
Icariside II, a Naturally Occurring SIRT3 Agonist, Protects against Myocardial Infarction through the AMPK/PGC-1α/Apoptosis Signaling Pathway


 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. MI Model and Drug Administration
2.3. Determination of Echocardiography
2.4. Assessment of Myocardial Infarct Size
2.5. Microarray Processing and Data Analysis
2.6. Hematoxylin–Eosin (HE) Staining
2.7. Transmission Electron Microscopy (TEM)
2.8. TdT-Mediated dUTP Nick-End Labeling (TUNEL) Staining
2.9. Cell Culture and OGD Model
2.10. Determination of Cell Viability and Cytotoxicity
2.11. Determination of LDH, Troponin (Tn), and TnT Levels
2.12. Measurement of ROS Generation
2.13. Determination of Oxidative Stress-Related Indicators
2.14. Flow Cytometric Analysis
2.15. Western Blot
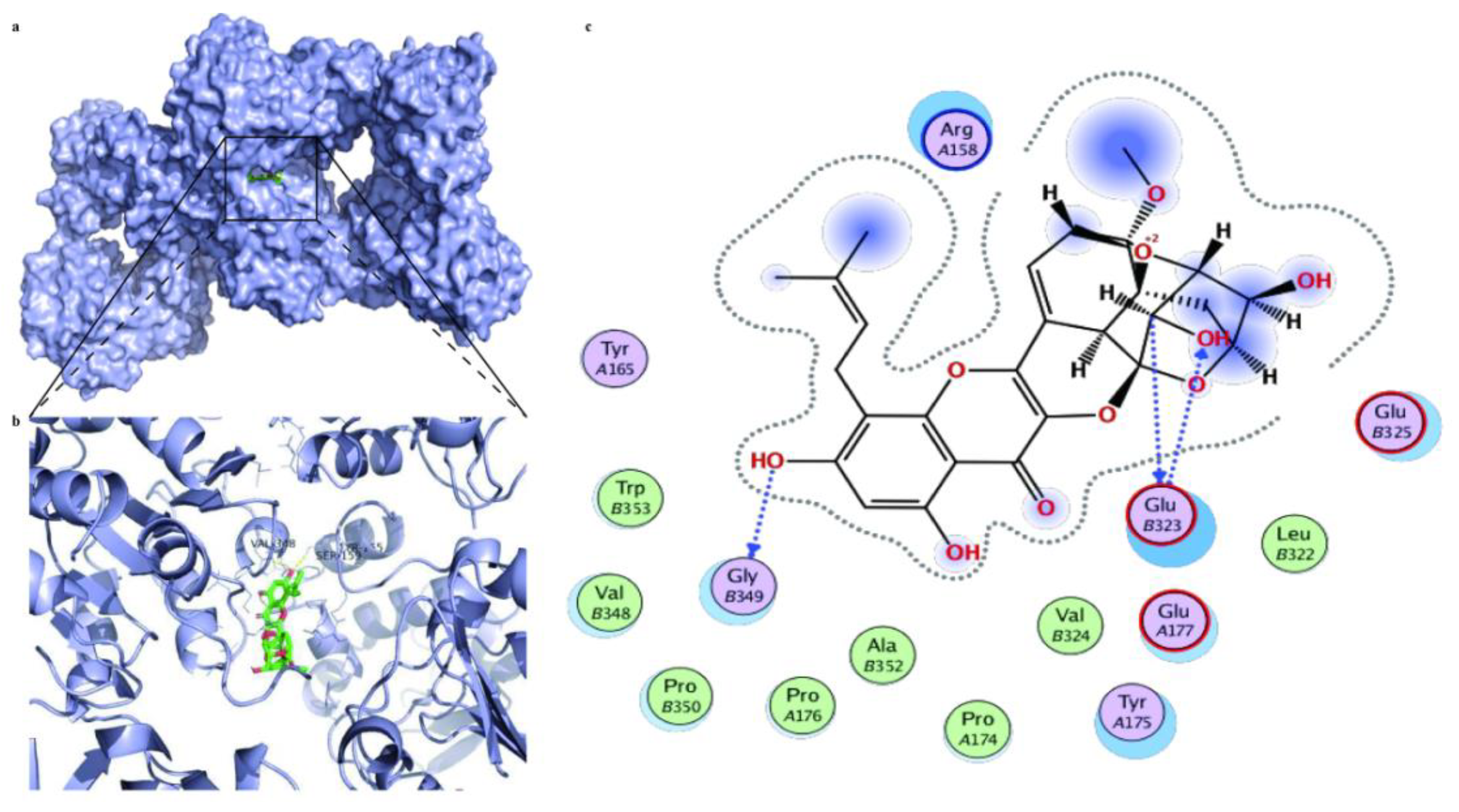
2.16. Molecular Docking Analysis
2.17. Statistical Analyses
3. Results
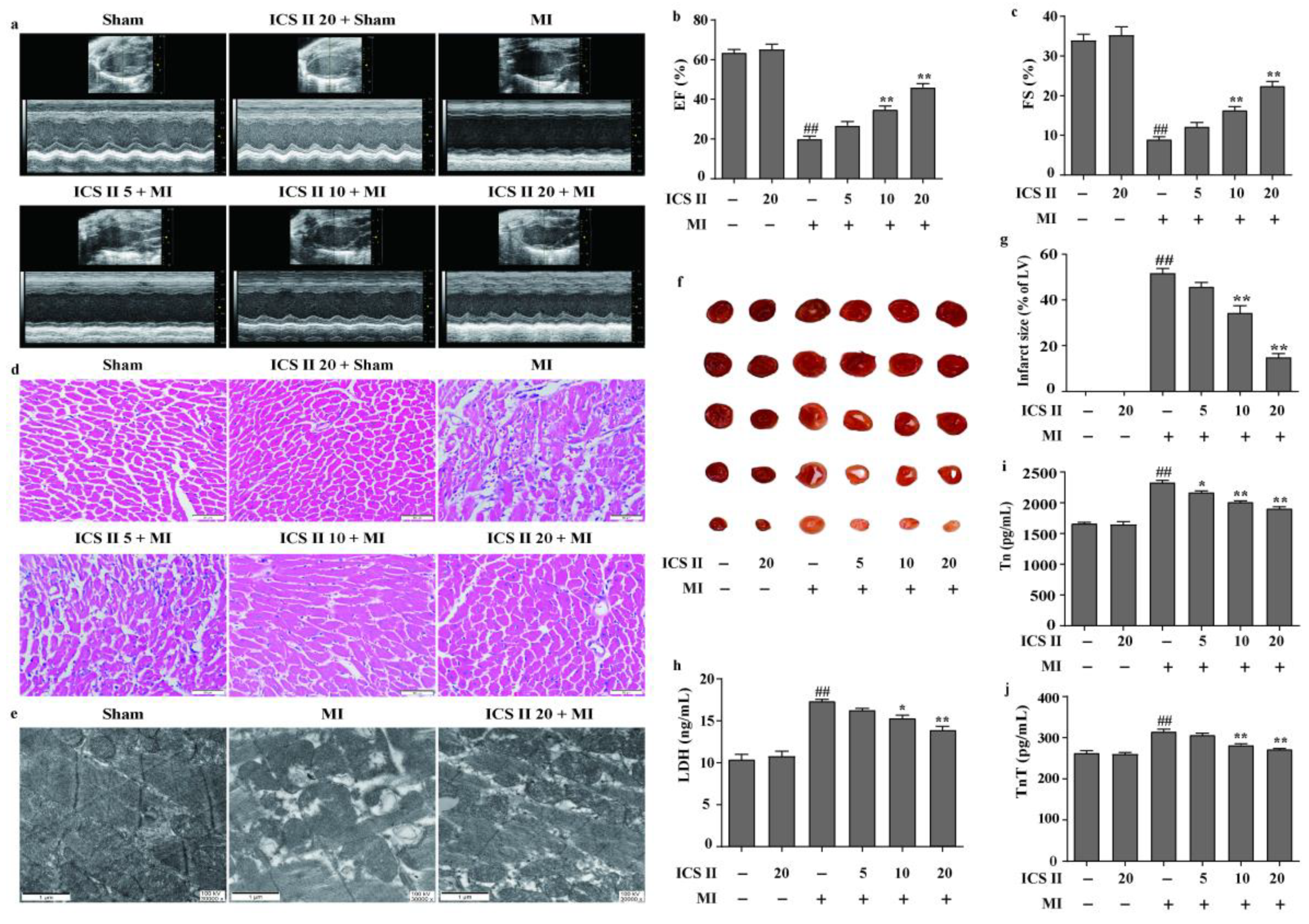
3.1. ICS II Mitigated MI-Induced Myocardial Injury in Mice
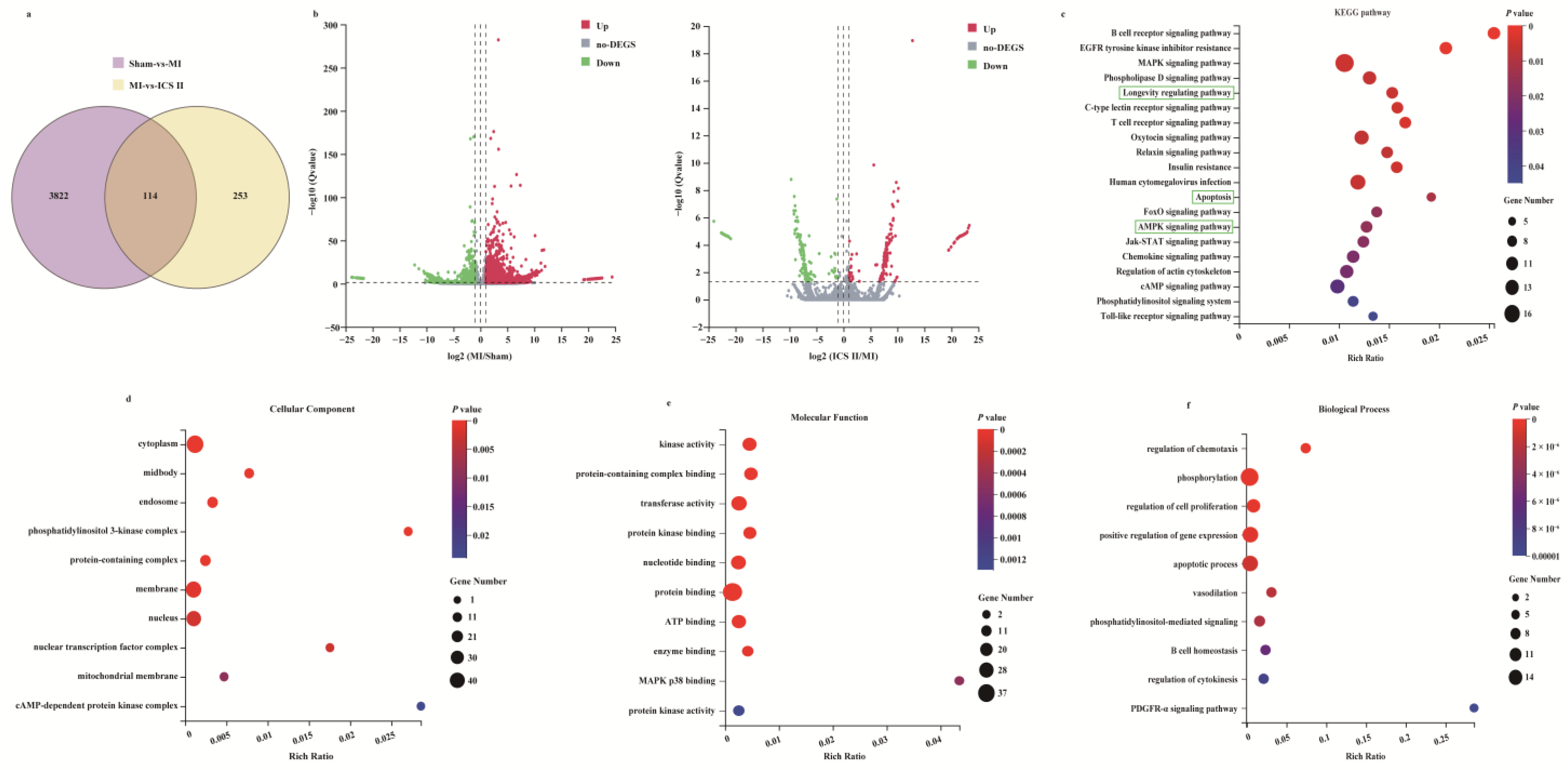
3.2. Microarray Data Analyses
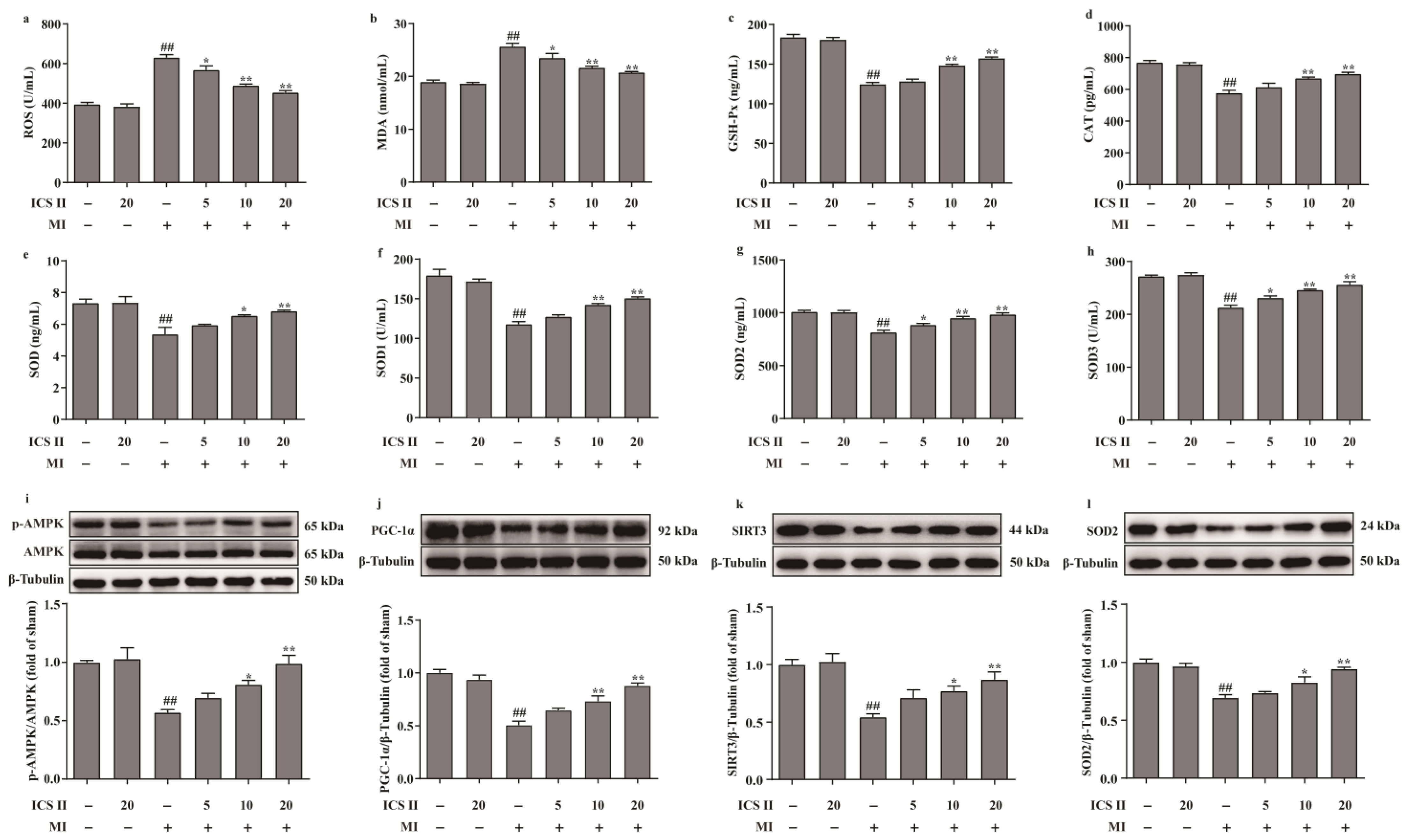
3.3. ICS II Alleviated Oxidative Stress and Activated AMPK/PGC-1α/SIRT3 Signaling Pathway after MI in Mice
3.4. ICS II Inhibited Myocardial Apoptosis in Mice after MI
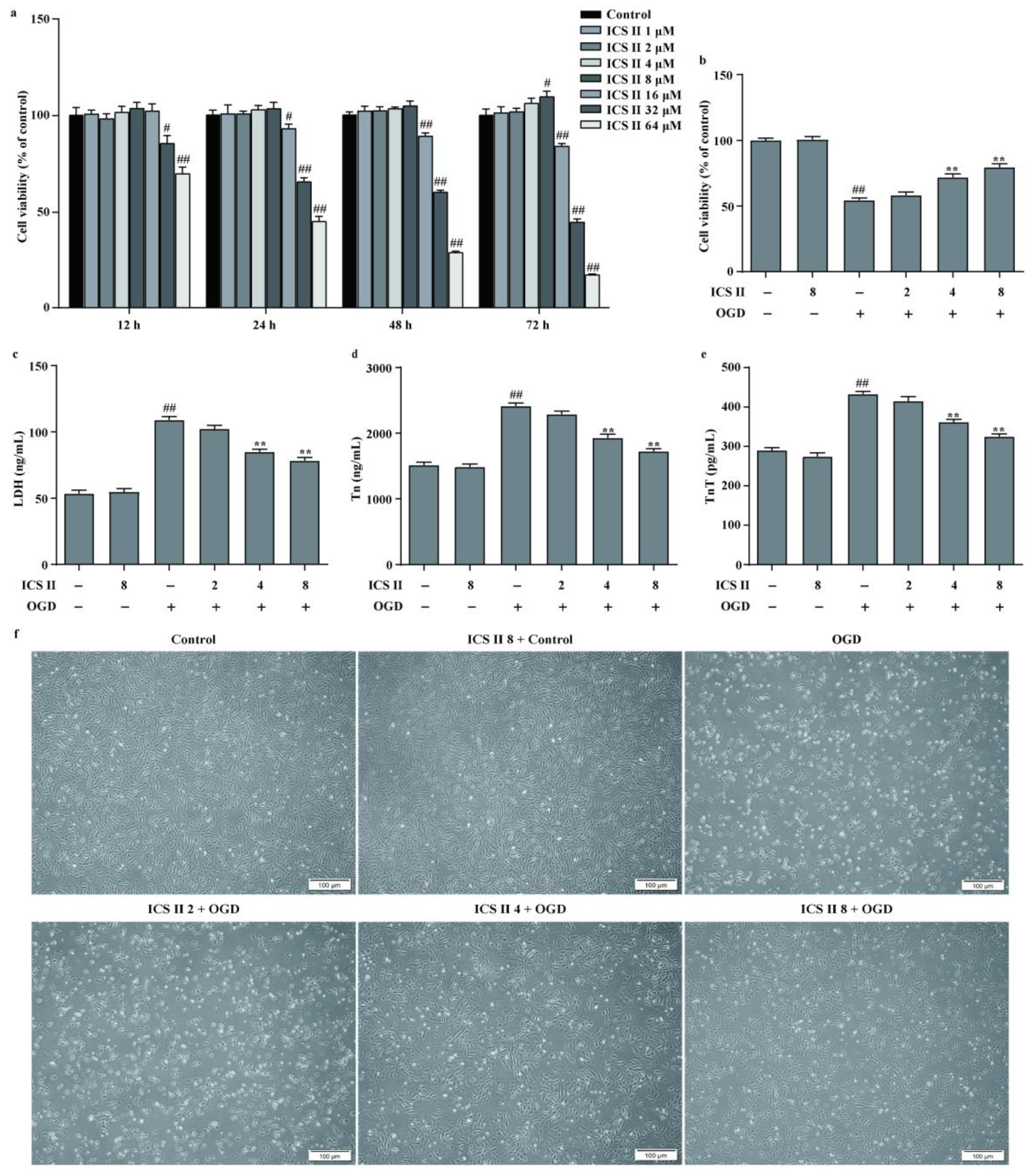
3.5. ICS II Protected against OGD-Induced Cardiomyocyte Injury
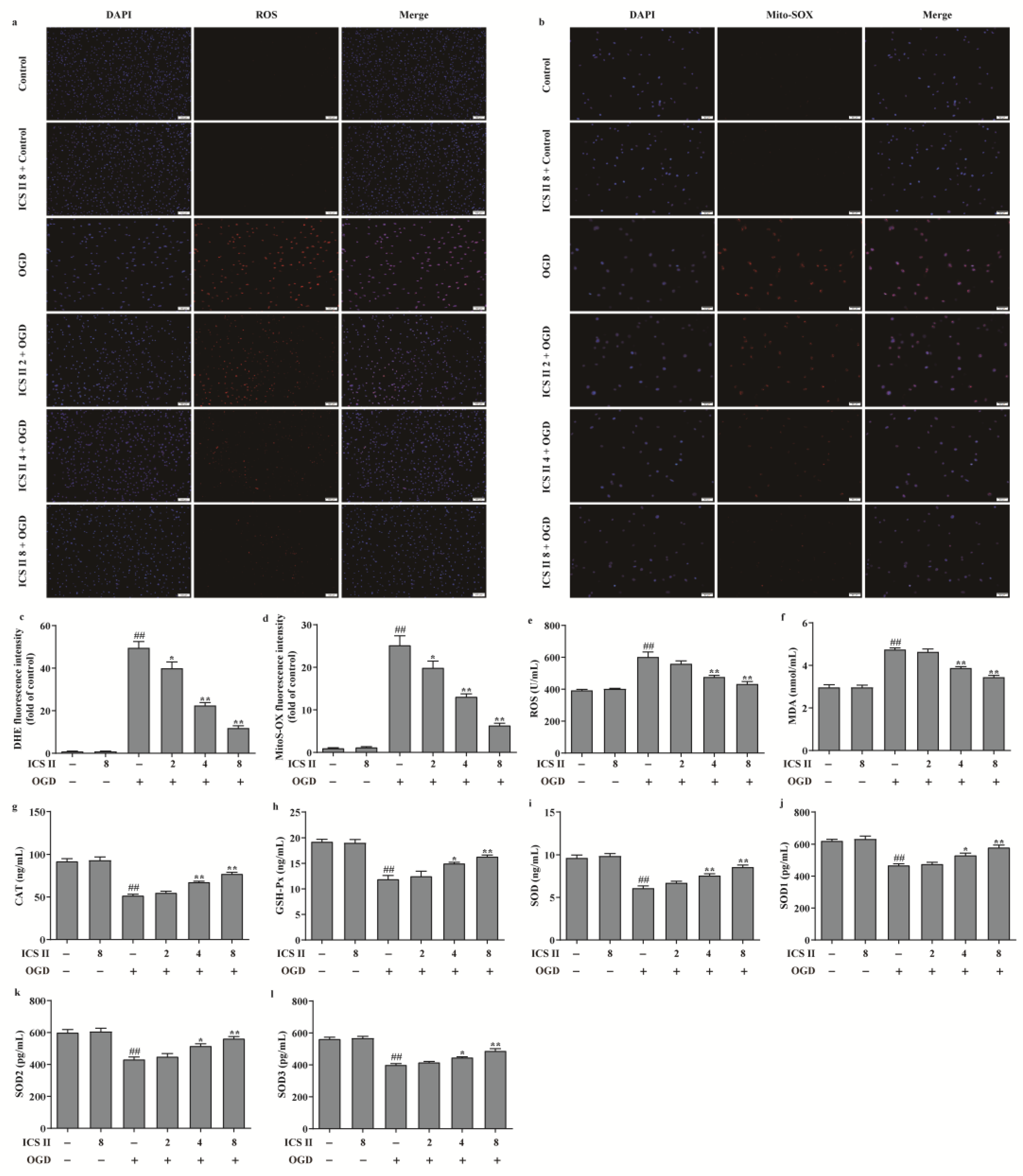
3.6. ICS II Suppressed OGD-Induced Mitochondrial Oxidative Stress by Activating AMPK/PGC-1α/SIRT3 Signaling Pathway
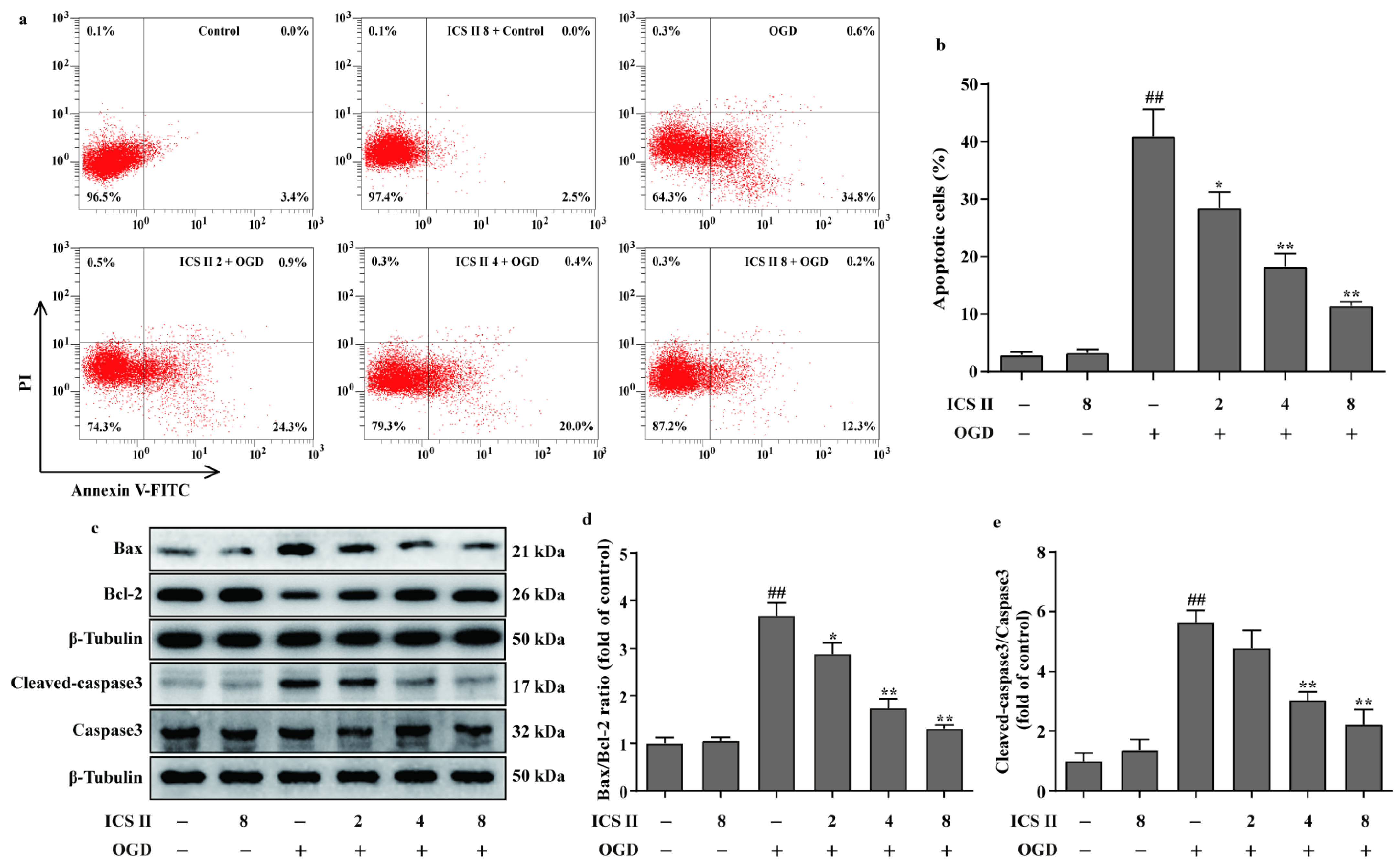
3.7. ICS II Reduced OGD-Induced Cardiomyocytes Apoptosis
3.8. Prediction of Therapeutic Targets of ICS II against MI
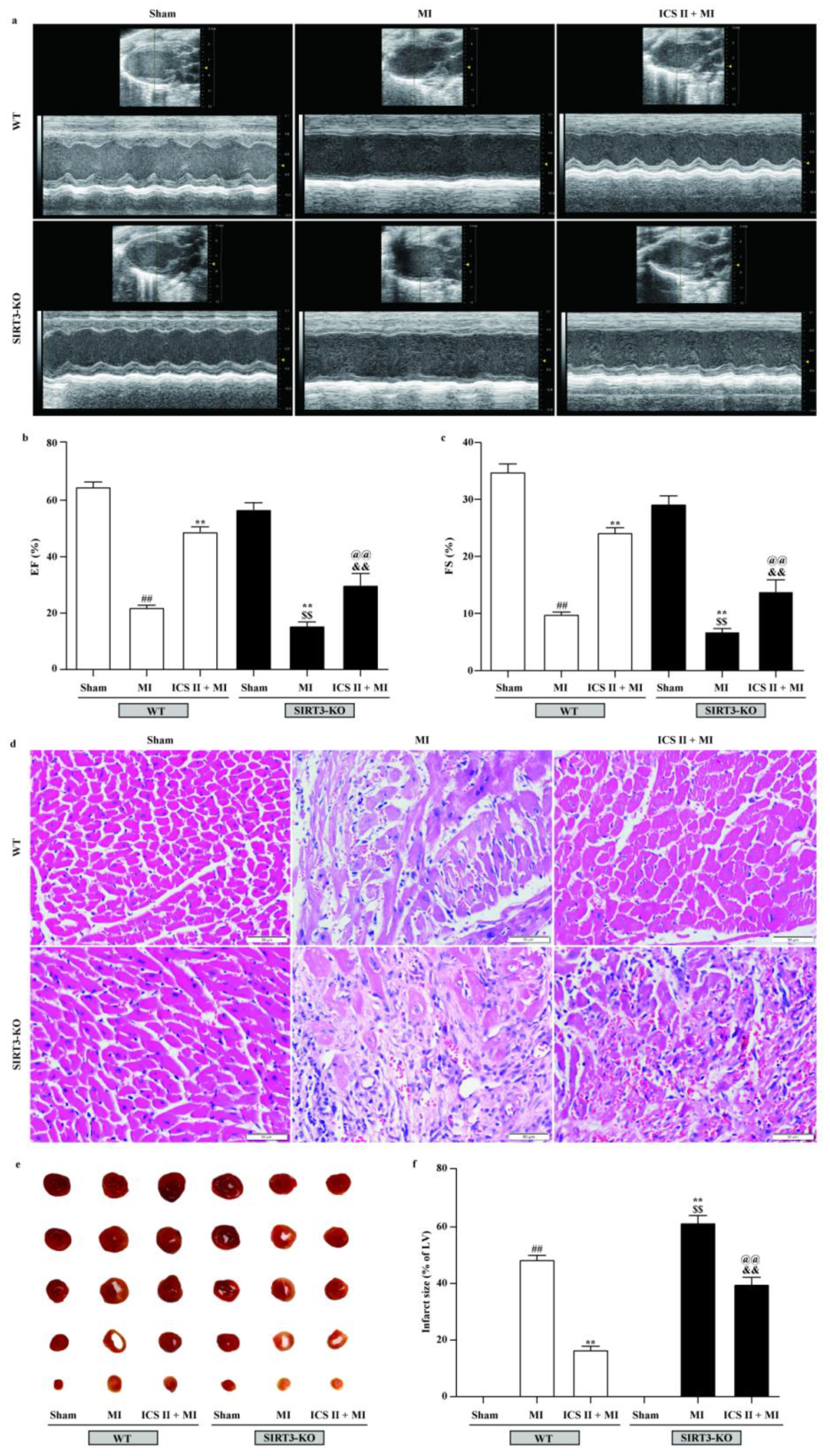
3.9. Effect of ICS II on SIRT3-KO Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramachandra, C.J.A.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Lin, Y.H.; Hausenloy, D.J. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 2020, 57, 102884. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Morrow, D.A. Acute Myocardial Infarction. N. Engl. J. Med. 2017, 376, 2053–2064. [Google Scholar] [CrossRef] [Green Version]
- Marin-Juez, R.; El-Sammak, H.; Helker, C.S.M.; Kamezaki, A.; Mullapuli, S.T.; Bibli, S.I.; Foglia, M.J.; Fleming, I.; Poss, K.D.; Stainier, D.Y.R. Coronary Revascularization during Heart Regeneration Is Regulated by Epicardial and Endocardial Cues and Forms a Scaffold for Cardiomyocyte Repopulation. Dev. Cell 2019, 51, 503–515.e4. [Google Scholar] [CrossRef] [PubMed]
- Rentrop, K.P.; Feit, F. Reperfusion therapy for acute myocardial infarction: Concepts and controversies from inception to acceptance. Am. Heart J. 2015, 170, 971–980. [Google Scholar] [CrossRef]
- Bonaca, M.P.; Sabatine, M.S. Antiplatelet Therapy for Long-Term Secondary Prevention after Myocardial Infarction. JAMA Cardiol. 2016, 1, 627–628. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, H.; Zhao, L.; Zhu, Y.; Bai, R.; Jin, Z.; Fu, Z.; Zhang, X.; Su, J.; Liu, H.; et al. Effects of carbon nanotube-mediated Caspase3 gene silencing on cardiomyocyte apoptosis and cardiac function during early acute myocardial infarction. Nanoscale 2020, 12, 21599–21604. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, L.; Wang, S.; Cheng, H.; Xu, L.; Pei, G.; Wang, Y.; Fu, C.; Jiang, Y.; He, C.; et al. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct. Target Ther. 2022, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.N.; Ju, J.M.; Shabanova, A.; Li, Y.; Fang, R.N.; Sun, J.B.; Guo, Y.Y.; Jin, T.Z.; Liu, Y.Y.; et al. Mzb1 protects against myocardial infarction injury in mice via modulating mitochondrial function and alleviating inflammation. Acta Pharmacol. Sin. 2021, 42, 691–700. [Google Scholar] [CrossRef]
- Wallert, M.; Ziegler, M.; Wang, X.; Maluenda, A.; Xu, X.; Yap, M.L.; Witt, R.; Giles, C.; Kluge, S.; Hortmann, M.; et al. alpha-Tocopherol preserves cardiac function by reducing oxidative stress and inflammation in ischemia/reperfusion injury. Redox Biol. 2019, 26, 101292. [Google Scholar] [CrossRef]
- Kibel, A.; Lukinac, A.M.; Dambic, V.; Juric, I.; Selthofer-Relatic, K. Oxidative Stress in Ischemic Heart Disease. Oxidative Med. Cell. Longev. 2020, 2020, 6627144. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Teringova, E.; Tousek, P. Apoptosis in ischemic heart disease. J. Transl. Med. 2017, 15, 87. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Liu, H.; Yang, D.; He, F.; Yuan, Y.; Guo, J.; Hu, J.; Yu, J.; Yan, X.; Wang, S.; et al. Aloe-emodin attenuates myocardial infarction and apoptosis via up-regulating miR-133 expression. Pharmacol. Res. 2019, 146, 104315. [Google Scholar] [CrossRef]
- Ye, L.; Li, M.; Wang, Z.; Yang, Z.; Zhang, J.; Fang, H.; He, Z.; Wang, X. Depression of Mitochondrial Function in the Rat Skeletal Muscle Model of Myofascial Pain Syndrome Is through Down-Regulation of the AMPK-PGC-1alpha-SIRT3 Axis. J. Pain Res. 2020, 13, 1747–1756. [Google Scholar] [CrossRef]
- Marino, A.; Hausenloy, D.J.; Andreadou, I.; Horman, S.; Bertrand, L.; Beauloye, C. AMP-activated protein kinase: A remarkable contributor to preserve a healthy heart against ROS injury. Free Radic. Biol. Med. 2021, 166, 238–254. [Google Scholar] [CrossRef]
- Kukidome, D.; Nishikawa, T.; Sonoda, K.; Imoto, K.; Fujisawa, K.; Yano, M.; Motoshima, H.; Taguchi, T.; Matsumura, T.; Araki, E. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 2006, 55, 120–127. [Google Scholar] [CrossRef]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef]
- Fu, S.; Li, Y.L.; Wu, Y.T.; Yue, Y.; Qian, Z.Q.; Yang, D.L. Icariside II attenuates myocardial fibrosis by inhibiting nuclear factor-kappaB and the TGF-beta1/Smad2 signalling pathway in spontaneously hypertensive rats. Biomed. Pharmacother. 2018, 100, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.S.; Thakur, K.; Hu, F.; Zhang, J.G.; Wei, Z.J. Icariside II inhibits tumorigenesis via inhibiting AKT/Cyclin E/CDK 2 pathway and activating mitochondria-dependent pathway. Pharmacol. Res. 2020, 152, 104616. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Deng, Y.; Gao, J.M.; Lv, C.; Lang, L.H.; Shi, J.S.; Yu, C.Y.; Gong, Q.H. Icariside II inhibits lipopolysaccharide-induced inflammation and amyloid production in rat astrocytes by regulating IKK/IkappaB/NF-kappaB/BACE1 signaling pathway. Acta Pharmacol. Sin. 2020, 41, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.B.; Wang, W.; Gao, J.M.; Li, F.; Shi, J.S.; Gong, Q.H. Icariside II attenuates cerebral ischemia/reperfusion-induced blood-brain barrier dysfunction in rats via regulating the balance of MMP9/TIMP1. Acta Pharmacol. Sin. 2020, 41, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Gao, J.; Liu, Y.; Shi, J.; Gong, Q. Icariside II alleviates oxygen-glucose deprivation and reoxygenation-induced PC12 cell oxidative injury by activating Nrf2/SIRT3 signaling pathway. Biomed. Pharmacother. 2018, 103, 9–17. [Google Scholar] [CrossRef]
- Wu, Y.; Qian, Z.; Fu, S.; Yue, Y.; Li, Y.; Sun, R.; Huang, B.; Yang, D. IcarisideII improves left ventricular remodeling in spontaneously hypertensive rats by inhibiting the ASK1-JNK/p38 signaling pathway. Eur. J. Pharmacol. 2018, 819, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.F.; Dai, X.F.; Huang, Q.B.; Zhao, D.; Shi, J.L.; Chen, C.; Zhu, Y.; Ai, F. Icariside II ameliorates myocardial ischemia and reperfusion injury by attenuating inflammation and apoptosis through the regulation of the PI3K/AKT signaling pathway. Mol. Med. Rep. 2020, 22, 3151–3160. [Google Scholar] [CrossRef]
- Gao, E.; Lei, Y.H.; Shang, X.; Huang, Z.M.; Zuo, L.; Boucher, M.; Fan, Q.; Chuprun, J.K.; Ma, X.L.; Koch, W.J. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ. Res. 2010, 107, 1445–1453. [Google Scholar] [CrossRef]
- Gao, J.; Long, L.; Xu, F.; Feng, L.; Liu, Y.; Shi, J.; Gong, Q. Icariside II, a phosphodiesterase 5 inhibitor, attenuates cerebral ischaemia/reperfusion injury by inhibiting glycogen synthase kinase-3beta-mediated activation of autophagy. Br. J. Pharmacol. 2020, 177, 1434–1452. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.L.; Ding, Z.W.; Yin, P.P.; Wu, J.; Hu, K.; Sun, A.J.; Zou, Y.Z.; Ge, J.B. Hypertrophic preconditioning cardioprotection after myocardial ischaemia/reperfusion injury involves ALDH2-dependent metabolism modulation. Redox Biol. 2021, 43, 101960. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, N.; Li, N.; Xu, F.; Wang, W.; Lei, Y.; Shi, J.; Gong, Q. Neuroprotective Effects of Trilobatin, a Novel Naturally Occurring Sirt3 Agonist from Lithocarpus polystachyus Rehd., Mitigate Cerebral Ischemia/Reperfusion Injury: Involvement of TLR4/NF-kappaB and Nrf2/Keap-1 Signaling. Antioxid. Redox Signal 2020, 33, 117–143. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Muntean, D.M.; Sturza, A.; Danila, M.D.; Borza, C.; Duicu, O.M.; Mornos, C. The Role of Mitochondrial Reactive Oxygen Species in Cardiovascular Injury and Protective Strategies. Oxidative Med. Cell. Longev. 2016, 2016, 8254942. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Li, J.; Li, J.; Pan, C.; Xiao, Y.; Lan, X.; Wang, M. Activation of AMPK/Sirt3 pathway by phloretin reduces mitochondrial ROS in vascular endothelium by increasing the activity of MnSOD via deacetylation. Food Funct. 2020, 11, 3073–3083. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, X.; Huang, D. Mfn2 Overexpression Attenuates Cardio-Cerebrovascular Ischemia-Reperfusion Injury Through Mitochondrial Fusion and Activation of the AMPK/Sirt3 Signaling. Front. Cell Dev. Biol. 2020, 8, 598078. [Google Scholar] [CrossRef] [PubMed]
- Caballero, E.P.; Mariz-Ponte, N.; Rigazio, C.S.; Santamaria, M.H.; Corral, R.S. Honokiol attenuates oxidative stress-dependent heart dysfunction in chronic Chagas disease by targeting AMPK/NFE2L2/SIRT3 signaling pathway. Free Radic. Biol. Med. 2020, 156, 113–124. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Feng, L.; Xie, D.; Lin, M.; Li, Y.; Chen, N.; Yang, D.; Gao, J.; Zhu, Y.; Gong, Q. Icariside II, a Naturally Occurring SIRT3 Agonist, Protects against Myocardial Infarction through the AMPK/PGC-1α/Apoptosis Signaling Pathway. Antioxidants 2022, 11, 1465. https://doi.org/10.3390/antiox11081465
Li Y, Feng L, Xie D, Lin M, Li Y, Chen N, Yang D, Gao J, Zhu Y, Gong Q. Icariside II, a Naturally Occurring SIRT3 Agonist, Protects against Myocardial Infarction through the AMPK/PGC-1α/Apoptosis Signaling Pathway. Antioxidants. 2022; 11(8):1465. https://doi.org/10.3390/antiox11081465
Chicago/Turabian StyleLi, Yeli, Linying Feng, Dianyou Xie, Mu Lin, Yiqi Li, Nana Chen, Danli Yang, Jianmei Gao, Yizhun Zhu, and Qihai Gong. 2022. "Icariside II, a Naturally Occurring SIRT3 Agonist, Protects against Myocardial Infarction through the AMPK/PGC-1α/Apoptosis Signaling Pathway" Antioxidants 11, no. 8: 1465. https://doi.org/10.3390/antiox11081465
APA StyleLi, Y., Feng, L., Xie, D., Lin, M., Li, Y., Chen, N., Yang, D., Gao, J., Zhu, Y., & Gong, Q. (2022). Icariside II, a Naturally Occurring SIRT3 Agonist, Protects against Myocardial Infarction through the AMPK/PGC-1α/Apoptosis Signaling Pathway. Antioxidants, 11(8), 1465. https://doi.org/10.3390/antiox11081465