Cellular and Mitochondrial Effects of Alcohol Consumption

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Alcoholism
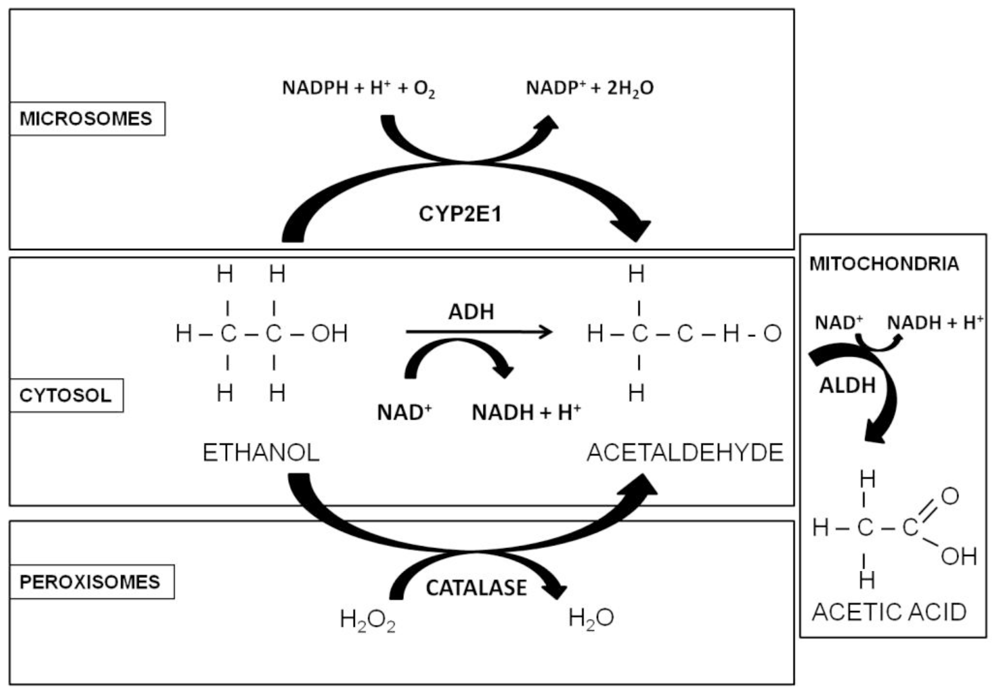
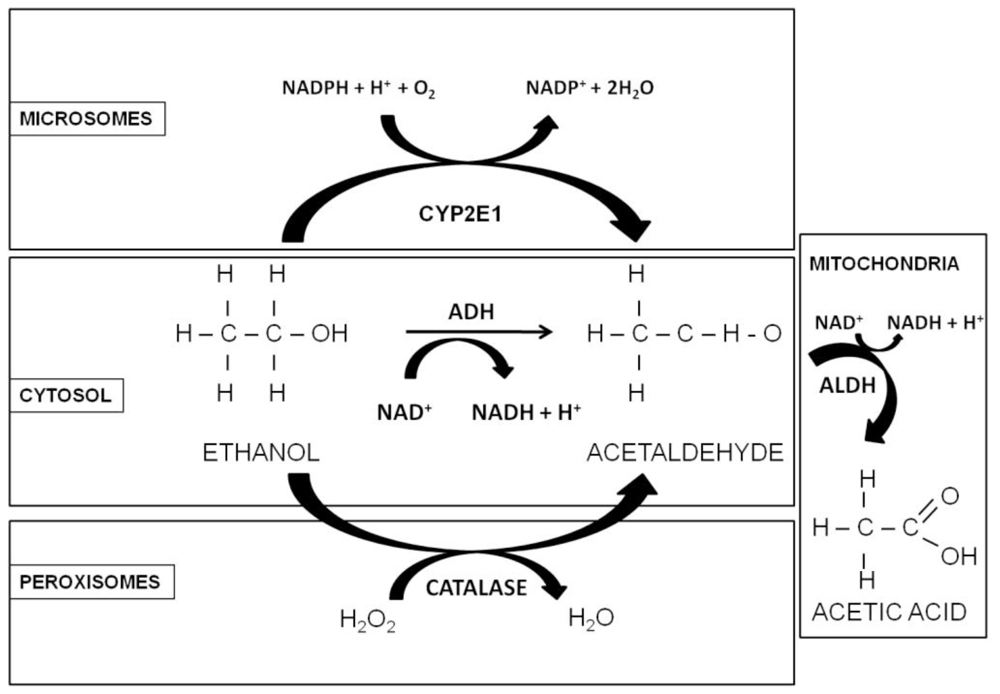
1.2. Alcohol Metabolism
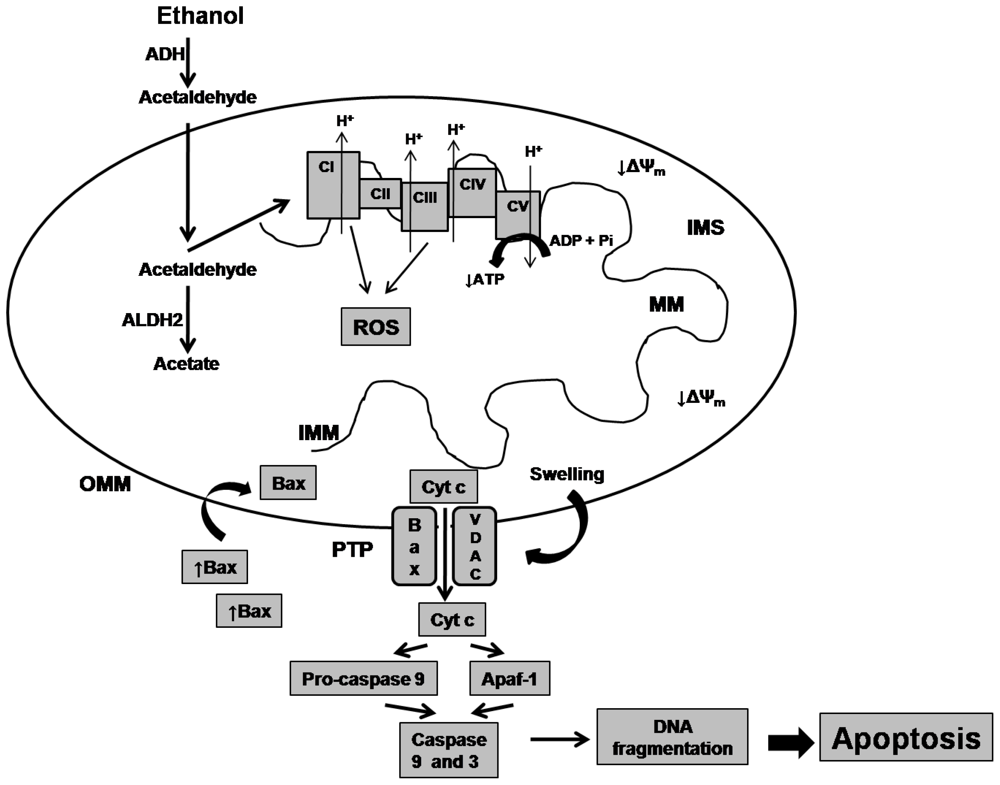
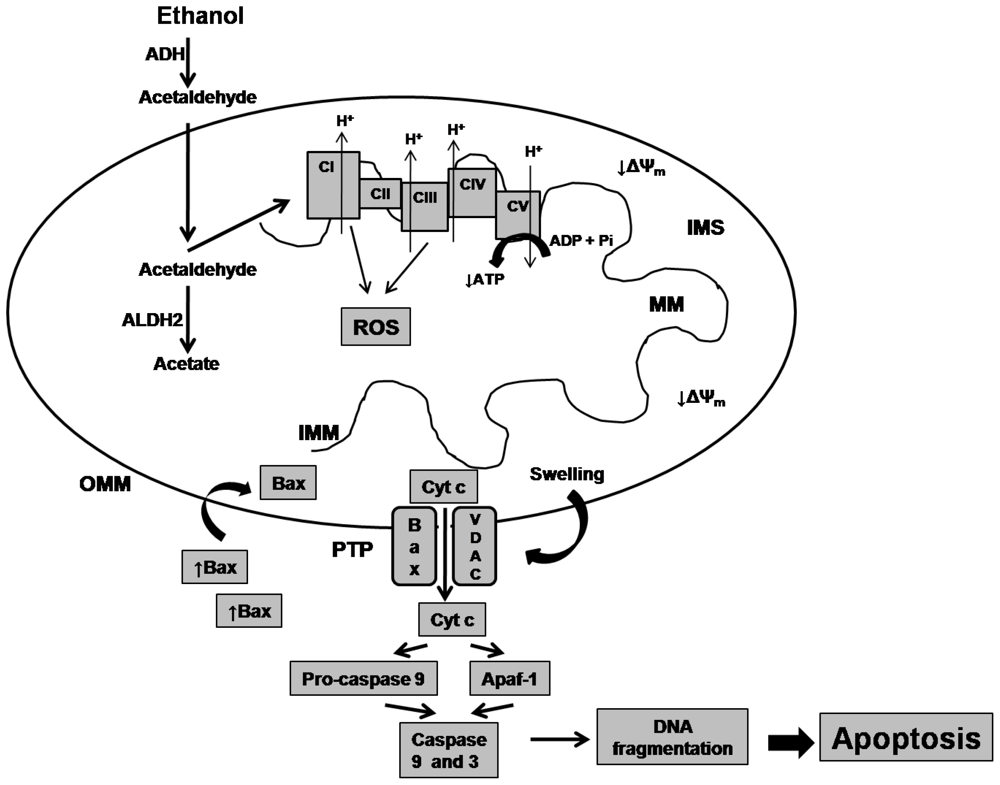
2. Effects of Alcohol on Mitochondrial Biomolecules
3. Alcohol Effects on the Heart
4. Alcohol Effects on the Stomach
5. Alcohol Effects on the Liver
6. Alcohol Effects on the Nervous System
7. Alcohol and Prenatal Effects
8. Alcoholism Therapeutics at the Mitochondrial Level
9. Concluding Remarks
Acknowledgements
References
- Guo, R; Ren, J. Alcohol and acetaldehyde in public health: From marvel to menace. Int. J. Environ. Res. Public Health 2010, 7, 1285–1301. [Google Scholar]
- Mokdad, AH; Marks, JS; Stroup, DF; Gerberding, JL. Actual causes of death in the United States. JAMA 2000, 291, 1238–1245. [Google Scholar]
- Diagnostic and Statistical Manual of Mental Disorders, 4th ed; American Psychiatric Association: Washington, DC, USA, 1994.
- Grant, BF; Stinson, FS; Harford, TC. Age at onset of alcohol use and DSM-IV alcohol abuse and dependence: A 12-year follow-up. J. Substain. Abuse 2001, 13, 493–504. [Google Scholar]
- George, A; Figueredo, VM. Alcohol and arrhythmias: A comprehensive review. J. Cardiov. Med. (Hagerstown) 2010, 11, 221–228. [Google Scholar]
- Marinho, V; Laks, J; Engelhardt, E; Conn, D. Alcohol abuse in an elderly woman taking donepezil for Alzheimer disease. J. Clin. Psychopharmacol 2006, 26, 683–685. [Google Scholar]
- Ohkubo, T; Metoki, H; Imai, Y. Alcohol intake, circadian blood pressure variation, and stroke. Hypertension 2009, 53, 4–5. [Google Scholar]
- Cederbaum, AI; Lu, Y; Wu, D. Role of oxidative stress in alcohol-induced liver injury. Arch. Toxicol 2009, 83, 519–548. [Google Scholar]
- Seitz, HK; Becker, P. Alcohol metabolism and cancer risk. Alcohol Res. Health 2007, 30, 38–47. [Google Scholar]
- Morris, MJ. Alcohol breath testing in patients with respiratory disease. Thorax 1990, 45, 717–721. [Google Scholar]
- Baliunas, DO; Taylor, BJ; Irving, H; Roerecke, M; Patra, J; Mohapatra, S; Rehm, J. Alcohol as a risk factor for type 2 diabetes: A systematic review and meta-analysis. Diabetes Care 2009, 32, 2123–2132. [Google Scholar]
- Chen, Y; Cui, L; Liao, J; huang, L. Effects of alcohol on bone metabolism and biomechanical property of mice. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi 2009, 26, 780–782. [Google Scholar]
- Rehm, J; Mathers, C; Popova, S; Thavorncharoensap, M; Teerawattananon, Y; Patra, J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet 2009, 373, 2223–2233. [Google Scholar]
- Gohlke, JM; Griffith, WC; Faustman, EM. Computational models of ethanol-induced neurodevelopmental toxicity across species: Implications for risk assessment. Birth Defects Res. B. Dev. Reprod. Toxicol 2008, 83, 1–11. [Google Scholar]
- Pietrzykowski, AZ; Friesen, RM; Martin, GE; Puig, SI; Nowak, CL; Wynne, PM; Siegelmann, HT; Treistman, SN. Posttranscriptional regulation of BK channel splice variant stability by miR-9 underlies neuroadaptation to alcohol. Neuron 2008, 59, 274–287. [Google Scholar]
- Roberto, M; Treistman, SN; Pietrzykowski, AZ; Weiner, J; Galindo, R; Mameli, M; Valenzuela, F; Zhu, PJ; Lovinger, D; Zhang, TA; Hendricson, AH; Morrisett, R; Siggins, GR. Actions of acute and chronic ethanol on presynaptic terminals. Alcohol Clin. Exp. Res 2006, 30, 222–232. [Google Scholar]
- Wilkie, MB; Besheer, J; Kelley, SP; Kumar, S; O’Buckley, TK; Morrow, AL; Hodge, CW. Acute ethanol administration rapidly increases phosphorylation of conventional protein kinase C in specific mammalian brain regions in vivo. Alcohol Clin. Exp. Res 2007, 31, 1259–1267. [Google Scholar]
- Le Marquand, D; Pihl, RO; Benkelfat, C. Serotonin and alcohol intake, abuse, and dependence: Clinical evidence. Biol. Psychiatry 1994, 36, 326–337. [Google Scholar]
- Prosser, RA; Mangrum, CA; Glass, JD. Acute ethanol modulates glutamatergic and serotonergic phase shifts of the mouse circadian clock in vitro. Neuroscience 2008, 152, 837–848. [Google Scholar]
- Wallner, M; Hanchar, HJ; Olsen, RW. Low-dose alcohol actions on alpha4beta3delta GABAA receptors are reversed by the behavioral alcohol antagonist Ro15-4513. Proc. Natl. Acad. Sci. USA 2006, 103, 8540–8545. [Google Scholar]
- Perra, S; Pillolla, G; Luchicchi, A; Pistis, M. Alcohol inhibits spontaneous activity of basolateral amigdala proyection neurons in the rat: Involvement of the endocannabinoid system. Alcohol Clin. Exp. Res 2008, 32, 443–449. [Google Scholar]
- Fisher, SJ; Lee, IJ; Swaan, PW; Eddington, ND. Evaluation of the effect of ethanol’s toxic metabolite acetaldehyde on the gastrointestinal oligopeptide transporter, PEPT1: In vitro and in vivo studies. Alcohol Clin. Exp. Res 2008, 32, 162–170. [Google Scholar]
- Karinch, AM; Martin, JH; Vary, TC. Acute and chronic ethanol consumption differentially impact pathways limiting hepatic protein synthesis. Am. J. Physiol. Endocrinol. Metab 2008, 295, E3–E9. [Google Scholar]
- Yang, AL; Vadhavkar, S; Singh, G; Omary, MB. Epidemiology of alcohol-related liver and pancreatic disease in the United States. Arch. Intern. Med 2008, 168, 649–656. [Google Scholar]
- Ting, JW; Lautt, WW. The effect of acute, chronic, and prenatal ethanol exposure on insulin sensitivity. Pharmacol. Ther 2006, 111, 346–373. [Google Scholar]
- Happel, KI; Rudner, X; Quinton, LJ; Movasshaghi, JL; Clark, C; Odden, AR; Zhang, P; Bagby, GJ; Nelson, S; Shellito, JE. Acute alcohol intoxication suppresses the pulmonary ELR-negative CXC chemokine response to lipopolysaccharide. Alcohol 2007, 41, 325–333. [Google Scholar]
- Greiffensstein, P; Mathis, KW; Stouwe, CV; Molina, PE. Alcohol binge before trauma/hemorrhage impairs integrity of host defense mechanisms during recovery. Alcohol Clin. Exp. Res 2007, 31, 704–715. [Google Scholar]
- Choudhry, MA; Li, X; Chaudry, IH. A role for corticosterone in impaired intestinal immunity and barrier function in a rodent model of acute alcohol intoxication and burn injury. J. Neuroimmune Pharmacol 2006, 1, 428–434. [Google Scholar]
- Radek, KA; Kovacs, EJ; DiPietro, LA. Matrix proteolytic activity during wound healing: Modulation by acute ethanol exposure. Alcohol Clin. Exp. Res 2007, 31, 1045–1052. [Google Scholar]
- Dolganiuc, A; Szabo, G. In vitro and in vivo models of acute alcohol exposure. World J. Gastroenterol 2009, 15, 1168–1177. [Google Scholar]
- Werner, DF; Swihart, AR; Ferguson, C; Lariviere, WR; Harrison, NL; Homanics, GE. Alcohol-induced tolerance and physical dependence in mice with ethanol insensitive α1 GABAA receptors. Alcohol Clin. Exp. Res 2009, 33, 289–299. [Google Scholar]
- Norberg, A; Jones, AW; Hahn, RG; Gabrielsson, JL. Role of variability in explaining ethanol pharmacokinetics: Research and forensic applications. Clin. Pharmacokinet 2003, 42, 1–31. [Google Scholar]
- Holford, NHG. Clinical pharmacokinetics of ethanol. Clin. Pharmacokinet 1987, 13, 273–292. [Google Scholar]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol Res. Health 2006, 29, 245–254. [Google Scholar]
- Agarwal, DP. Genetic polymorphisms of alcohol metabolizing enzymes. Pathol. Biol 2001, 49, 703–709. [Google Scholar]
- Gemma, S; Vichi, S; Testai, E. Individual susceptibility and alcohol effects: Biochemical and genetic aspects. Ann. Ist. Super. Sanita 2006, 42, 8–16. [Google Scholar]
- Weiner, H. Subcellular localization of acetaldehyde oxidation on liver. Ann. NY Acad. Sci 1987, 492, 25–34. [Google Scholar]
- Klein, H; Harmjanz, D. Effect of ethanol infusion on the ultrastructure of human myocardium. Postgrad. Med. J 1975, 51, 325–329. [Google Scholar]
- Regan, TJ. Alcohol and the cardiovascular system. JAMA 1990, 264, 377–381. [Google Scholar]
- Pachinger, O; Mao, J; Fauvel, J-M; Bing, RJ. Fleckstein, A, Dhalla, NS, Eds.; Mitochondrial function and excitation-contraction coupling in the development of alcoholic cardiomypathy. In Recent Advances in Studies on Cardiac Structure and Metabolism; University Park Press: Baltimore, MD, USA, 1975; Volume 5, pp. 423–429. [Google Scholar]
- Albano, E. Sherman, VR, Watson, RR, Eds.; Free radicals and alcohol-induced liver injury. In Ethanol and the Liver; Taylor and Francis: London, UK, 2002; pp. 153–190. [Google Scholar]
- Robin, MA; Sauvage, I; Grandperret, T; Descatoire, V; Pessayre, D; Fromenty, B. Ethanol increases mitochondrial cytochrome P450 2E1 in mouse liver and rat hepatocytes. FEBS Lett 2005, 579, 6895–6902. [Google Scholar]
- Cederbaum, AI. Microsomal generation of reactive oxygen species and their possible role in alcohol hepatoxicity. Alcohol Alcohol 1991, (Suppl 1), 291–296. [Google Scholar]
- Adam-Vizi, V. Production of reactive oxygen species in brain mitochondria: Contribution by electron transport chain and non-electron transport chain sources. Antioxid. Redox Signal 2005, 7, 1140–1149. [Google Scholar]
- Ronis, MJJ; Lindros, KO; Ingelman-Sundberg, M. Ioannides, C, Ed.; The CYP2E family. In Cytochromes P450: Metabolic and Toxicological Aspects; CRC Press: Boca Raton, FL, USA, 1996; pp. 211–239. [Google Scholar]
- Liangpunsakul, S; Kolwankar, D; Pinto, A; Gorski, CJ; Hall, SD; Chalasani, N. Activity of CYP2E1 and CYP3A enzymes in adults with moderate alcohol consumption: A comparison with nonalcoholics. Hepatology 2005, 41, 1144–1150. [Google Scholar]
- Albano, E. Alcohol, oxidative stress and free radical damage. Proc. Nutr. Soc 2006, 65, 278–290. [Google Scholar]
- Bailey, SM; Pietsch, EC; Cunningham, CC. Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III. Free Radic. Biol. Med 1999, 27, 891–900. [Google Scholar]
- Cunningham, CC; Coleman, WB; Spach, PI. The effects of chronic ethanol consumption on hepatic mitochondrial energy metabolism. Alcohol Alcohol 1990, 25, 127–136. [Google Scholar]
- Bailey, SM; Cunningham, CC. Effect of dietary fat on chronic ethanol-induced oxidative stress in hepatocytes. Alcohol Clin. Exp. Res 1999, 23, 1210–1218. [Google Scholar]
- Bailey, SM. A review of the role of reactive oxygen and nitrogen species in alcohol-induced mitochondrial energy metabolism. Free Radic. Res 2003, 37, 585–596. [Google Scholar]
- Mantena, SK; King, AL; Andringa, KK; Landar, A; Darley-Usmar, V; Bailey, SM. Novel interactions of mitochondria and reactive oxygen/nitrogen species in alcohol mediated liver disease. World J. Gastroenterol 2007, 13, 4967–4973. [Google Scholar]
- Hoek, JB; Cahill, A; Pastorino, JG. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology 2002, 122, 2049–2063. [Google Scholar]
- Coleman, WB; Cunningham, CC. Effect of chronic ethanol consumption on hepatic mitochondrial transcription and translation. Biochim. Biophys. Acta 1991, 1058, 178–186. [Google Scholar]
- Venkatraman, A; Landar, A; Davis, AJ; Chamlee, L; Sanderson, T; Kim, H; Page, G; Pompilius, M; Ballinger, S; Darley-Usmar, V; Bailey, SM. Modification of the mitochondrial proteome in response to the stress of ethanol-dependent hepatotoxicity. J. Biol. Chem 2004, 279, 22092–22101. [Google Scholar]
- Bailey, SM; Robinson, G; Pinner, A; Chamlee, L; Ulasova, E; Pompilius, M; Page, GP; Chhieng, D; Jhala, N; Landar, A; Kharbanda, KK; Ballinger, S; Darley-Usmar, V. S-adenosylmethionine prevents chronic alcohol-induced mitochondrial dysfunction in the rat liver. Am. J. Physiol. Gastrointest. Liver Physiol 2006, 291, G857–G867. [Google Scholar]
- Radi, R; Cassina, A; Hodara, R; Quijano, C; Castro, L. Peroxynitrite reactions and formation in mitocondria. Free Radic. Biol. Med 2002, 33, 1451–1464. [Google Scholar]
- Brookes, PS; Kraus, DW; Shiva, S; Doeller, JE; Barone, MC; Patel, RP; Lancaster, JR, Jr; Darley-Usmar, V. Control of mitochondrial respiration by NO., effects of low oxygen and respiratory state. J. Biol. Chem 2003, 278, 31603–31609. [Google Scholar]
- Moon, KH; Hood, BL; Kim, BJ; Hardwick, JP; Conrads, TP; Veenstra, TD; Song, BJ. Inactivation of oxidized and S-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology 2006, 44, 1218–1230. [Google Scholar]
- Matsuhashi, T; Karbowski, M; Liu, X; Usukura, J; Wozniak, M; Wakabayashi, T. Complete suppression of ethanol-induced formation of megamitochondria by 4-hydroxy-2,2,6,6,-tetramethyl-piperidine-1-oxyl (4-OH-TEMPO). Free Radic. Biol. Med 1998, 24, 139–147. [Google Scholar]
- Meager, EA; Barry, OP; Burke, A; Lucey, MR; Lawson, JA; Rokach, J; FitzGerald, GA. Alcohol-induced generation of lipid peroxidation products in humans. J. Clin. Invest 1999, 104, 805–813. [Google Scholar]
- Niemela, O; Parkkila, S; Yla-Herttuala, S; Halsted, C; Witztum, JL; Lanca, A; Israel, Y. Covalent protein adducts in the liver as a result of ethanol metabolism and lipid peroxidation. Lab. Invest 1994, 70, 537–546. [Google Scholar]
- Slater, TF. Free-radical mechanisms in tissue injury. Biochem. J 1984, 222, 1–15. [Google Scholar]
- Cortes-Rojo, C; Calderon-Cortes, E; Clemente-Guerrero, M; Estrada-Villagomez, M; Manzo-Avalos, S; Mejia-Zepeda, R; Boldogh, I; Saavedra-Molina, A. Elucidation of the effects of lipoperoxidation on the mitochondrial electron transport chain using yeast mitochondria with manipulated fatty acid content. J. Bioenerg. Biomembr 2009, 41, 15–28. [Google Scholar]
- Fukumura, A; Tsutsumi, M; Tsuchishima, M; Takase, S. Correlation between adenosine triphosphate content and apoptosis in liver of rats treated with alcohol. Alcohol Clin. Exp. Res 2003, 27, 12S–15S. [Google Scholar]
- Mansouri, A; Fromenty, B; Berson, A; Robin, MA; Grimbert, S; Beaugrand, M; Erlinger, S; Pessayre, D. Multiple hepatic mitochondrial DNA deletions suggest premature oxidative aging in alcoholic patients. J. Hepatol 1997, 27, 96–102. [Google Scholar]
- Tsuchishima, M; Tsutsumi, M; Shiroeda, H; Yano, H; Ueshima, Y; Shimanaka, K; Takase, S. Study of mitochondrial DNA deletion in alcoholics. Alcohol Clin. Exp. Res 2000, 24, 12S–15S. [Google Scholar]
- Von Wurmb-Schwark, N; Ringleb, A; Schwark, T; Broese, T; Weirich, S; Schlaefke, D; Wegener, R; Oehmichen, M. The effect of chronic alcohol consumption on mitochondrial DNA mutagenesis in human blood. Mutat. Res 2008, 637, 73–79. [Google Scholar]
- Hirano, T; Kaplowitz, N; Kamimura, T; Tsukamoto, H; Fernandez-Checa, JC. Hepatic mitocondrial GSH depletion and progression of experimental alcoholic liver disease in rats. Hepatology 1992, 16, 1423–1428. [Google Scholar]
- Fernandez-Checa, JC; Kaplowitz, N. Hepatic mitochondrial glutathione: Transport and role in disease and toxicity. Toxicol. App. Pharmacol 2005, 204, 263–273. [Google Scholar]
- Rouach, H; Fattaccioli, V; Gentil, M; French, SW; Morimoto, M; Nordmann, R. Effect of chronic ethanol feeding on lipid peroxidation and protein oxidation in relation to liver pathology. Hepatology 1997, 25, 351–355. [Google Scholar]
- Kowaltowski, AJ; Castillo, RF; Vercesi, AF. Mitochondrial permeability transition and oxidative stress. FEBS Lett 2001, 495, 12–15. [Google Scholar]
- Cortes-Rojo, C; Clemente-Guerrero, M; Saavedra-Molina, A. Effects of D-amino acids on lipoperoxidation in rat liver and kidney mitochondria. Amino Acids 2006, 32, 31–37. [Google Scholar]
- Kim, JS; He, L; Lemasters, JJ. Mitochondrial permeability transition: A common pathway to necrosis and apoptosis. Biochem. Biophys. Res. Commun 2003, 304, 463–470. [Google Scholar]
- Kim, WH; Hong, F; Jaruga, B; Zhang, ZS; Fan, SJ; Liang, TJ; Gao, B. Hepatitis B virus X protein sensitizes primary mouse hepatocytes to ethanol- and TNF-alpha-induced apoptosis by a caspase-3-dependent mechanism. Cell Mol. Immunol 2005, 2, 40–48. [Google Scholar]
- Urbano-Marquez, A; Fernandez-Sola, J. Effects of alcohol on skeletal and cardiac muscle. Muscle Nerve 2004, 30, 689–707. [Google Scholar]
- Capasso, JM; Li, P; Guideri, G; Malhotra, A; Cortese, R; Anversa, P. Myocardial mechanical, biochemical, and structural alterations induced by chronic ethanol ingestion in rats. Circ. Res 1992, 71, 346–356. [Google Scholar]
- Perfilova, VN; Ostrovskii, OV; Verovskii, VE; Popova, TA; Lebedeva, SA; Dib, H. Effect of citrocard on functional activity of cardiomyocyte mitochondria during chronic alcohol intoxication. Bull. Exp. Biol. Med 2007, 143, 341–343. [Google Scholar]
- Montgomery, RI; Coleman, WB; Eble, KS; Cunningham, CC. Ethanol-elicited alterations in the oligomycin sensitivity and structural stability of the mitochondrial F0.F1 ATPase. J. Biol. Chem 1987, 262, 13285–13289. [Google Scholar]
- Usha, R; Pendurthi, J; Todd, W; Vijya, M. Resveratrol a polyphenolic compound found in wine inhibits tissue factor expression in vascular cells. A possible mechanism for the cardiovascular benefits associated with moderate consumption of wine. Arterioscler. Thromb. Vasc. Biol 1999, 19, 419–426. [Google Scholar]
- Pace-Asciak, CR; Hahn, S; Diamandis, EP; Soleas, G; Goldberg, DM. The red wine phenolics trans-resveratrol and quercetin block human platelet aggregation and eicosanoid synthesis: Implications for protection against coronary heart disease. Clin. Chim. Acta 1995, 235, 207–219. [Google Scholar]
- Bertelli, AAE; Giovannini, L; Giannessi, D; Migliori, M; Bernini, W; Fregoni, M; Bertelli, A. Antiplatelet activity of synthetic and natural resveratrol in red wine. Int. J. Tiss. Reac 1995, 17, 1–3. [Google Scholar]
- Baur, JA; Sinclair, DA. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov 2006, 5, 493–506. [Google Scholar]
- Howitz, KT; Bitterman, KJ; Cohen, HY; Lamming, DW; Lavu, S; Wood, JG; Zipkin, RE; Chung, P; Kisielewski, A; Zhang, LL; Scherer, B; Sinclair, DA. Small molecule activators of sirtuins extended Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar]
- Belguendouz, L; Fremont, L; Gozzelino, MT. Interaction of transresveratrol with plasma lipoproteins. Biochem. Pharmacol 1998, 55, 811–816. [Google Scholar]
- Das, S; Falchi, M; Bertelli, A; Maulik, N; Das, DK. Attenuation of ischemia/reperfusion injury in rats by the anti-inflammatory action of resveratrol. Arzneimittelforschung 2006, 56, 700–706. [Google Scholar]
- Das, S; Alagappan, VK; Bagchi, D; Sharma, HS; Maulik, N; Das, DK. Coordinated induction of iNOS-VEGF-KDR-eNOS after resveratrol consumption: A potential mechanism for resveratrol preconditioning of the heart. Vascul. Pharmacol 2005, 42, 281–289. [Google Scholar]
- Hattori, R; Otani, H; Maulik, N; Das, DK. Pharmacological preconditioning with resveratrol: Role of nitric oxide. Am. J. Physiol 2002, 282, H1988–H1995. [Google Scholar]
- Li, HL; Wang, AB; Huang, Y; Liu, DP; Wei, C; Williams, GM; Zhang, CN; Liu, G; Liu, YQ; Hao, DL; Hui, RT; Lin, M; Liang, CC. Isorhapontigenin, a new resveratrol analog, attenuates cardiac hypertrophy via blocking signaling transduction pathways. Free Radic. Biol. Med 2005, 38, 243–257. [Google Scholar]
- Kaga, S; Zhan, L; Matsumoto, M; Maulik, N. Resveratrol enhances neovascularization in the infarcted rat myocardium through the induction of thioredoxin-1, heme oxygenase-1 and vascular endothelial growth factor. J. Mol. Cell Cardiol 2005, 39, 813–822. [Google Scholar]
- Zhang, Y; Liu, Y; Wang, T; Li, H; Wang, Z; Yang, B. Resveratrol, a natural ingredient of grape skin: Antiarrhyhtmic efficacy and ionic mechanisms. Biochem. Biophys. Res. Commun 2006, 340, 1192–1199. [Google Scholar]
- Wang, S; Wang, X; Yan, J; Xie, X; Fan, F; Zhou, X; Han, L; Chen, J. Resveratrol inhibits proliferation of cultured rat cardiac fibroblasts: Correlated with NO-cGMP signaling pathway. Eur. J. Pharmacol 2007, 567, 26–35. [Google Scholar]
- Hayek, T; Fuhrman, B; Vaya, J; Rosenblat, M; Belinky, P; Coleman, R; Elis, A; Aviram, M. Reduced progression of atherosclerosis in apolipoprotein E-deficient mice following consumption of red wine, or its polyphenols quercetin or catechin, is associated with reduced susceptibility of LDL to oxidation and aggregation. Arterioscler. Thromb. Vasc. Biol 1997, 17, 2744–2752. [Google Scholar]
- Rosenkranz, S; Knirel, D; Dietrich, H; Flesch, M; Erdmann, E; Bohm, M. Inhibition of the PDGF receptor by red wine flavonoids provides a molecular explanation for the “French paradox”. FASEB J 2002, 16, 1958–1960. [Google Scholar]
- Halder, UG; Roos, TU; Kontaridis, MI; Neel, BG; Sorescu, D; Griedling, KK; Vollmar, AM; Dirsch, VM. Resveratrol inhibits angiotensin II and epidermal growth factor-mediated Akt activation: Role of Gab1 and Shp2. Mol. Pharmacol 2005, 68, 41–48. [Google Scholar]
- Karatzi, KN; Papamichael, CM; Karatzis, EN; Papaioannou, TG; Aznaouridis, KA; Katsichti, PP; Stamatelopoulos, KS; Zampelas, A; Lekakis, JP; Mavrikakis, ME. Red wine acutely induces favorable effects on wave reflections and central pressures in coronary artery diseases patients. Am. J. Hypertens 2005, 18, 1161–1167. [Google Scholar]
- Baur, JA; Pearson, KJ; Price, NL; Jamieson, HA; Lerin, C; Kalra, A; Prabhu, VV; Allard, JS; Lopez-Lluch, G; Lewis, K; Pistell, PJ; Poosala, S; Becker, KG; Boss, O; Gwinn, D; Wang, M; Ramaswamy, S; Fishbein, KW; Spencer, RG; Lakatta, EG; Le Couteur, D; Shaw, RJ; Navas, P; Puigserver, P; Ingram, DK; de Cabo, R; Sinclair, DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar]
- Penumathsa, SV; Thirunavukkarasu, M; Koneru, S; Juhasz, B; Zhan, L; Pant, R; Menon, VP; Otani, H; Maulik, N. Statin and resveratrol in combination induces cardioprotection against myocardial infarction in hypercholesterolemic rat. J. Mol. Cell Cardiol 2007, 42, 508–516. [Google Scholar]
- Penumathsa, SV; Maulik, N. Resveratrol: A promising agent in promoting cardioprotection against coronary heart disease. Can. J. Physiol. Pharmacol 2009, 87, 275–286. [Google Scholar]
- Wagner, TM; Mullally, JE; Fitzpatrick, FA. Reactive lipid species from cyclooxygenase-2 inactivate tumor suppressor LKB1/STK11: Cyclopentenone prostaglandins and 4-hydroxy-2-nonenal covalently modify and inhibits the AMP kinase kinase that modulates cellular energy homeostasis and protein translation. J. Biol. Chem 2006, 281, 2598–2604. [Google Scholar]
- Pan, JS; He, SZ; Xu, HZ; Zhan, XJ; Yang, XN; Xiao, HM; Shi, HX. Oxidative stress disturbs energy metabolism of mitochondria in ethanol-induced gastric mucosa injury. World J. Gastroenterol 2008, 14, 5857–5867. [Google Scholar]
- Yin, GY; Zhang, WN; Shen, XJ; Chen, Y; He, XF. Ultrastructure and molecular biological changes of chronic gastritis, gastric cancer and gastric precancerous lesions: A comparative study. World J. Gastroenterol 2003, 9, 851–857. [Google Scholar]
- Hoek, JB; Cahill, A; Pastorino, JG. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology 2002, 122, 2049–2063. [Google Scholar]
- Rong, Q; Utevskaya, O; Ramilo, M; Chow, DC; Forte, JG. Nucleotide metabolism by gastric glands and H(+)-k(+)-ATPase-enriched membranes. Am. J. Physiol 1998, 274, G103–G110. [Google Scholar]
- Giannessi, F; Giambelluca, MA; Grasso, L; Scavuzzo, MC; Ruffoli, R. Curcumin protects Leydig cells of mice from damage induced by chronic alcohol administration. Med. Sci. Monit 2008, 14, 237–242. [Google Scholar]
- Wakabayashi, T. Megamitochondria formation-physiology and pathology. J. Cell Mol. Med 2002, 6, 497–538. [Google Scholar]
- Dey, A; Cederbaum, AI. Alcohol and oxidative liver injury. Hepatology 2006, 43, S63–S74. [Google Scholar]
- Mills, SJ; Harrison, SH. Comparison of the natural history of alcoholic and nonalcoholic fatty liver disease. Curr. Gastroenterol. Reports 2010, 7, 32–36. [Google Scholar]
- Stewart, S; Jones, D; Day, CP. Alcoholic liver disease: New insights into mechanisms and preventative strategies. Trends Mol. Med 2001, 7, 408–413. [Google Scholar]
- Morgan, TR; Mandayam, S; Jamal, MM. Alcohol and hepatocellular carcinoma. Gastroenterology 2004, 127, S87–96. [Google Scholar]
- Miranda-Mendez, A; Lugo-Baruqui, A; Armendariz-Borunda, J. Molecular basis and current treatment for alcoholic liver disease. Int. J. Environ. Res. Public Health 2010, 7, 1872–1888. [Google Scholar]
- Malhi, H; Gores, GJ. Cellular and molecular mechanisms of liver injury. Gastroenterology 2008, 134, 1641–1654. [Google Scholar]
- Song, BJ; Moon, KH; Olsson, NU; Salem, N, Jr. Prevention of alcoholic fatty liver and mitochondrial dysfunction in the rat by long-chain polyunsaturated fatty acids. J. Hepatol 2008, 49, 262–273. [Google Scholar]
- Friedman, SL. Molecular regulation of hepatic fibrosis, and integrated cellular response to tissue injury. J. Biol. Chem 2000, 275, 2247–2250. [Google Scholar]
- Bronchal, S; Nain, CK; Prasad, KK; Nada, R; Sharma, AK; Sinha, SK; Singh, K. Functional and morphological alterations in small intestine mucosa of chronic alcoholics. J. Gastroenterol. Hepatol 2008, 23, e43–e48. [Google Scholar]
- Kravos, M; Malesic, I. Kinetics and isoforms of serum glutamate dehydrogenase in alcoholics. Alcohol Alcohol 2008, 43, 281–286. [Google Scholar]
- Lakshmi, DS; Anuradha, CV. Mitochondrial damage, cytotoxicity and apoptosis in iron-potentiated alcoholic liver fibrosis: Amelioration by taurine. Amino Acids 2010, 38, 869–879. [Google Scholar]
- Sastre, J; Serviddio, G; Pereda, J; Minana, JB; Arduini, A; Vendemiale, G; Poli, G; Pallardo, FV; Vina, J. Mitochondrial function in liver disease. Front. Biosci 2007, 12, 1200–1209. [Google Scholar]
- Hajnoczky, G; Buzas, CJ; Pacher, P; Hoek, JB; Rubin, E. Alcohol and mitochondria in cardiac apoptosis: Mechanisms and visualization. Alcohol Clin. Exp. Res 2005, 29, 693–701. [Google Scholar]
- White, AM. What happened? Alcohol, memory blackouts, and the brain. Alcohol Res. Health 2003, 27, 186–196. [Google Scholar]
- Davis, KM; Wu, JY. Role of glutamatergic and GABAergic systems in alcoholism. J. Biomed. Sci 2001, 8, 7–19. [Google Scholar]
- Koch-Weser, J; Sellers, EM; Kalent, HL. Alcohol intoxication and withdrawal. N. Engl. J. Med 1976, 294, 757–762. [Google Scholar]
- Kosten, TR; O’Connor, PG. Management of drug and alcohol withdrawal. N. Engl. J. Med 2003, 348, 1786–1795. [Google Scholar]
- Brust, JCM. Ethanol and cognition: Indirect effects, neurotoxicity and neuroprotection: A review. Int. J. Environ. Res. Public Health 2010, 7, 1540–1557. [Google Scholar]
- Obenier, JA; Bouldin, TW; Crews, FT. Binge ethanol exposure in adult rats causes necrotic cell death. Alcohol Clin. Exp. Res 2002, 26, 547–557. [Google Scholar]
- Silvers, JM; Tokunaga, S; Berry, RB; White, AM; Matthews, DB. Impairment in spatial learning and memory: Ethanol, allopregnanolone, and the hippocampus. Brain Res. Brain Res. Rev 2003, 43, 275–284. [Google Scholar]
- Grant, I. Alcohol and the brain: Neuropsychological correlates. J. Consult. Clin. Psychol 1987, 55, 310–324. [Google Scholar]
- Israel, Y; Kalant, H. Effect of ethanol on the transport of sodium in frog skin. Nature 1963, 200, 476–478. [Google Scholar]
- Blum, K; Noble, EP; Sheridan, PJ; Montgomery, A; Ritchie, T; Jagadeesawaran, P; Nogami, H; Briggs, AH; Cohn, JB. Allelic association of human dopamine D2 and Gaba receptor gene in alcoholism. JAMA 1990, 263, 2055–2060. [Google Scholar]
- Kluge, H; Neumann, J; Seidel, K. Biochemical mechanisms for the effect of alcohol on the brain. Psychiatr. Neurol. Med. Psychol (Leipz) 1979, 31, 65–73. [Google Scholar]
- Kharchenko, NK. Dopamine content in blood and activity of alcohol-transforming enzymes in alcoholism. Ukr. Biokhim. Zh 1997, 69, 87–92. [Google Scholar]
- Smith, AA. Interaction of biogenic amines with ethanol. Adv. Exp. Med. Biol 1975, 56, 265–275. [Google Scholar]
- Weiner, H. Relationship between 3,4-dihydroxyphenylacetaldehyde levels and tetrahydropapaveroline formation. Alcohol Clin. Exp. Res 1978, 2, 127–131. [Google Scholar]
- Haorah, J; Ramírez, SH; Floreani, N; Gorantla, S; Morsey, B; Persidsky, Y. Mechanism of alcohol-induced stress and neuronal injury. Free Radic. Biol. Med 2008, 45, 1542–1550. [Google Scholar]
- Mansouri, A; Demeilliers, C; Amsellen, S; Pessayre, D; Fromenty, B. Acute ethanol administration oxidatively damages and depletes mitochondrial DNA in mouse liver, brain, heart, and skeletal muscles: Protective effects of antioxidants. J. Pharmacol. Exp. Ther 2001, 298, 737–743. [Google Scholar]
- Jung, ME; Agarwal, R; Simpkins, JW. Ethanol withdrawal posttranslationally decreases the activity of cytochrome c oxidase in an estrogen reversible manner. Neurosci. Lett 2007, 41, 160–164. [Google Scholar]
- Jung, ME; Simpkins, JW; Wilson, AM; Downey, HF; Mallet, RT. Intermittent hypoxia conditioning prevents behavioral deficit and brain oxidative stress in ethanol-withdrawn rats. J. Appl. Physiol 2008, 105, 510–517. [Google Scholar]
- Lee do, Y; Lee, KS; Lee, HJ; Jung, HY; Lee, JY; Lee, SH; Youn, YC; Seo, KM; Lee, JH; Lee, WB; Kim, SS. Alcohol enhances Abeta42-induced neuronal cell death through mitochondrial dysfunction. FEBS Lett 2008, 582, 4185–4190. [Google Scholar]
- Streissguth, AP; Landersman-Dwyer, S; Martin, JC; Smith, DW. Teratogenic effects of alcohol in humans and laboratory animals. Science 1980, 209, 353–361. [Google Scholar]
- Clarren, SK; Alvord, EC; Sumi, SM; Streissguth, AP; Smith, DW. Brain malformations related to prenatal exposure to ethanol. J. Pediatr 1978, 92, 64–67. [Google Scholar]
- Riikonen, RS; Nokelainen, P; Valkonen, K; Kolehmainen, AI; Kumpulainen, KI; Könönen, M; Vanninen, RL; Kuikka, JT. Deep serotonergic and dopaminergic structures in fetal alcoholic syndrome: A study with nor-beta-CIT-single-photon emission computed tomography and magnetic resonance imaging volumetry. Biol. Psychiatry 2005, 57, 1565–1572. [Google Scholar]
- Druse, MJ; Tajuddin, NF; Gillespie, RA; Le, P. The effects of ethanol and the serotonin(1A) agonist ipsapirone on the expression of the serotonin(1A) receptor and several antiapoptotic proteins in fetal rhombencephalic neurons. Brain Res 2006, 1092, 79–86. [Google Scholar]
- Sari, Y; Powrozek, T; Zhou, FC. Alcohol deters the outgrowth of serotonergic neurons at midgestation. J. Biomed. Sci 2001, 8, 119–125. [Google Scholar]
- Mayordomo, F; Renau-Piqueras, J; Megias, L; Guerri, C; Iborra, FJ; Azorin, I; Ledig, M. Cytochemical and stereological analysis of rat cortical astrocytes during development in primary culture. Effect of prenatal exposure to ethanol. Int. J. Dev. Biol 1992, 36, 311–321. [Google Scholar]
- Devi, BG; Henderson, GI; Frosto, TA; Schenker, S. Effect of ethanol on rat fetal hepatocytes: Studies on cell replication, lipid peroxidation and glutathione. Hepatology 1993, 18, 648–659. [Google Scholar]
- Sanchis, R; Guerri, C. Alcohol-metabolizing enzymes in placenta and fetal liver: Effect of chronic ethanol intake. Alcohol Clin. Exp. Res 1986, 10, 39–44. [Google Scholar]
- Maheshwari, RK; Singh, AK; Gaddipati, J; Srimal, RC. Multiple biological activities of curcumin: A short review. Life Sci 2006, 78, 2081–2087. [Google Scholar]
- Priyadarsini, KI; Maity, DK; Naik, GH; Kumar, MS; Unnikrishnan, MK; Satav, JG; Mohan, H. Role of phenolic O-H and methylene hydrogen on the free radical reactions and antioxidant activity of curcumin. Free Radic. Biol. Med 2003, 35, 475–484. [Google Scholar]
- Churchill, EN; Disatnik, MH; Mochly-Rosen, D. Time-dependent and ethanol-induced cardiac protection from ischemia mediated by mitochondrial translocation of varepsilon PKC and activation of aldehyde dehydrogenase2. J. Mol. Cell Cardiol 2009, 46, 278–284. [Google Scholar]
- Cannon, CP; Braunwald, E; McCabe, CH; Rader, DJ; Rouleau, JL; Belder, R; Joyal, SV; Hill, KA; Skene, AM. Pravastatin or atorvastatin evaluation and infection therapy-thrombolysis in myocardial infarction 22 investigators. N. Engl. J. Med 2004, 350, 1495–1504. [Google Scholar]
- Blake, GJ; Ridker, PM. Novel clinical markers of vascular wall inflammation. Circulation Res 2001, 89, 763–771. [Google Scholar]
- Clarke, R; Daly, L; Robinson, K; Naughten, E; Cahalane, S; Fowler, B; Graham, I. Hyperhomocysteinemia: An independent risk factor for vascular disease. N. Engl. J. Med 1991, 324, 1149–1155. [Google Scholar]
- Brisdelli, F; D’Andrea, G; Bozzi, A. Resveratrol: A natural polyphenol with multiple chemopreventive properties. Curr. Drug Metab 2009, 10, 530–46. [Google Scholar]
- Cheng, TH; Liu, JC; Lin, H; Shih, NL; Chen, YL; Huang, MT; Chan, P; Cheng, CF; Chen, JJ. Inhibitory effect of resveratrol on angiotensin II-induced cardiomyocyte hypertrophy. Naunyn Schmiedebergs Arch. Pharmacol 2004, 369, 239–244. [Google Scholar]
- Chan, AY; Dolinsky, VW; Soltys, CL; Viollet, B; Baksh, S; Light, PE; Dyck, JR. Resveratrol inhibits cardiac hypertrophy via AMP-activated protein kinase and Akt. J. Biol. Chem 2008, 283, 24194–24201. [Google Scholar]
- Serafini, M; Maiani, G; Ferro-Luzzi, A. Alcohol-free red wine enhances plasma antioxidant capacity in humans. J. Nutr 1998, 128, 1003–1007. [Google Scholar]
- Opie, LH; Lecour, S. The red wine hypothesis: From concepts to protective signalling molecules. Eur Heart J 2007, 28, 1683–1693. [Google Scholar]
- Hung, LM; Chen, JK; Lee, RS; Liang, HC; Su, MJ. Beneficial effects of astringinin, a resveratrol analogue, on the ischemia and reperfusion damage in rat heart. Free Radic. Biol. Med 2001, 30, 877–883. [Google Scholar]
- Fauconneau, B; Waffo-Teguo, P; Huguet, F; Barrier, L; Decendit, A; Merillon, JM. Comparative study of radical scavenger and antioxidant properties of phenolic compounds from Vitis vinifera cell cultures using in vitro tests. Life Sci 1997, 61, 2103–2110. [Google Scholar]
- El-Assal, O; Hong, F; Kim, W-H; Radaeva, S; Gao, B. IL-6 deficient mice are susceptible to ethanol-induced hepatic steatosis: IL-6 protects against ethanol-induced oxidative stress and mitochondrial permeability transition in the liver. Cell Mol. Immunol 2004, 1, 205–211. [Google Scholar]
- Hong, F; Kim, WH; Tian, Z; Jaruga, B; Ishac, E; Shen, X; Gao, B. Elevated interleukin-6 during ethanol consumption acts as a potential endogenous protective cytokine against ethanol-induced apoptosis in the liver: Involvement of induction of Bcl-2 and Bcl-x(L) proteins. Oncogene 2002, 21, 32–43. [Google Scholar]
- Lowe, ED; Gao, GY; Johnson, LN; Keung, WM. Structure of didzin, a naturally occurring anti-alcohol-addiction agent, in complex with human mitochondrial aldehyde dehydrogenase. J. Med. Chem 2008, 51, 4482–4487. [Google Scholar]
- Kim, YC; Jung, YS; Kim, SK. Effect of betaine supplementation on changes in hepatic metabolism of sulfur-containing amino acids and experimental cholestasis induced by alphanaphthylisothiocyanate. Food Chem. Toxicol 2005, 43, 663–670. [Google Scholar]
- Craig, SA. Betaine in human nutrition. Am. J. Clin. Nutr 2004, 80, 539–549. [Google Scholar]
- Zhang, P; Gong, ZJ; Wang, LW; Sun, XM; Zhou, XR. Effects of Betaine on hyperhomocysteinemia and lipid peroxidation in rats with ethanol-induced liver injury. Zhongxiyi Jiehe Ganbing Zazhi 2006, 16, 30–32. [Google Scholar]
- Ji, C; Shinohara, M; Vance, D; Than, TA; Ookhtens, M; Chan, C; Kaplowitz, N. Effect of transgenic extrahepatic expression of betaine-homocysteine methyltransferase on alcohol or homocysteine-induced fatty liver. Alcohol Clin. Exp. Res 2008, 32, 1049–1058. [Google Scholar]
- Cantoni, GL. Activation of methionine for transmethylation. J. Biol. Chem 1951, 189, 745–754. [Google Scholar]
- Mato, JM; Alvarez, L; Ortiz, P; Pajares, MA. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol. Ther 1997, 73, 265–280. [Google Scholar]
- Santamaria, E; Avila, MA; Latasa, MU; Rubio, A; Martin-Duce, A; Lu, SC; Mato, JM; Corrales, FJ. Functional proteomics of nonalcoholic steatohepatitis: Mitochondrial proteins as targets of S-adenosylmethionine. Proc. Natl. Acad. Sci. USA 2003, 100, 3065–3070. [Google Scholar]
- Bailey, SM; Robinson, G; Pinner, A; Chamlee, L; Ulasova, E; Pompilius, M; Page, GP; Chhieng, D; Jhala, N; Landar, A; Kharbanda, KK; Ballinger, S; Darley-Usmar, V. S-adenosylmethionine prevents chronic alcohol-induced mitochondrial dysfunction in the rat liver. Am. J. Gastrointest. Liver Physiol 2006, 291, G857–G867. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Manzo-Avalos, S.; Saavedra-Molina, A. Cellular and Mitochondrial Effects of Alcohol Consumption. Int. J. Environ. Res. Public Health 2010, 7, 4281-4304. https://doi.org/10.3390/ijerph7124281
Manzo-Avalos S, Saavedra-Molina A. Cellular and Mitochondrial Effects of Alcohol Consumption. International Journal of Environmental Research and Public Health. 2010; 7(12):4281-4304. https://doi.org/10.3390/ijerph7124281
Chicago/Turabian StyleManzo-Avalos, Salvador, and Alfredo Saavedra-Molina. 2010. "Cellular and Mitochondrial Effects of Alcohol Consumption" International Journal of Environmental Research and Public Health 7, no. 12: 4281-4304. https://doi.org/10.3390/ijerph7124281
APA StyleManzo-Avalos, S., & Saavedra-Molina, A. (2010). Cellular and Mitochondrial Effects of Alcohol Consumption. International Journal of Environmental Research and Public Health, 7(12), 4281-4304. https://doi.org/10.3390/ijerph7124281