
Mechanisms of Transmission and Processing of Pain: A Narrative Review

 , , ,
, , ,  , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Characteristics and Properties of Nociceptors
2.1. Structural Characteristics
2.2. Stimuli
2.3. Nociceptor Population
2.4. Sensitization
2.5. Efferent Function
3. Clinical and Preclinical Studies on Ion Channel Signaling Modulation
4. Immune System and Pain
5. Immune Regulation of Pain
6. Cytokines Involved in Pain
7. Lipids Involved in Pain
8. Nerve Injury and Neuropathic Pain
9. Neuronal Regulation of Vasculature
10. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Maruyama, K. Senso-immunology: Crosstalk between nociceptive and immune systems. FEBS J. 2021, 289, 4132–4145. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Waxman, S.G. Translational pain research: Lessons from genetics and genomics. Sci. Transl. Med. 2014, 6, 249sr4. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J.; Ma, Q. Nociceptors—Noxious stimulus detectors. Neuron 2007, 55, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Gebhart, G.F. Inside Information: The Unique Features of Visceral Sensation. Mol. Interv. 2008, 8, 242–253. [Google Scholar] [CrossRef]
- Le Bars, D.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharmacol. Rev. 2001, 53, 597–652. [Google Scholar]
- Schmidt, R.; Schmelz, M.; Forster, C.; Ringkamp, M.; Torebjork, E.; Handwerker, H. Novel classes of responsive and unresponsive C nociceptors in human skin. J. Neurosci. 1995, 15, 333–341. [Google Scholar] [CrossRef]
- Adelman, P.C.; Baumbauer, K.M.; Friedman, R.; Shah, M.; Wright, M.; Young, E.; Jankowski, M.P.; Albers, K.M.; Koerber, H.R. Single-cell q-PCR derived expression profiles of identified sensory neurons. Mol. Pain 2019, 15. [Google Scholar] [CrossRef]
- Dallos, P.; Oertel, D. The Senses a Comprehensive Reference; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Dubin, A.E.; Patapoutian, A. Nociceptors: The sensors of the pain pathway. J. Clin. Investig. 2010, 120, 3760–3772. [Google Scholar] [CrossRef]
- Lewin, G.R.; Moshourab, R. Mechanosensation and pain. J. Neurobiol. 2004, 61, 30–44. [Google Scholar] [CrossRef]
- Takayama, Y.; Derouiche, S.; Maruyama, K.; Tominaga, M. Emerging Perspectives on Pain Management by Modulation of TRP Channels and ANO1. Int. J. Mol. Sci. 2019, 20, 3411. [Google Scholar] [CrossRef]
- Tsagareli, M.G.; Nozadze, I.; Tsiklauri, N.; Carstens, M.I.; Gurtskaia, G.; Carstens, E. Thermal Hyperalgesia and Mechanical Allodynia Elicited by Histamine and Non-histaminergic Itch Mediators: Respective Involvement of TRPV1 and TRPA1. Neuroscience 2020, 449, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Brenneis, C.; Kistner, K.; Puopolo, M.; Segal, D.; Roberson, D.; Sisignano, M.; Labocha, S.; Ferreirós, N.; Strominger, A.; Cobos, E.J.; et al. Phenotyping the function of TRPV1-expressing sensory neurons by targeted axonal silencing. J. Neurosci. 2013, 33, 315–326. [Google Scholar] [CrossRef]
- Zurborg, S.; Yurgionas, B.; A Jira, J.; Caspani, O.; A Heppenstall, P. Direct activation of the ion channel TRPA1 by Ca2+. Nat. Neurosci. 2007, 10, 277–279. [Google Scholar] [CrossRef]
- Díaz-Franulic, I.; Raddatz, N.; Castillo, K.; González-Nilo, F.D.; Latorre, R. A folding reaction at the C-terminal domain drives temperature sensing in TRPM8 channels. Proc. Natl. Acad. Sci. USA 2020, 117, 20298–20304. [Google Scholar] [CrossRef] [PubMed]
- Correction for Cavanaugh et al., Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proc. Natl. Acad. Sci. USA 2009, 106, 11424. [CrossRef]
- Arcourt, A.; Gorham, L.; Dhandapani, R.; Prato, V.; Taberner, F.J.; Wende, H.; Gangadharan, V.; Birchmeier, C.; Heppenstall, P.A.; Lechner, S.G. Touch Receptor-Derived Sensory Information Alleviates Acute Pain Signaling and Fine-Tunes Nociceptive Reflex Coordination. Neuron 2016, 93, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Giniatullin, R. Ion Channels of Nociception. Int. J. Mol. Sci. 2020, 21, 3553. [Google Scholar] [CrossRef]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 Are Essential Components of Distinct Mechanically Activated Cation Channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef]
- Gottlieb, P. A Tour de Force. Curr. Top. Membr. 2017, 79, 1–36. [Google Scholar] [CrossRef]
- Della Pietra, A.; Mikhailov, N.; Giniatullin, R. The Emerging Role of Mechanosensitive Piezo Channels in Migraine Pain. Int. J. Mol. Sci. 2020, 21, 696. [Google Scholar] [CrossRef]
- Li, W.; Gong, Y.; Liu, J.; Guo, Y.; Tang, H.; Qin, S.; Zhao, Y.; Wang, S.; Xu, Z.; Chen, B. Peripheral and Central Pathological Mechanisms of Chronic Low Back Pain: A Narrative Review. J. Pain Res. 2021, 14, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Kuner, R. Central mechanisms of pathological pain. Nat. Med. 2010, 16, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Julius, D. Sense and specificity: A molecular identity for nociceptors. Curr. Opin. Neurobiol. 1999, 9, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Diószegi, J.; Kurshed, A.A.M.; Pikó, P.; Kósa, Z.; Sándor, J.; Ádány, R. Association of single nucleotide polymorphisms with taste and food preferences of the Hungarian general and Roma populations. Appetite 2021, 164, 105270. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, D.; Xu, C.; Li, H.; Caron, K.M.; Frenette, P.S. Nociceptive nerves regulate haematopoietic stem cell mobilization. Nature 2020, 589, 591–596. [Google Scholar] [CrossRef]
- Richardson, J.D.; Vasko, M.R. Cellular Mechanisms of Neurogenic Inflammation. Experiment 2002, 302, 839–845. [Google Scholar] [CrossRef]
- Lee, S.; Jo, S.; Talbot, S.; Zhang, H.-X.B.; Kotoda, M.; Andrews, N.A.; Puopolo, M.; Liu, P.W.; Jacquemont, T.; Pascal, M.; et al. Novel charged sodium and calcium channel inhibitor active against neurogenic inflammation. eLife 2019, 8, e48118. [Google Scholar] [CrossRef]
- Li, Y.; Yang, M.; Wu, F.; Cheng, K.; Chen, H.; Shen, X.; Lao, L. Mechanism of electroacupuncture on inflammatory pain: Neural-immune-endocrine interactions. J. Tradit. Chin. Med. 2019, 39, 740–749. [Google Scholar]
- La Marra, M.; Villano, I.; Ilardi, C.R.; Carosella, M.; Staiano, M.; Iavarone, A.; Chieffi, S.; Messina, G.; Polito, R.; Porro, C.; et al. Executive Functions in Overweight and Obese Treatment-Seeking Patients: Cross-Sectional Data and Longitudinal Perspectives. Brain Sci. 2022, 12, 777. [Google Scholar] [CrossRef]
- Shakhanbeh, J.; Lynn, B. Morphine inhibits antidromic vasodilation without affecting the excitability of C-polymodal nociceptors in the skin of the rat. Brain Res. 1993, 607, 314–318. [Google Scholar] [CrossRef]
- Chieffi, S.; Castaldi, C.; Di Maio, G.; La Marra, M.; Messina, A.; Monda, V.; Villano, I. Attentional bias in the radial and vertical dimensions of space. C. R. Biol. 2019, 342, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Francavilla, V.C.; Genovesi, F.; Asmundo, A.; Di Nunno, N.R.; Ambrosi, A.; Tartaglia, N.; Tafuri, D.; Monda, V.; Monda, M.; Messina, A.; et al. Fascia and Movement: The Primary Link in the Prevention of Accidents in Soccer. Revision and Models of Intervention. Med Sport 2020, 73, 291–301. [Google Scholar] [CrossRef]
- Knezevic, N.N.; Yekkirala, A.; Yaksh, T.L. Basic/Translational Development of Forthcoming Opioid and Nonopioid-Targeted Pain Therapeutics. Anesth. Analg. 2017, 125, 1714–1732. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; McLellan, A.T. Opioid Abuse in Chronic Pain–Misconceptions and Mitigation Strategies. N. Engl. J. Med. 2016, 374, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Skerratt, S.E.; West, C.W. Ion channel therapeutics for pain. Channels 2015, 9, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar] [CrossRef]
- Fertleman, C.R.; Baker, M.D.; Parker, K.A.; Moffatt, S.; Elmslie, F.V.; Abrahamsen, B.; Ostman, J.; Klugbauer, N.; Wood, J.N.; Gardiner, R.M.; et al. SCN9A Mutations in Paroxysmal Extreme Pain Disorder: Allelic Variants Underlie Distinct Channel Defects and Phenotypes. Neuron 2006, 52, 767–774. [Google Scholar] [CrossRef]
- Bahar, E.; Yoon, H. Lidocaine: A Local Anesthetic, Its Adverse Effects and Management. Medicina 2021, 57, 782. [Google Scholar] [CrossRef]
- Pal, R.; Kumar, B.; Akhtar, J.; Chawla, P.A. Voltage gated sodium channel inhibitors as anticonvulsant drugs: A systematic review on recent developments and structure activity relationship studies. Bioorganic Chem. 2021, 115, 105230. [Google Scholar] [CrossRef]
- Backonja, M.; Glanzman, R.L. Gabapentin dosing for neuropathic pain: Evidence from randomized, placebo-controlled clinical trials. Clin. Ther. 2003, 25, 81–104. [Google Scholar] [CrossRef]
- Reddy, D.S. An enigmatic role of tonic inhibition in gabapentin therapy. Ebiomedicine 2019, 42, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Bagal, S.K.; Brown, A.D.; Cox, P.J.; Omoto, K.; Owen, R.M.; Pryde, D.C.; Sidders, B.; Skerratt, S.E.; Stevens, E.B.; Storer, R.I.; et al. Ion Channels as Therapeutic Targets: A Drug Discovery Perspective. J. Med. Chem. 2012, 56, 593–624. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, Y.P.; Price, N.; Namdari, R.; Cohen, C.J.; Lamers, M.H.; Winters, C.; Price, J.; Young, C.E.; Verschoof, H.; Sherrington, R.; et al. Treatment of Nav1.7-mediated pain in inherited erythromelalgia using a novel sodium channel blocker. Pain 2012, 153, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, M.F.; Honore, P.; Shieh, C.-C.; Chapman, M.; Joshi, S.; Zhang, X.-F.; Kort, M.; Carroll, W.; Marron, B.; Atkinson, R.; et al. A-803467, a potent and selective Na v 1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. USA 2007, 104, 8520–8525. [Google Scholar] [CrossRef]
- Scanio, M.J.; Shi, L.; Drizin, I.; Gregg, R.J.; Atkinson, R.N.; Thomas, J.B.; Johnson, M.S.; Chapman, M.L.; Liu, D.; Krambis, M.J.; et al. Discovery and biological evaluation of potent, selective, orally bioavailable, pyrazine-based blockers of the Nav1.8 sodium channel with efficacy in a model of neuropathic pain. Bioorganic Med. Chem. 2010, 18, 7816–7825. [Google Scholar] [CrossRef] [PubMed]
- Kashio, M.; Tominaga, M. TRP channels in thermosensation. Curr. Opin. Neurobiol. 2022, 75, 102591. [Google Scholar] [CrossRef] [PubMed]
- Iftinca, M.; Defaye, M.; Altier, C. TRPV1-Targeted Drugs in Development for Human Pain Conditions. Drugs 2020, 81, 7–27. [Google Scholar] [CrossRef]
- Ferreira, L.G.B.; Faria, J.V.; dos Santos, J.P.S.; Faria, R.X. Capsaicin: TRPV1-independent mechanisms and novel therapeutic possibilities. Eur. J. Pharmacol. 2020, 887, 173356. [Google Scholar] [CrossRef]
- Andrews, M.D.; Forselles, K.A.; Beaumont, K.; Galan, S.R.G.; Glossop, P.A.; Grenie, M.; Jessiman, A.; Kenyon, A.S.; Lunn, G.; Maw, G.; et al. Discovery of a Selective TRPM8 Antagonist with Clinical Efficacy in Cold-Related Pain. ACS Med. Chem. Lett. 2015, 6, 419–424. [Google Scholar] [CrossRef]
- Yamamoto, T.; Takahara, A. Recent Updates of N-Type Calcium Channel Blockers with Therapeutic Potential for Neuropathic Pain and Stroke. Curr. Top. Med. Chem. 2009, 9, 377–395. [Google Scholar] [CrossRef]
- Altier, C.; Dale, C.S.; Kisilevsky, A.E.; Chapman, K.; Castiglioni, A.J.; Matthews, E.A.; Evans, R.M.; Dickenson, A.H.; Lipscombe, D.; Vergnolle, N.; et al. Differential Role of N-Type Calcium Channel Splice Isoforms in Pain. J. Neurosci. 2007, 27, 6363–6373. [Google Scholar] [CrossRef] [PubMed]
- Deer, T.; Hagedorn, J.M. How has ziconotide impacted non-cancer pain management? Expert Opin. Pharmacother. 2020, 21, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Kreir, M.; De Bondt, A.; Van den Wyngaert, I.; Teuns, G.; Lu, H.R.; Gallacher, D.J. Role of Kv7. 2/Kv7. 3 and M1 muscarinic receptors in the regulation of neuronal excitability in hiPSC-derived neurons. Eur. J. Pharmacol. 2019, 858, 172474. [Google Scholar] [CrossRef] [PubMed]
- Mruk, K.; Kobertz, W.R. Discovery of a Novel Activator of KCNQ1-KCNE1 K+ Channel Complexes. PLoS ONE 2009, 4, e4236. [Google Scholar] [CrossRef] [PubMed]
- Blackburn-Munro, G.; Jensen, B.S. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur. J. Pharmacol. 2003, 460, 109–116. [Google Scholar] [CrossRef]
- Gogas, K.R. Glutamate-based therapeutic approaches: NR2B receptor antagonists. Curr. Opin. Pharmacol. 2006, 6, 68–74. [Google Scholar] [CrossRef]
- Ismail, C.A.N.; Suppian, R.; Aziz, C.B.A.; Haris, K.; Long, I. Increased Nociceptive Responses in Streptozotocin-Induced Diabetic Rats and the Related Expression of Spinal NR2B Subunit of N-Methyl-D-Aspartate Receptors. Diabetes Metab. J. 2019, 43, 222–235. [Google Scholar] [CrossRef]
- Becker, R.; Gass, N.; Kußmaul, L.; Schmid, B.; Scheuerer, S.; Schnell, D.; Dorner-Ciossek, C.; Weber-Fahr, W.; Sartorius, A. NMDA receptor antagonists traxoprodil and lanicemine improve hippocampal-prefrontal coupling and reward-related networks in rats. Psychopharmacology 2019, 236, 3451–3463. [Google Scholar] [CrossRef]
- Polito, R.; Francavilla, V.C.; Ambrosi, A.; Tartaglia, N.; Tafuri, D.; Monda, M.; Messina, A.; Sessa, F.; Di Maio, G.; Ametta, A.; et al. The Orexin-A serum levels are strongly modulated by physical activity intervention in diabetes mellitus patients. J. Hum. Sport Exerc. 2020, 15, 244–251. [Google Scholar] [CrossRef]
- Raghavendra, V.; Tanga, F.Y.; DeLeo, J.A. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur. J. Neurosci. 2004, 20, 467–473. [Google Scholar] [CrossRef]
- Zhao, P.; Waxman, S.G.; Hains, B.C.; Carter, A.G.; Soler-Llavina, G.J.; Sabatini, B.L. Modulation of Thalamic Nociceptive Processing after Spinal Cord Injury through Remote Activation of Thalamic Microglia by Cysteine Cysteine Chemokine Ligand 21. J. Neurosci. 2007, 27, 8893–8902. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Guo, W.; Zou, S.; Ren, K.; Dubner, R. Supraspinal Glial-Neuronal Interactions Contribute to Descending Pain Facilitation. J. Neurosci. 2008, 28, 10482–10495. [Google Scholar] [CrossRef] [PubMed]
- Villano, I.; Ilardi, C.R.; Arena, S.; Scuotto, C.; Gleijeses, M.G.; Messina, G.; Messina, A.; Monda, V.; Monda, M.; Iavarone, A.; et al. Obese Subjects without Eating Disorders Experience Binge Episodes Also Independently of Emotional Eating and Personality Traits among University Students of Southern Italy. Brain Sci. 2021, 11, 1145. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wang, H.; Watanabe, M.; Shimizu, K.; Zou, S.; LaGraize, S.C.; Wei, F.; Dubner, R.; Ren, K. Glial-Cytokine-Neuronal Interactions Underlying the Mechanisms of Persistent Pain. J. Neurosci. 2007, 27, 6006–6018. [Google Scholar] [CrossRef]
- Wen, Y.-R.; Suter, M.R.; Kawasaki, Y.; Huang, J.; Pertin, M.; Kohno, T.; Berde, C.B.; Decosterd, I.; Ji, R.-R. Nerve Conduction Blockade in the Sciatic Nerve Prevents but Does Not Reverse the Activation of p38 Mitogen-activated Protein Kinase in Spinal Microglia in the Rat Spared Nerve Injury Model. Anesthesiology 2007, 107, 312–321. [Google Scholar] [CrossRef]
- Xie, W.; Strong, J.; Zhang, J.-M. Early blockade of injured primary sensory afferents reduces glial cell activation in two rat neuropathic pain models. Neuroscience 2009, 160, 847–857. [Google Scholar] [CrossRef]
- Oka, Y.; Ibuki, T.; Matsumura, K.; Namba, M.; Yamazaki, Y.; Poole, S.; Tanaka, Y.; Kobayashi, S. Interleukin-6 is a candidate molecule that transmits inflammatory information to the CNS. Neuroscience 2007, 145, 530–538. [Google Scholar] [CrossRef]
- Costigan, M.; Moss, A.; Latremoliere, A.; Johnston, C.; Verma-Gandhu, M.; Herbert, T.A.; Barrett, L.; Brenner, G.; Vardeh, D.; Woolf, C.J.; et al. T-Cell Infiltration and Signaling in the Adult Dorsal Spinal Cord Is a Major Contributor to Neuropathic Pain-Like Hypersensitivity. J. Neurosci. 2009, 29, 14415–14422. [Google Scholar] [CrossRef]
- Cao, L.; DeLeo, J.A. CNS-infiltrating CD4+ T lymphocytes contribute to murine spinal nerve transection-induced neuropathic pain. Eur. J. Immunol. 2008, 38, 448–458. [Google Scholar] [CrossRef]
- Chacur, M.; Lambertz, D.; Hoheisel, U.; Mense, S. Role of spinal microglia in myositis-induced central sensitisation: An immunohistochemical and behavioural study in rats. Eur. J. Pain 2009, 13, 915–923. [Google Scholar] [CrossRef]
- Shan, S.; Hong, C.; Mei, H.; Ting-Ting, L.; Hai-Li, P.; Zhi-Qi, Z.; Yu-Qiu, Z. New evidence for the involvement of spinal fractalkine receptor in pain facilitation and spinal glial activation in rat model of monoarthritis. Pain 2007, 129, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Ledeboer, A.; Sloane, E.M.; Milligan, E.D.; Frank, M.G.; Mahony, J.H.; Maier, S.F.; Watkins, L.R. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain 2005, 115, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Riazi, K.; Galic, M.A.; Kuzmiski, J.B.; Ho, W.; Sharkey, K.A.; Pittman, Q.J. Microglial activation and TNFα production mediate altered CNS excitability following peripheral inflammation. Proc. Natl. Acad. Sci. USA 2008, 105, 17151–17156. [Google Scholar] [CrossRef] [PubMed]
- La Marra, M.; Ilardi, C.R.; Villano, I.; Carosella, M.; Staiano, M.; Iavarone, A.; Chieffi, S.; Messina, G.; Polito, R.; Scarinci, A.; et al. Functional Relationship between Inhibitory Control, Cognitive Flexibility, Psychomotor Speed and Obesity. Brain Sci. 2022, 12, 1080. [Google Scholar] [CrossRef] [PubMed]
- Schöbitz, B.; de Kloet, E.R.; Sutanto, W.; Holsboer, F. Cellular Localization of Interleukin 6 mRNA and Interleukin 6 Receptor mRNA in Rat Brain. Eur. J. Neurosci. 1993, 5, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Vallières, L.; Rivest, S. Regulation of the Genes Encoding Interleukin-6, Its Receptor, and gp130 in the Rat Brain in Response to the Immune Activator Lipopolysaccharide and the Proinflammatory Cytokine Interleukin-1β. J. Neurochem. 1997, 69, 1668–1683. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, X.Q.; Echeverry, S.; Mogil, J.S.; De Koninck, Y.; Rivest, S. Expression of CCR2 in Both Resident and Bone Marrow-Derived Microglia Plays a Critical Role in Neuropathic Pain. J. Neurosci. 2007, 27, 12396–12406. [Google Scholar] [CrossRef] [PubMed]
- Deleo, J.A.; Tanga, F.Y.; Tawfik, V.L. Neuroimmune Activation and Neuroinflammation in Chronic Pain and Opioid Tolerance/Hyperalgesia. Neuroscientist 2004, 10, 40–52. [Google Scholar] [CrossRef]
- Watkins, L.R.; Maier, S.F. Immune regulation of central nervous system functions: From sickness responses to pathological pain. J. Intern. Med. 2005, 257, 139–155. [Google Scholar] [CrossRef]
- Scholz, J.; Woolf, C.J. The neuropathic pain triad: Neurons, immune cells and glia. Nat. Neurosci. 2007, 10, 1361–1368. [Google Scholar] [CrossRef]
- Ren, K.; Dubner, R. Neuron–glia crosstalk gets serious: Role in pain hypersensitivity. Curr. Opin. Anaesthesiol. 2008, 21, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Thacker, M.A.; Clark, A.K.; Marchand, F.; McMahon, S.B. Pathophysiology of Peripheral Neuropathic Pain: Immune Cells and Molecules. Obstet. Anesthesia Dig. 2007, 105, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Chieffi, S.; Villano, I.; Messina, A.; Monda, V.; La Marra, M.; Messina, G.; Monda, M. Involvement of orexin in sleep disorders and neurodegenerative diseases. Curr. Top. Pept. 2015, 16, 49–54. [Google Scholar]
- Milligan, E.D.; Watkins, L.R. Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci. 2009, 10, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Calvo, M.; Zhu, N.; Tsantoulas, C.; Ma, Z.; Grist, J.; Loeb, J.A.; Bennett, D.L.H. Neuregulin-ErbB Signaling Promotes Microglial Proliferation and Chemotaxis Contributing to Microgliosis and Pain after Peripheral Nerve Injury. J. Neurosci. 2010, 30, 5437–5450. [Google Scholar] [CrossRef] [PubMed]
- Verge, G.M.; Milligan, E.D.; Maier, S.F.; Watkins, L.R.; Naeve, G.S.; Foster, A.C. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur. J. Neurosci. 2004, 20, 1150–1160. [Google Scholar] [CrossRef]
- Zhuang, Z.-Y.; Kawasaki, Y.; Tan, P.-H.; Wen, Y.-R.; Huang, J.; Ji, R.-R. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav. Immun. 2007, 21, 642–651. [Google Scholar] [CrossRef]
- Coull, J.A.M.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; De Koninck, Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 2005, 438, 1017–1021. [Google Scholar] [CrossRef]
- Hamilton-Whitaker, N.; Attwell, D. Do astrocytes really exocytose neurotransmitters? Nat. Rev. Neurosci. 2010, 11, 227–238. [Google Scholar] [CrossRef]
- Sung, B.; Lim, G.; Mao, J. Altered Expression and Uptake Activity of Spinal Glutamate Transporters after Nerve Injury Contribute to the Pathogenesis of Neuropathic Pain in Rats. J. Neurosci. 2003, 23, 2899–2910. [Google Scholar] [CrossRef]
- Chiang, C.-Y.; Wang, J.; Xie, Y.-F.; Zhang, S.; Hu, J.W.; Dostrovsky, J.O.; Sessle, B.J. Astroglial Glutamate Glutamine Shuttle Is Involved in Central Sensitization of Nociceptive Neurons in Rat Medullary Dorsal Horn. J. Neurosci. 2007, 27, 9068–9076. [Google Scholar] [CrossRef] [PubMed]
- Okada-Ogawa, A.; Suzuki, I.; Sessle, B.J.; Chiang, C.-Y.; Salter, M.W.; Dostrovsky, J.O.; Tsuboi, Y.; Kondo, M.; Kitagawa, J.; Kobayashi, A.; et al. Astroglia in Medullary Dorsal Horn (Trigeminal Spinal Subnucleus Caudalis) Are Involved in Trigeminal Neuropathic Pain Mechanisms. J. Neurosci. 2009, 29, 11161–11171. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Weng, H.-R. Glutamate Transporters Prevent Excessive Activation of NMDA Receptors and Extrasynaptic Glutamate Spillover in the Spinal Dorsal Horn. J. Neurophysiol. 2009, 101, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Ren, K. Emerging role of astroglia in pain hypersensitivity. Jpn. Dent. Sci. Rev. 2010, 46, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Samad, T.A.; Moore, K.A.; Sapirstein, A.; Billet, S.; Allchorne, A.; Poole, S.; Bonventre, J.V.; Woolf, C.J. Interleukin-1β-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 2001, 410, 471–475. [Google Scholar] [CrossRef]
- Clark, A.; Staniland, A.A.; Marchand, F.; Kaan, T.K.Y.; McMahon, S.; Malcangio, M. P2X7-Dependent Release of Interleukin-1 and Nociception in the Spinal Cord following Lipopolysaccharide. J. Neurosci. 2010, 30, 573–582. [Google Scholar] [CrossRef]
- Choi, J.I.; Svensson, C.I.; Koehrn, F.J.; Bhuskute, A.; Sorkin, L.S. Peripheral inflammation induces tumor necrosis factor dependent AMPA receptor trafficking and Akt phosphorylation in spinal cord in addition to pain behavior. Pain 2010, 149, 243–253. [Google Scholar] [CrossRef]
- Cheng, X.; Choi, J.-S.; Waxman, S.G.; Dib-Hajj, S.D. Mini-review—Sodium channels and beyond in peripheral nerve disease: Modulation by cytokines and their effector protein kinases. Neurosci. Lett. 2020, 741, 135446. [Google Scholar] [CrossRef]
- Kiguchi, N.; Kobayashi, Y.; Maeda, T.; Fukazawa, Y.; Tohya, K.; Kimura, M.; Kishioka, S. Epigenetic Augmentation of the Macrophage Inflammatory Protein 2/C-X-C Chemokine Receptor Type 2 Axis through Histone H3 Acetylation in Injured Peripheral Nerves Elicits Neuropathic Pain. Experiment 2011, 340, 577–587. [Google Scholar] [CrossRef]
- Cunha, T.M.; Verri, W.A.; Schivo, I.R.; Napimoga, M.H.; Parada, C.A.; Poole, S.; Teixeira, M.M.; Ferreira, S.H.; Cunha, F.Q. Crucial role of neutrophils in the development of mechanical inflammatory hypernociception. J. Leukoc. Biol. 2008, 83, 824–832. [Google Scholar] [CrossRef]
- Ghasemlou, N.; Chiu, I.M.; Julien, J.-P.; Woolf, C.J. CD11b+ Ly6G− myeloid cells mediate mechanical inflammatory pain hypersensitivity. Proc. Natl. Acad. Sci. USA 2015, 112, E6808–E6817. [Google Scholar] [CrossRef] [PubMed]
- Aich, A.; Afrin, L.B.; Gupta, K. Mast Cell-Mediated Mechanisms of Nociception. Int. J. Mol. Sci. 2015, 16, 29069–29092. [Google Scholar] [CrossRef] [PubMed]
- Chatterjea, D.; Martinov, T. Mast cells: Versatile gatekeepers of pain. Mol. Immunol. 2015, 63, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Chieffi, S.; Messina, A.; Villano, I.; Valenzano, A.A.; Nigro, E.; La Marra, M.; Cibelli, G.; Monda, V.; Salerno, M.; Tafuri, D.; et al. The Use of Velocity Information in Movement Reproduction. Front. Psychol. 2017, 8, 983. [Google Scholar] [CrossRef]
- Oliveira, S.M.; Drewes, C.C.; Silva, C.R.; Trevisan, G.; Boschen, S.L.; Moreira, C.G.; Cabrini, D.D.A.; Da Cunha, C.; Ferreira, J. Involvement of mast cells in a mouse model of postoperative pain. Eur. J. Pharmacol. 2011, 672, 88–95. [Google Scholar] [CrossRef]
- Li, W.-W.; Guo, M.T.-Z.; Liang, D.-Y.; Sun, Y.; Kingery, M.W.S.; Clark, M.J.D. Substance P Signaling Controls Mast Cell Activation, Degranulation, and Nociceptive Sensitization in a Rat Fracture Model of Complex Regional Pain Syndrome. Anesthesiology 2012, 116, 882–895. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kiguchi, N.; Fukazawa, Y.; Saika, F.; Maeda, T.; Kishioka, S. Macrophage-T Cell Interactions Mediate Neuropathic Pain through the Glucocorticoid-induced Tumor Necrosis Factor Ligand System. J. Biol. Chem. 2015, 290, 12603–12613. [Google Scholar] [CrossRef]
- Old, E.A.; Nadkarni, S.; Grist, J.; Gentry, C.; Bevan, S.J.; Kim, K.-W.; Mogg, A.J.; Perretti, M.; Malcangio, M. Monocytes expressing CX3CR1 orchestrate the development of vincristine-induced pain. J. Clin. Investig. 2014, 124, 2023–2036. [Google Scholar] [CrossRef]
- Chieffi, S.; Iavarone, A.; La Marra, M.; Messina, G.; Villano, I.; Ranucci, S.; Monda, M. Memory for proprioceptive targets in bulimia nervosa. J. Psychiatry 2015, 18, 297. [Google Scholar]
- Villano, I.; La Marra, M.; Messina, A.; Di Maio, G.; Moscatelli, F.; Chieffi, S.; Monda, M.; Messina, G.; Monda, V. Effects of vegetarian and vegan nutrition on body composition in competitive futsal athletes. Prog. Nutr. 2021, 23, e2021126. [Google Scholar] [CrossRef]
- Trevisan, G.; Benemei, S.; Materazzi, S.; De Logu, F.; De Siena, G.; Fusi, C.; Rossato, M.F.; Coppi, E.; Marone, I.M.; Ferreira, J.; et al. TRPA1 mediates trigeminal neuropathic pain in mice downstream of monocytes/macrophages and oxidative stress. Brain 2016, 139, 1361–1377. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.F.; Moalem-Taylor, G. Interleukin-17 Contributes to Neuroinflammation and Neuropathic Pain Following Peripheral Nerve Injury in Mice. J. Pain 2011, 12, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, G.; Alessio, N.; Demirsoy, I.; Peluso, G.; Perrotta, S.; Monda, M.; Di Bernardo, G. Evaluation of Browning Agents on the White Adipogenesis of Bone Marrow Mesenchymal Stromal Cells: A Contribution to Fighting Obesity. Cells 2021, 10, 403. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, G.; Alessio, N.; Peluso, G.; Perrotta, S.; Monda, M.; Di Bernardo, G. Molecular and Physiological Effects of Browning Agents on White Adipocytes from Bone Marrow Mesenchymal Stromal Cells. Int. J. Mol. Sci. 2022, 23, 12151. [Google Scholar] [CrossRef] [PubMed]
- Najar, M.; Raicevic, G.; Fayyad-Kazan, H.; Bron, D.; Toungouz, M.; Lagneaux, L. Mesenchymal stromal cells and immunomodulation: A gathering of regulatory immune cells. Cytotherapy 2016, 18, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Marefati, N.; Ghorani, V.; Shakeri, F.; Boskabady, M.; Kianian, F.; Rezaee, R.; Boskabady, M.H. A review of anti-inflammatory, antioxidant, and immunomodulatory effects of Allium cepa and its main constituents. Pharm. Biol. 2021, 59, 285–300. [Google Scholar] [CrossRef]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 Beta—A Friend or Foe in Malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef]
- Ferreira, S.H.; Lorenzetti, B.B.; Bristow, A.F.; Poole, S. Interleukin-1β as a potent hyperalgesic agent antagonized by a tripeptide analogue. Nature 1988, 334, 698–700. [Google Scholar] [CrossRef]
- Verri, W.A., Jr.; Cunha, T.M.; Parada, C.A.; Poole, S.; Cunha, F.Q.; Ferreira, S.H. Hypernociceptive role of cytokines and chemokines: Targets for analgesic drug development? Pharmacol. Ther. 2006, 112, 116–138. [Google Scholar] [CrossRef]
- Polito, R.; Scarinci, A.; Ambrosi, A.; Tartaglia, N.; Tafuri, D.; Monda, M.; Messina, A.; Cimmino, F.; Catapano, A.; Sessa, F.; et al. The beneficial effects of physical activity and weight loss on human colorectal carcinoma cell lines. In Proceedings of the Winter Conferences of Sports Science: Costa Blanca Sports Science Events, Alicante, Spain, 24 April 2020. [Google Scholar] [CrossRef]
- Binshtok, A.M.; Wang, H.; Zimmermann, K.; Amaya, F.; Vardeh, D.; Shi, L.; Brenner, G.J.; Ji, R.-R.; Bean, B.P.; Woolf, C.J.; et al. Nociceptors Are Interleukin-1 Sensors. J. Neurosci. 2008, 28, 14062–14073. [Google Scholar] [CrossRef] [PubMed]
- Ebbinghaus, M.; Uhlig, B.; Richter, F.; von Banchet, G.S.; Gajda, M.; Bräuer, R.; Schaible, H.-G. The role of interleukin-1β in arthritic pain: Main involvement in thermal, but not mechanical, hyperalgesia in rat antigen-induced arthritis. Arthritis Rheum. 2012, 64, 3897–3907. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Kong, L.-Y.; Cai, J.; Li, S.; Liu, X.-D.; Han, J.-S.; Xing, G.-G. Interleukin-6-mediated functional upregulation of TRPV1 receptors in dorsal root ganglion neurons through the activation of JAK/PI3K signaling pathway. Pain 2015, 156, 1124–1144. [Google Scholar] [CrossRef] [PubMed]
- Malsch, P.; Andratsch, M.; Vogl, C.; Link, A.S.; Alzheimer, C.; Brierley, S.; Hughes, P.; Kress, M. Deletion of Interleukin-6 Signal Transducer gp130 in Small Sensory Neurons Attenuates Mechanonociception and Down-Regulates TRPA1 Expression. J. Neurosci. 2014, 34, 9845–9856. [Google Scholar] [CrossRef]
- Cunha, F.; Poole, S.; Lorenzetti, B.; Ferreira, S. The pivotal role of tumour necrosis factor α in the development of inflammatory hyperalgesia. Br. J. Pharmacol. 1992, 107, 660–664. [Google Scholar] [CrossRef]
- Cunha, T.M.; Verri, W.A., Jr.; Silva, J.S.; Poole, S.; Cunha, F.Q.; Ferreira, S.H. A cascade of cytokines mediates mechanical inflammatory hypernociception in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 1755–1760. [Google Scholar] [CrossRef]
- Gudes, S.; Barkai, O.; Caspi, Y.; Katz, B.; Lev, S.; Binshtok, A.M. The role of slow and persistent TTX-resistant sodium currents in acute tumor necrosis factor-α-mediated increase in nociceptors excitability. J. Neurophysiol. 2015, 113, 601–619. [Google Scholar] [CrossRef]
- Ilardi, C.R.; Iavarone, A.; La Marra, M.; Iachini, T.; Chieffi, S. Hand movements in Mild Cognitive Impairment: Clinical implications and insights for future research. J. Integr. Neurosci. 2022, 21, 67. [Google Scholar] [CrossRef]
- Pinto, L.G.; Cunha, T.M.; Vieira, S.M.; Lemos, H.P.; Verri, W.A.; Cunha, F.Q.; Ferreira, S.H. IL-17 mediates articular hypernociception in antigen-induced arthritis in mice. Pain 2010, 148, 247–256. [Google Scholar] [CrossRef]
- Cilenti, F.; Barbiera, G.; Caronni, N.; Iodice, D.; Montaldo, E.; Barresi, S.; Lusito, E.; Cuzzola, V.; Vittoria, F.M.; Mezzanzanica, L.; et al. A PGE2-MEF2A axis enables context-dependent control of inflammatory gene expression. Immunity 2021, 54, 1665–1682.e14. [Google Scholar] [CrossRef]
- Samuchiwal, S.K.; Balestrieri, B.; Raff, H.; Boyce, J.A. Endogenous prostaglandin E2 amplifies IL-33 production by macrophages through an E prostanoid (EP)2/EP4-cAMP-EPAC-dependent pathway. J. Biol. Chem. 2017, 292, 8195–8206. [Google Scholar] [CrossRef]
- Vuilleumier, P.H.; Schliessbach, J.; Curatolo, M. Current evidence for central analgesic effects of NSAIDs: An overview of the literature. Minerva Anestesiol. 2018, 84, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Souza, G.R.; Cunha, T.M.; Silva, R.L.; Lotufo, C.M.; Verri, W.A.; Funez, M.I.; Villarreal, C.F.; Talbot, J.; Sousa, L.P.; Parada, C.A.; et al. Involvement of nuclear factor kappa B in the maintenance of persistent inflammatory hypernociception. Pharmacol. Biochem. Behav. 2015, 134, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Langeslag, M.; Quarta, S.; Leitner, M.G.; Kress, M.; Mair, N. Sphingosine 1-Phosphate to p38 Signaling via S1P1 Receptor and Gαi/o Evokes Augmentation of Capsaicin-Induced Ionic Currents in Mouse Sensory Neurons. Mol. Pain 2014, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Posadas, A.; Picazo-Juárez, G.; Llorente, I.; Jara-Oseguera, A.; Morales-Lázaro, S.; Escalante-Alcalde, D.; Islas, L.D.; Rosenbaum, T. Lysophosphatidic acid directly activates TRPV1 through a C-terminal binding site. Nat. Chem. Biol. 2011, 8, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.-X.; Li, K.-X.; Hong, F.-F.; Yang, S.-L. Pain and bone damage in rheumatoid arthritis: Role of leukotriene B4. Ann. Rheum. Dis. 2019, 37, 872–878. [Google Scholar]
- Martin, H.; Basbaum, A.; Kwiat, G.; Goetzl, E.; Levine, J. Leukotriene and prostaglandin sensitization of cutaneous high-threshold C- and A-delta mechanonociceptors in the hairy skin of rat hindlimbs. Neuroscience 1987, 22, 651–659. [Google Scholar] [CrossRef]
- Andoh, T.; Kuraishi, Y. Expression of BLT1 leukotriene B4 receptor on the dorsal root ganglion neurons in mice. Mol. Brain Res. 2005, 137, 263–266. [Google Scholar] [CrossRef]
- Napimoga, M.H.; Souza, G.R.; Cunha, T.M.; Ferrari, L.F.; Clemente-Napimoga, J.T.; Parada, C.A.; Verri, W.A.; Cunha, F.Q.; Ferreira, S.H. 15d-Prostaglandin J2 Inhibits Inflammatory Hypernociception: Involvement of Peripheral Opioid Receptor. Experiment 2007, 324, 313–321. [Google Scholar] [CrossRef]
- Ji, R.-R.; Xu, Z.-Z.; Strichartz, G.; Serhan, C.N. Emerging roles of resolvins in the resolution of inflammation and pain. Trends Neurosci. 2011, 34, 599–609. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Pinho-Ribeiro, F.A.; Verri, W.A., Jr.; Chiu, I.M. Nociceptor Sensory Neuron–Immune Interactions in Pain and Inflammation. Trends Immunol. 2017, 38, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Park, C.-K.; Xu, Z.-Z.; Liu, T.; Lü, N.; Serhan, C.N.; Ji, R.-R. Resolvin D2 Is a Potent Endogenous Inhibitor for Transient Receptor Potential Subtype V1/A1, Inflammatory Pain, and Spinal Cord Synaptic Plasticity in Mice: Distinct Roles of Resolvin D1, D2, and E1. J. Neurosci. 2011, 31, 18433–18438. [Google Scholar] [CrossRef] [PubMed]
- Van Neerven, S.G.A.; Mouraux, A. Capsaicin-Induced Skin Desensitization Differentially Affects A-Delta and C-Fiber-Mediated Heat Sensitivity. Front. Pharmacol. 2020, 11, e00615. [Google Scholar] [CrossRef]
- Yoo, S.; Lim, J.Y.; Hwang, S.W. Sensory TRP Channel Interactions with Endogenous Lipids and Their Biological Outcomes. Molecules 2014, 19, 4708–4744. [Google Scholar] [CrossRef]
- Jancsó, G.; Kiraly, E.; Jancsó-Gábor, A. Pharmacologically induced selective degeneration of chemosensitive primary sensory neurones. Nature 1977, 270, 741–743. [Google Scholar] [CrossRef]
- Maggi, C.A. Tachykinins and calcitonin gene-related peptide (CGRP) as co-transmitters released from peripheral endings of sensory nerves. Prog. Neurobiol. 1995, 45, 1–98. [Google Scholar] [CrossRef]
- Neri, M.; Frati, A.; Turillazzi, E.; Cantatore, S.; Cipolloni, L.; Di Paolo, M.; Frati, P.; La Russa, R.; Maiese, A.; Scopetti, M.; et al. Immunohistochemical Evaluation of Aquaporin-4 and its Correlation with CD68, IBA-1, HIF-1α, GFAP, and CD15 Expressions in Fatal Traumatic Brain Injury. Int. J. Mol. Sci. 2018, 19, 3544. [Google Scholar] [CrossRef]
- Chieffi, S.; Messina, G.; La Marra, M.; Iavarone, A.; Viggiano, A.; De Luca, V.; Monda, M. Distractor interference in visual motor tasks. In Horizon in Neuroscience Research; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2014. [Google Scholar]
- Ilie, M.A.; Caruntu, C.; Tampa, M.; Georgescu, S.-R.; Matei, C.; Negrei, C.; Ion, R.-M.; Constantin, C.; Neagu, M.; Boda, D. Capsaicin: Physicochemical properties, cutaneous reactions and potential applications in painful and inflammatory conditions. Exp. Ther. Med. 2019, 18, 916–925. [Google Scholar] [CrossRef]
- Frey, E.; Karney-Grobe, S.; Krolak, T.; Milbrandt, J.; DiAntonio, A. TRPV1 Agonist, Capsaicin, Induces Axon Outgrowth after Injury via Ca2+/PKA Signaling. Eneuro 2018, 5. [Google Scholar] [CrossRef]
- Taylor, B.K. Spinal inhibitory neurotransmission in neuropathic pain. Curr. Pain Headache Rep. 2009, 13, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M. Pathobiology of neuropathic pain. Eur. J. Pharmacol. 2001, 429, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Murnion, B.P. Neuropathic pain: Current definition and review of drug treatment. Aust. Prescr. 2018, 41, 60–63. [Google Scholar] [CrossRef]
- Gracely, R.H.; Lynch, S.A.; Bennett, G.J. Painful neuropathy: Altered central processing maintained dynamically by peripheral input. Pain 1992, 51, 175–194. [Google Scholar] [CrossRef]
- Tsuzuki, K.; Kondo, E.; Fukuoka, T.; Yi, D.; Tsujino, H.; Sakagami, M.; Noguchi, K. Differential regulation of P2X3 mRNA expression by peripheral nerve injury in intact and injured neurons in the rat sensory ganglia. Pain 2001, 91, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Ji, R.-R.; Ito, N.; Allchorne, A.J.; Befort, K.; Karchewski, L.A.; Woolf, C.J. Peripheral axonal injury results in reduced μ opioid receptor pre- and post-synaptic action in the spinal cord. Pain 2005, 117, 77–87. [Google Scholar] [CrossRef]
- Emery, E.C.; Ernfors, P. Dorsal root ganglion neuron types and their functional specialization. In The Oxford Handbook of the Neurobiology of Pain; Oxford University Press: Oxford, UK, 2018; pp. 1–30. [Google Scholar]
- Jänig, W.; Grossmann, L.; Gorodetskaya, N. Mechano- and thermosensitivity of regenerating cutaneous afferent nerve fibers. Exp. Brain Res. 2009, 196, 101–114. [Google Scholar] [CrossRef]
- McLachlan, E.M.; Jänig, W.; Devor, M.; Michaelis, M. Peripheral nerve injury triggers noradrenergic sprouting within dorsal root ganglia. Nature 1993, 363, 543–546. [Google Scholar] [CrossRef]
- Amir, R.; Argoff, C.E.; Bennett, G.J.; Cummins, T.R.; Durieux, M.E.; Gerner, P.; Gold, M.S.; Porreca, F.; Strichartz, G.R. The Role of Sodium Channels in Chronic Inflammatory and Neuropathic Pain. J. Pain 2006, 7, S1–S29. [Google Scholar] [CrossRef]
- Sousa-Valente, J.; Brain, S.D. A historical perspective on the role of sensory nerves in neurogenic inflammation. Semin. Immunopathol. 2018, 40, 229–236. [Google Scholar] [CrossRef]
- Brain, S.; Williams, T.J. Inflammatory oedema induced by synergism between calcitonin gene-related peptide (CGRP) and mediators of increased vascular permeability. Br. J. Pharmacol. 1985, 86, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Messina, A.; Monda, V.; Avola, R.; Moscatelli, F.; Avalenzano AA, V.; Villano, I.; Messina, G. Role Of The Orexin System on Arousal, Attention, Feeding Behaviour And Sleep Disorders. Acta Med. Mediterr. 2017, 4, 645–649. [Google Scholar]
- Zieglgänsberger, W. Substance P and pain chronicity. Cell Tissue Res. 2018, 375, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S. Substance P. Int. J. Biochem. Cell Biol. 2001, 33, 555–576. [Google Scholar] [CrossRef] [PubMed]
- Severini, C. The Tachykinin Peptide Family. Pharmacol. Rev. 2002, 54, 285–322. [Google Scholar] [CrossRef] [PubMed]
- La Marra, M.; Caviglia, G.; Perrella, R. Using Smartphones When Eating Increases Caloric Intake in Young People: An Overview of the Literature. Front. Psychol. 2020, 11, 587886. [Google Scholar] [CrossRef]
- Siniscalco, D.; Giordano, C.; Rossi, F.; Maione, S.; De Novellis, V. Role of Neurotrophins in Neuropathic Pain. Curr. Neuropharmacol. 2011, 9, 523–529. [Google Scholar] [CrossRef]
- Hökfelt, T.; Pernow, B.; Wahren, J. Substance P: A pioneer amongst neuropeptides. J. Intern. Med. 2001, 249, 27–40. [Google Scholar] [CrossRef]
- Maggi, C.A. The mammalian tachykinin receptors. Gen. Pharmacol. Vasc. Syst. 1995, 26, 911–944. [Google Scholar] [CrossRef]
- Vennekens, R.; Menigoz, A.; Nilius, B. TRPs in the Brain. Rev. Physiol. Biochem. Pharmacol. 2012, 163, 27–64. [Google Scholar] [CrossRef]
- Saffroy, M.; Torrens, Y.; Glowinski, J.; Beaujouan, J.-C. Autoradiographic distribution of tachykinin NK2 binding sites in the rat brain: Comparison with NK1 and NK3 binding sites. Neuroscience 2003, 116, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Terrón, J.A. Is the 5-HT7 receptor involved in the pathogenesis and prophylactic treatment of migraine? Eur. J. Pharmacol. 2002, 439, 1–11. [Google Scholar] [CrossRef]
- Messina, A.; Bitetti, I.; Precenzano, F.; Iacono, D.; Messina, G.; Roccella, M.; Parisi, L.; Salerno, M.; Valenzano, A.; Maltese, A.; et al. Non-Rapid Eye Movement Sleep Parasomnias and Migraine: A Role of Orexinergic Projections. Front. Neurol. 2018, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Villano, I.; La Marra, M.; Di Maio, G.; Monda, V.; Chieffi, S.; Guatteo, E.; Messina, G.; Moscatelli, F.; Monda, M.; Messina, A. Physiological Role of Orexinergic System for Health. Int. J. Environ. Res. Public Heal. 2022, 19, 8353. [Google Scholar] [CrossRef]
- Chakraborty, S.; Nepiyushchikh, Z.; Davis, M.J.; Zawieja, D.C.; Muthuchamy, M. Substance P Activates Both Contractile and Inflammatory Pathways in Lymphatics Through the Neurokinin Receptors NK1R and NK3R. Microcirculation 2010, 18, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Ilardi, C.R.; Garofalo, E.; Chieffi, S.; Gamboz, N.; La Marra, M.; Iavarone, A. Daily exposure to digital displays may affect the clock-drawing test: From psychometrics to serendipity. Neurol. Sci. 2020, 41, 3683–3690. [Google Scholar] [CrossRef]
- Lorusso, L.; Salerno, M.; Sessa, F.; Nicolosi, D.; Longhitano, L.; Loreto, C.; Carotenuto, M.; Messina, A.; Monda, V.; Villano, I.; et al. Autoalgometry: An Important Tool for Pressure Pain Threshold Evaluation. J. Clin. Med. 2018, 7, 273. [Google Scholar] [CrossRef]

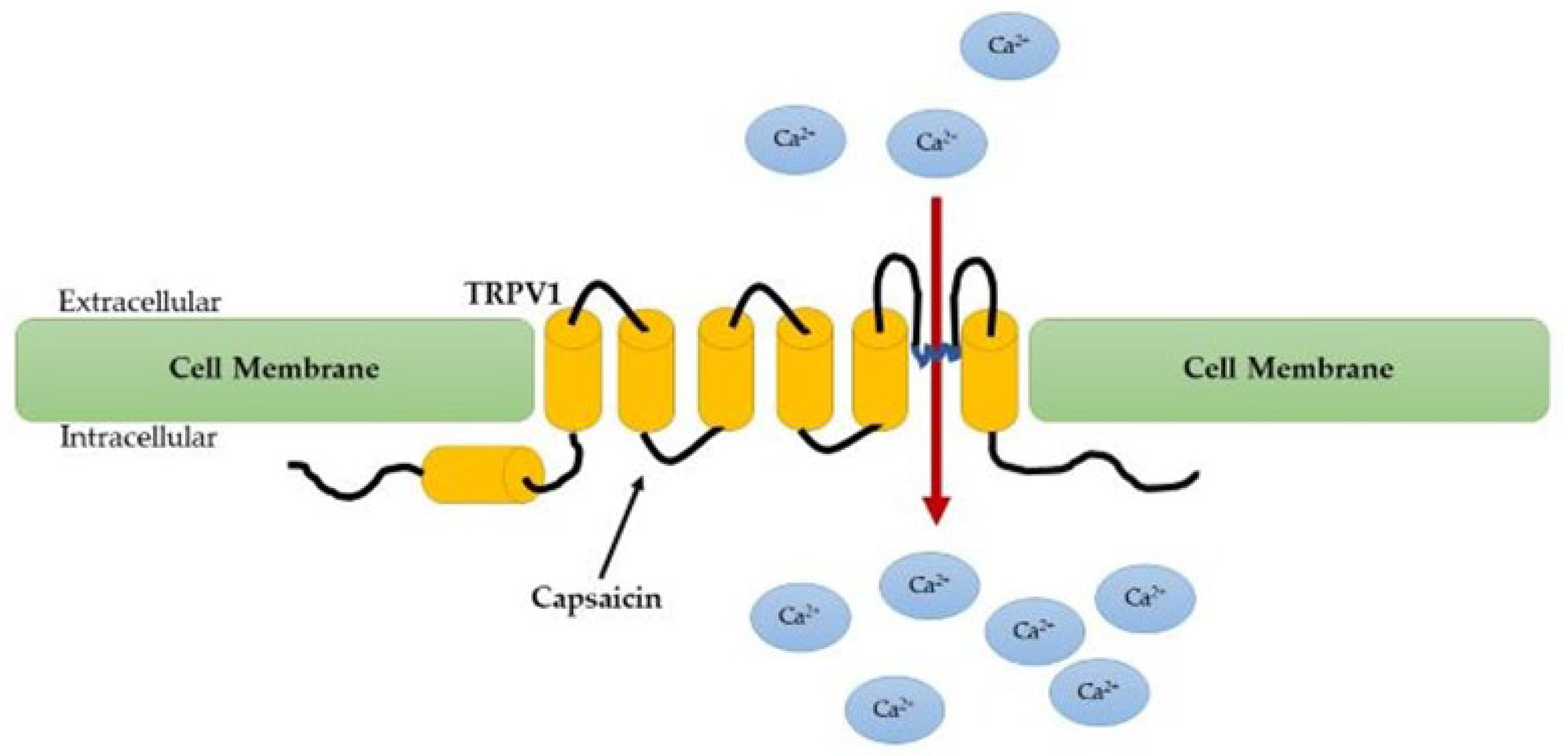

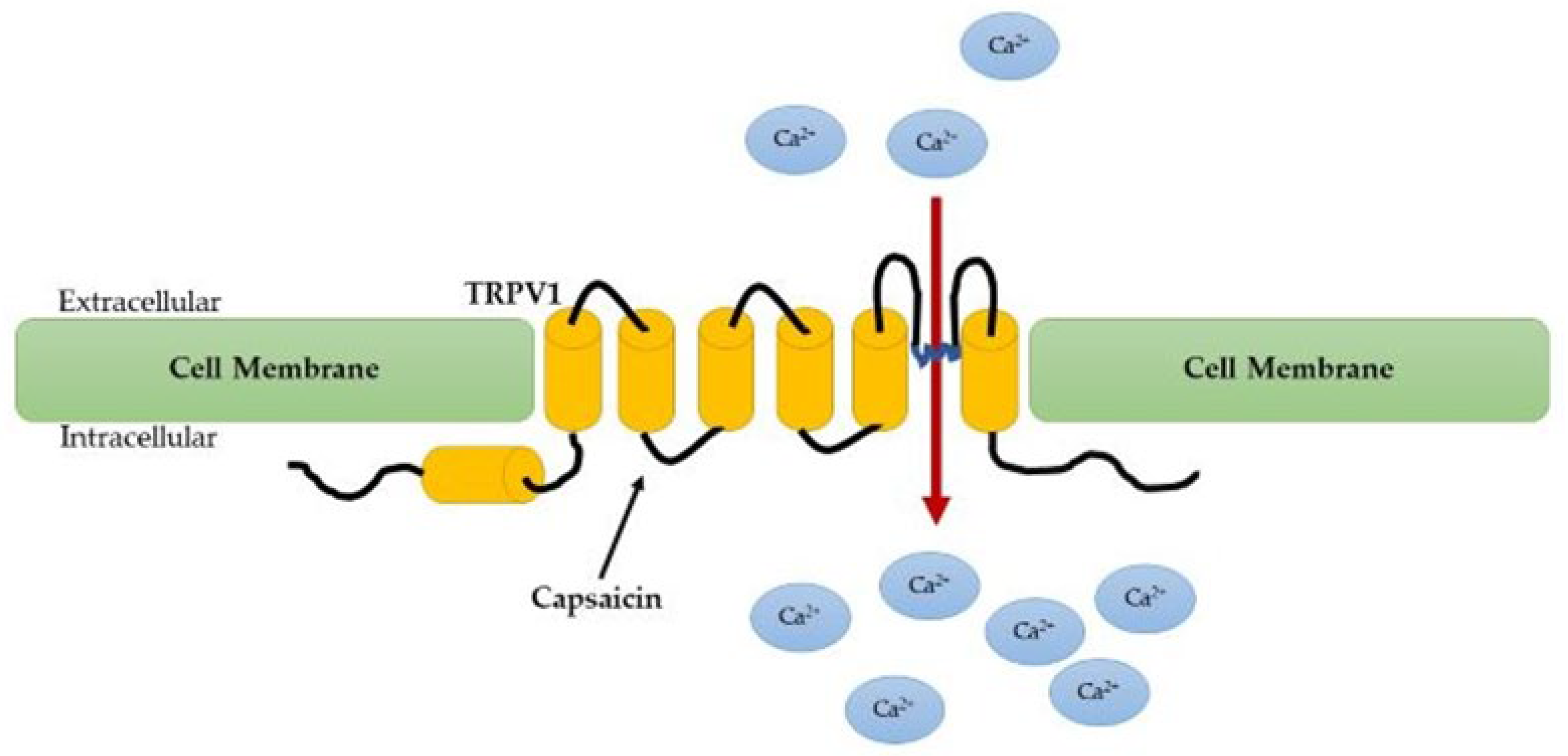
{kind=link}
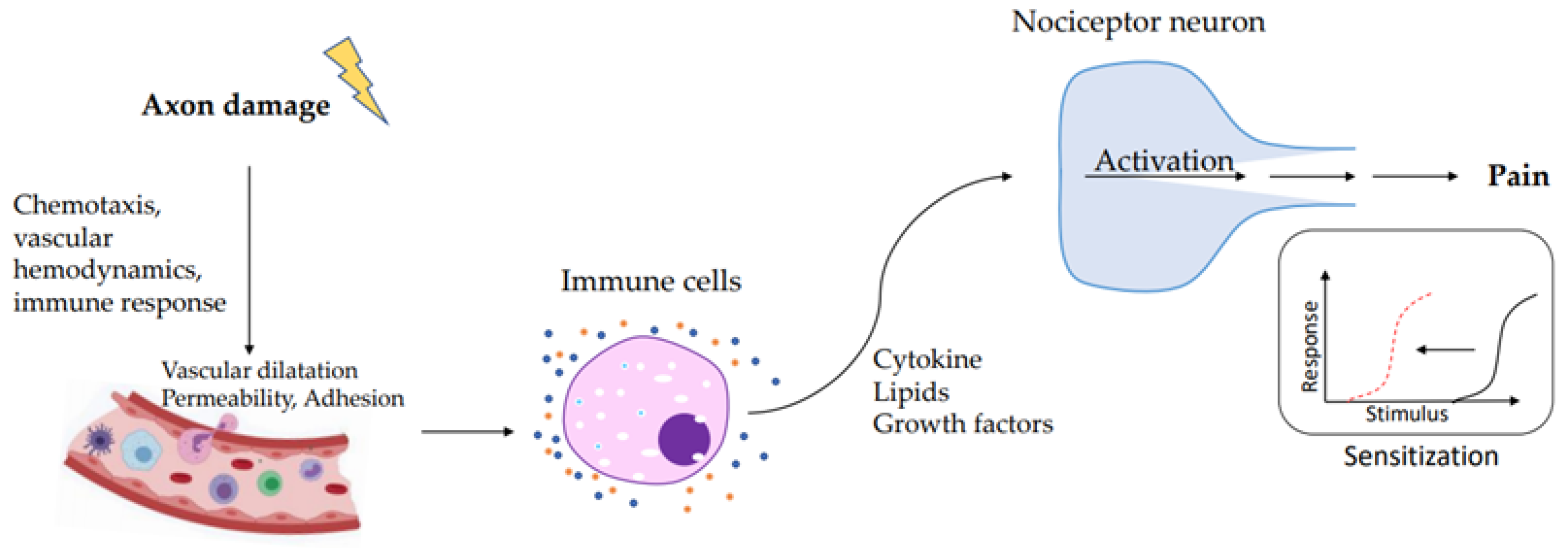
{kind=link}
| Immune Cell | Mediator | Receptor |
|---|---|---|
| Mast cell | IL-5 | IL-5R |
| 5-HT | 5-HT2 | |
| IL-6 | Gp130 | |
| IL-1β | IL-1R1 | |
| TNF/α | TNFR1 | |
| Histamine | H1/2 | |
| NGF | TrkA | |
| T cell | IL-17A | IL-17AR |
| IFN-γ | IFN-γR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Maio, G.; Villano, I.; Ilardi, C.R.; Messina, A.; Monda, V.; Iodice, A.C.; Porro, C.; Panaro, M.A.; Chieffi, S.; Messina, G.; et al. Mechanisms of Transmission and Processing of Pain: A Narrative Review. Int. J. Environ. Res. Public Health 2023, 20, 3064. https://doi.org/10.3390/ijerph20043064
Di Maio G, Villano I, Ilardi CR, Messina A, Monda V, Iodice AC, Porro C, Panaro MA, Chieffi S, Messina G, et al. Mechanisms of Transmission and Processing of Pain: A Narrative Review. International Journal of Environmental Research and Public Health. 2023; 20(4):3064. https://doi.org/10.3390/ijerph20043064
Chicago/Turabian StyleDi Maio, Girolamo, Ines Villano, Ciro Rosario Ilardi, Antonietta Messina, Vincenzo Monda, Ashlei Clara Iodice, Chiara Porro, Maria Antonietta Panaro, Sergio Chieffi, Giovanni Messina, and et al. 2023. "Mechanisms of Transmission and Processing of Pain: A Narrative Review" International Journal of Environmental Research and Public Health 20, no. 4: 3064. https://doi.org/10.3390/ijerph20043064
APA StyleDi Maio, G., Villano, I., Ilardi, C. R., Messina, A., Monda, V., Iodice, A. C., Porro, C., Panaro, M. A., Chieffi, S., Messina, G., Monda, M., & La Marra, M. (2023). Mechanisms of Transmission and Processing of Pain: A Narrative Review. International Journal of Environmental Research and Public Health, 20(4), 3064. https://doi.org/10.3390/ijerph20043064