during oxidative stress. FEBS Lett. 2003, 543, 144–147. [Google Scholar] [CrossRef]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M. Oxidants, Antioxidants and Thiol Redox Switches in the Control of Regulated Cell Death Pathways. Antioxidants 2020, 9, 309. [Google Scholar] [CrossRef] [PubMed]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef]
- Jackson, J.B. Proton translocation by transhydrogenase. FEBS Lett. 2003, 545, 18–24. [Google Scholar] [CrossRef]
- Zhang, Q.; Padayatti, P.S.; Leung, J.H. Proton-Translocating Nicotinamide Nucleotide Transhydrogenase: A Structural Perspective. Front. Physiol. 2017, 8, 1089. [Google Scholar] [CrossRef]
- Nonn, L.; Williams, R.R.; Erickson, R.P.; Powis, G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol. Cell Biol. 2003, 23, 916–922. [Google Scholar] [CrossRef]
- Jaganjac, M.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anticancer Therapies. Antioxidants 2020, 9, 1151. [Google Scholar] [CrossRef]
- Mohammadi, F.; Soltani, A.; Ghahremanloo, A.; Javid, H.; Hashemy, S.I. The thioredoxin system and cancer therapy: A review. Cancer Chemother. Pharmacol. 2019, 84, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Ma, W.; Liu, P.; Zhou, F. Overview of thioredoxin system and targeted therapies for acute leukemia. Mitochondrion 2019, 47, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; Sevier, C.S.; Bulleid, N.J. How Are Proteins Reduced in the Endoplasmic Reticulum? Trends Biochem. Sci. 2018, 43, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P. Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid. Redox Signal. 2013, 18, 1623–1641. [Google Scholar] [CrossRef]
- Oka, O.B.V.; Bulleid, N.J. Forming disulfides in the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2425–2429. [Google Scholar] [CrossRef]
- Go, Y.-M.; Jones, D.P. Redox clamp model for study of extracellular thiols and disulfides in redox signaling. Methods Enzymol. 2010, 474, 165–179. [Google Scholar] [CrossRef]
- Li, H.; Hou, J. Entropy, Free Radical and Life System. Open J. Biophys. 2016, 6, 83–86. [Google Scholar] [CrossRef][Green Version]
- Payen, V.L.; Zampieri, L.X.; Porporato, P.E.; Sonveaux, P. Pro- and antitumor effects of mitochondrial reactive oxygen species. Cancer Metastasis Rev. 2019, 38, 189–203. [Google Scholar] [CrossRef]
- Lee, K.H.; Cha, M.; Lee, B.H. Crosstalk between Neuron and Glial Cells in Oxidative Injury and Neuroprotection. Int. J. Mol. Sci. 2021, 22, 13315. [Google Scholar] [CrossRef]
- Bellanti, F.; Lo Buglio, A.; Vendemiale, G. Redox Homeostasis and Immune Alterations in Coronavirus Disease-19. Biology 2022, 11, 159. [Google Scholar] [CrossRef]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Aljada, A.; Chaudhuri, A.; Mohanty, P.; Garg, R. Metabolic Syndrome. Circulation 2005, 111, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Tanrıkulu-Küçük, S.; Başaran-Küçükgergin, C.; Söğüt, İ.; Tunçdemir, M.; Doğru-Abbasoğlu, S.; Seyithanoğlu, M.; Koçak, H.; Öner-İyidoğan, Y. Dietary curcumin and capsaicin: Relationship with hepatic oxidative stress and apoptosis in rats fed a high fat diet. Adv. Clin. Exp. Med. 2019, 28, 1013–1020. [Google Scholar] [CrossRef]
- Chenna, S.; Koopman, W.J.H.; Prehn, J.H.M.; Connolly, N.M.C. Mechanisms and mathematical modeling of ROS production by the mitochondrial electron transport chain. Am. J. Physiol. Cell Physiol. 2022, 323, C69–C83. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Vizi, V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J. Neurosci. 2004, 24, 7771–7778. [Google Scholar] [CrossRef]
- Nemeria, N.S.; Gerfen, G.; Guevara, E.; Nareddy, P.R.; Szostak, M.; Jordan, F. The Human Krebs Cycle 2-Oxoglutarate Dehydrogenase Complex Creates an Additional Source of Superoxide/Hydrogen Peroxide from 2-Oxoadipate as Alternative Substrate. Free Radic. Biol. Med. 2017, 108, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Nimse, S.B.; Pal, D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Ciesielska, S.; Slezak-Prochazka, I.; Bil, P.; Rzeszowska-Wolny, J. Micro RNAs in Regulation of Cellular Redox Homeostasis. Int. J. Mol. Sci. 2021, 22, 6022. [Google Scholar] [CrossRef]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.-A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef]
- Niki, E. Oxidative stress and antioxidants: Distress or eustress? Free Radic. Biol. Med. 2018, 124, 564. [Google Scholar] [CrossRef]
- Maity, S.; Rajkumar, A.; Matai, L.; Bhat, A.; Ghosh, A.; Agam, G.; Kaur, S.; Bhatt, N.R.; Mukhopadhyay, A.; Sengupta, S.; et al. Oxidative Homeostasis Regulates the Response to Reductive Endoplasmic Reticulum Stress through Translation Control. Cell Rep. 2016, 16, 851–865. [Google Scholar] [CrossRef] [PubMed]
- Ido, Y.; Kilo, C.; Williamson, J.R. Cytosolic NADH/NAD+, free radicals, and vascular dysfunction in early diabetes mellitus. Diabetologia 1997, 40 (Suppl. S2), S115–S117. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
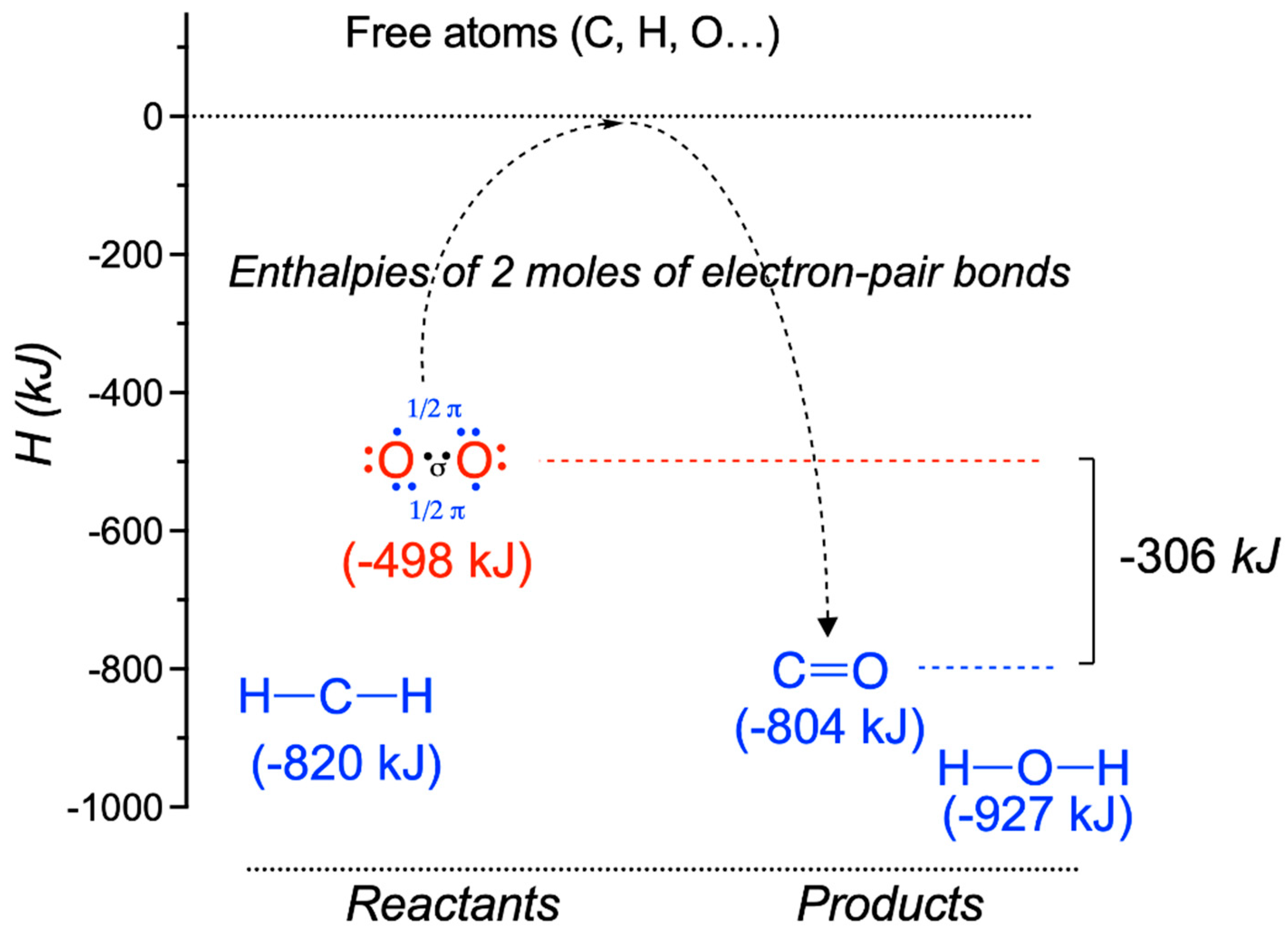{kind=link}
{kind=link}
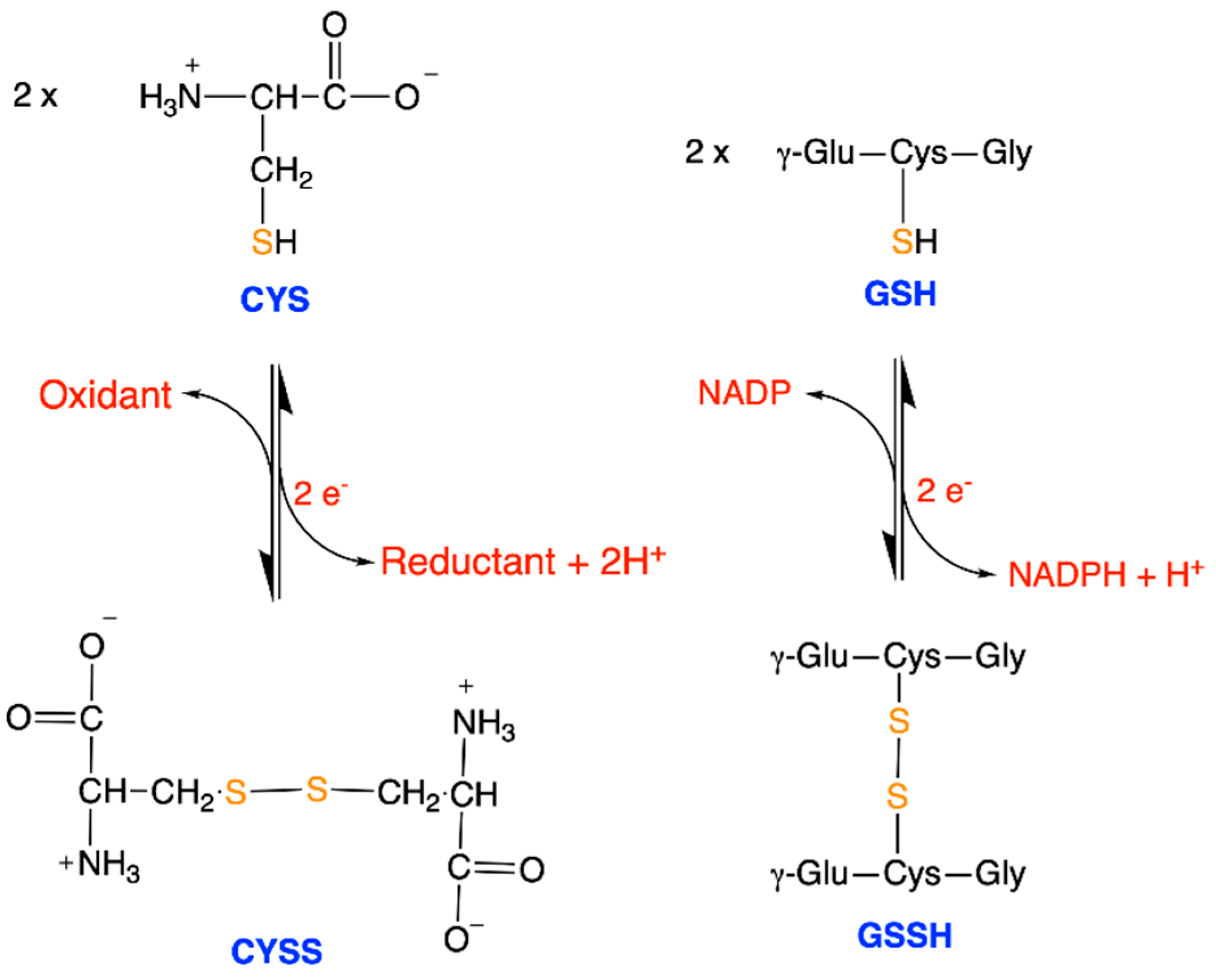{kind=link}
{kind=link}
{kind=link}
| Driving Force (J.mol−1) | Source of Driving Force |
|---|---|
| ∆G = ∆G° + RT ln Q | Differences in the structural arrangement between initial and final reactants (∆G°) and concentrations (Q) |
| F∆E | ln Q) |
| Denomination | Energy Source | Hydrogen (e−) Source | Carbon Source | Organism |
|---|---|---|---|---|
| Phototrophs | Light | Green plants, cyanobacteria, purple bacteria | ||
| Chemotrophs | Oxidation of organic or inorganic substrates | All others | ||
| Lithotrophs | Inorganic compounds (H2O, NH3, H2S) | Green plants, cyanobacteria, purple bacteria, nitrifying bacteria, thiobacilli | ||
| Organotrophs | Organic compounds | Animals and most microorganisms | ||
| Autotrophs | CO2 fixation | Green plants, cyanobacteria, purple bacteria, nitrifying bacteria, thiobacilli | ||
| Heterotrophs | Assimilation of organic compounds | Animals and most microorganisms | ||
| Photolithoauxotrophs | Light | H2O | CO2 fixation | Green plants, cyanobacteria, purple bacteria * |
| Chemolithoauxotrophs | Oxidation of inorganic compounds | Inorganic compounds | Generally, CO2 fixation | Nitrifying bacteria (NH3), Thiobacilli (H2S) ** |
| Chemoorganoheterotrophs | Oxidation of organic compounds | Organic compounds | Assimilation of organic compounds | Animals and most microorganisms |
| Oxidant (Oxidized Form) | Reductant (Reduced Form) | z | E′° (V) |
|---|---|---|---|
| Succinate + CO2 | Oxoglutarate | 2 | −0.67 |
| Acetate + 3 H+ | Acetaldehyde + H2O | 2 | −0.60 |
| Ferredoxin oxidized | Ferredoxin reduced | 1 | −0.43 |
| 2 H+ | H2 | 2 | −0.42 |
| NAD+ + H+ | NADH | 2 | −0.32 |
| NADP+ + H+ | NADPH | 2 | −0.32 |
| Lipoate oxidized + 2 H+ | Lipoate reduced | 2 | −0.29 |
| Glutathione oxidized ** + 2 H+ | Glutathione reduced | 2 | −0.24 |
| FAD + 2 H+ | FADH2 | 2 | −0.22 * |
| Acetaldehyde + 2 H+ | Ethanol | 2 | −0.20 |
| Pyruvate + H+ | Lactate | 2 | −0.19 |
| Fumarate + 2 H+ | Succinate | 2 | 0.03 |
| Ubiquinone (Q) + 2 H+ | Ubiquinol (QH2) | 2 | 0.06 |
| Cytochrome b (Fe3+) | Cytochrome b (Fe2+) | 2 | 0.07 |
| Dehydroascorbate + 2 H+ | Ascorbate | 2 | 0.08 |
| Cytochrome c (Fe3+) | Cytochrome c (Fe2+) | 1 | 0.22 |
| Fe3+ | Fe2+ | 1 | 0.77 |
| 1/2 O2 + 2 H+ | H2O | 2 | 0.82 |
| P680+ | P680 | 1 | 1.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bettendorff, L. Reduced Nucleotides, Thiols and O2 in Cellular Redox Balance: A Biochemist’s View. Antioxidants 2022, 11, 1877. https://doi.org/10.3390/antiox11101877
Bettendorff L. Reduced Nucleotides, Thiols and O2 in Cellular Redox Balance: A Biochemist’s View. Antioxidants. 2022; 11(10):1877. https://doi.org/10.3390/antiox11101877
Chicago/Turabian StyleBettendorff, Lucien. 2022. "Reduced Nucleotides, Thiols and O2 in Cellular Redox Balance: A Biochemist’s View" Antioxidants 11, no. 10: 1877. https://doi.org/10.3390/antiox11101877
APA StyleBettendorff, L. (2022). Reduced Nucleotides, Thiols and O2 in Cellular Redox Balance: A Biochemist’s View. Antioxidants, 11(10), 1877. https://doi.org/10.3390/antiox11101877