Circulating Peptidome Is Strongly Altered in COVID-19 Patients

 ,
,  ,
,  , ,
, ,  ,
, 

Abstract
1. Introduction
2. Materials and Methods
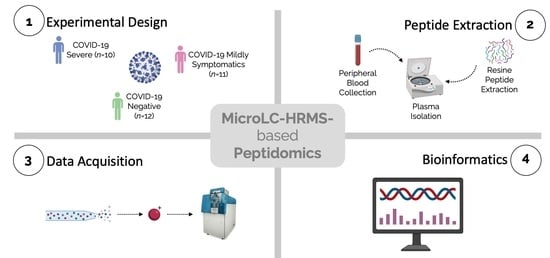
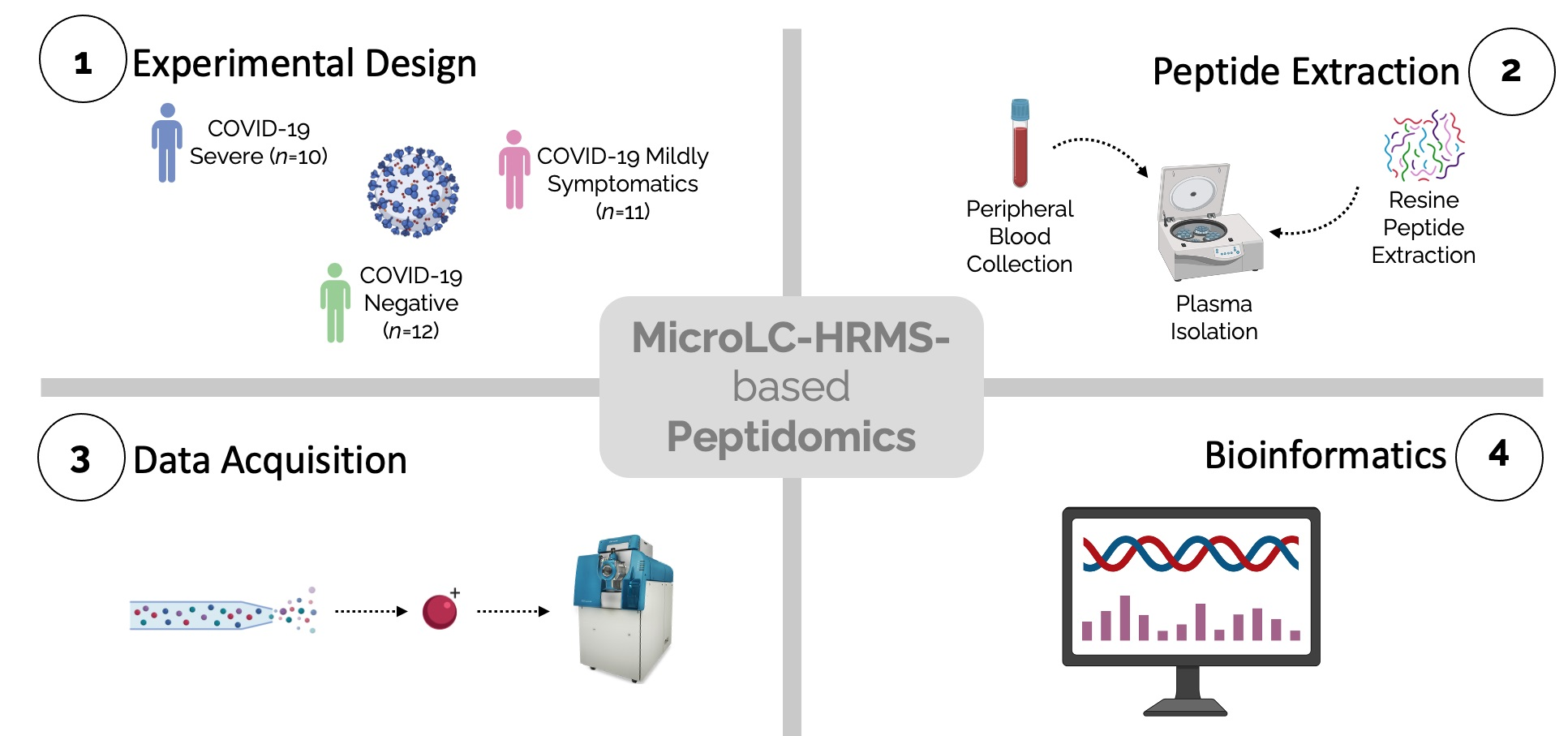
2.1. Patients
2.2. Peptide Extraction
2.3. LC-MS/MS Analyses
2.4. Peptide Database Search
2.5. Statistical and Bioinformatic Analysis
3. Results
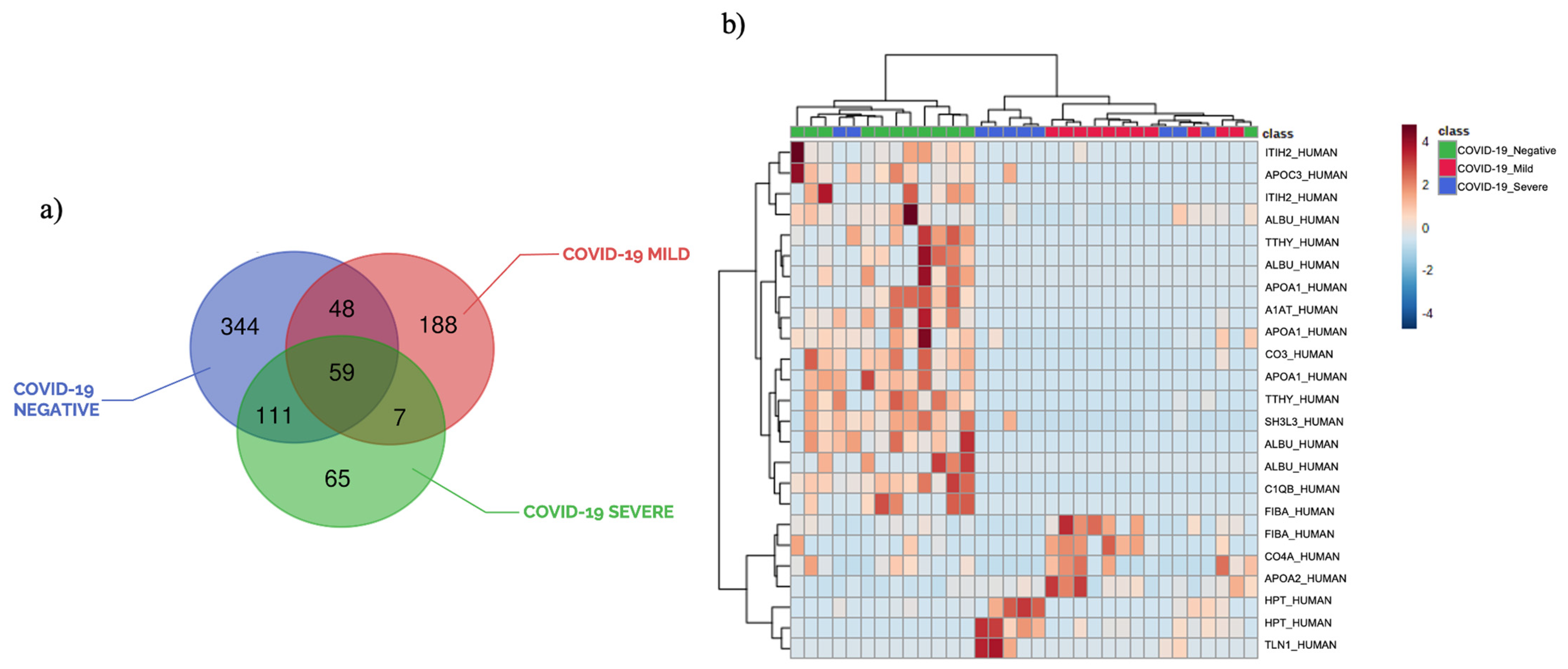
3.1. Plasma Peptide Alterations in COVID-19 Patients
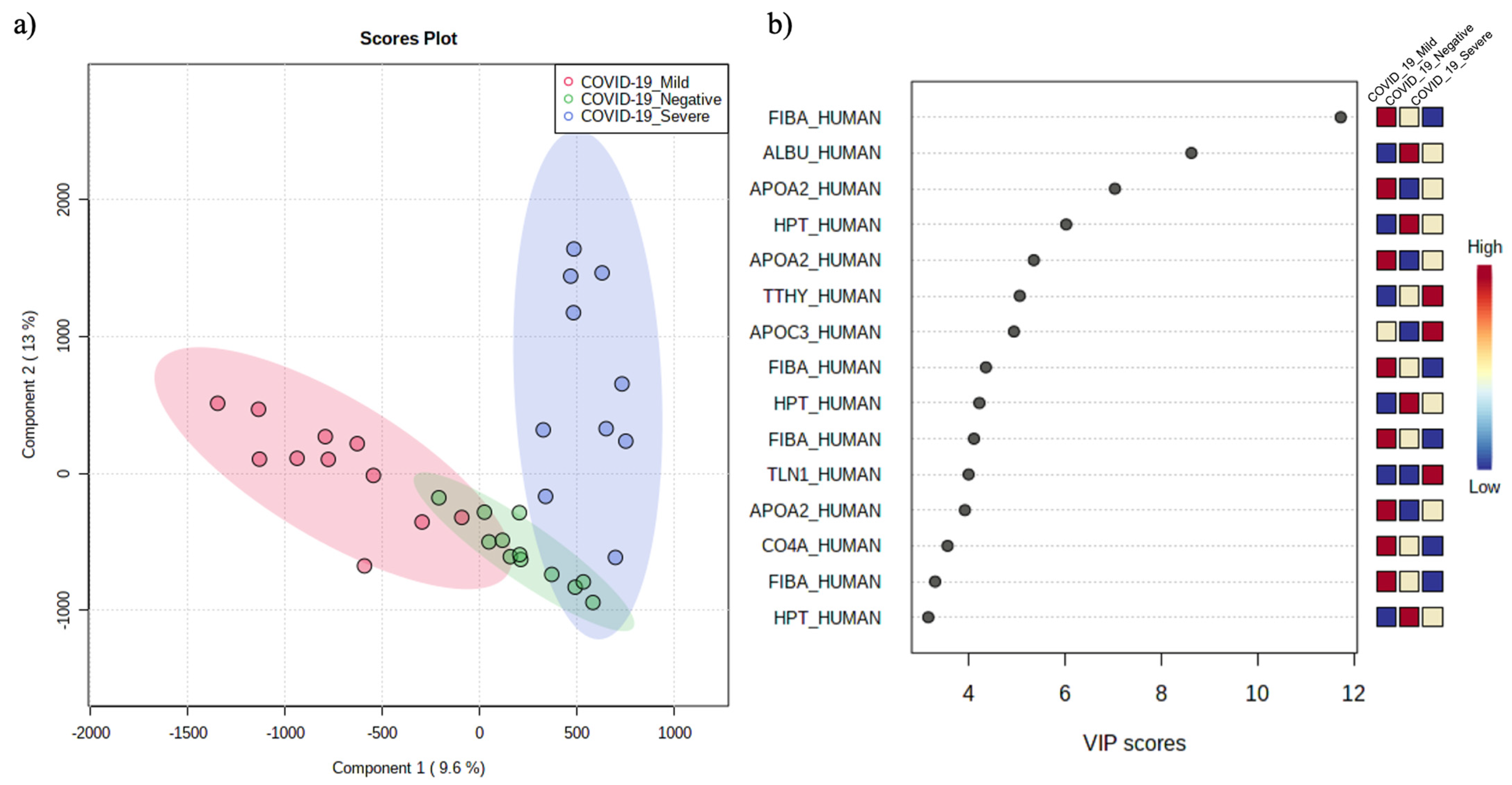
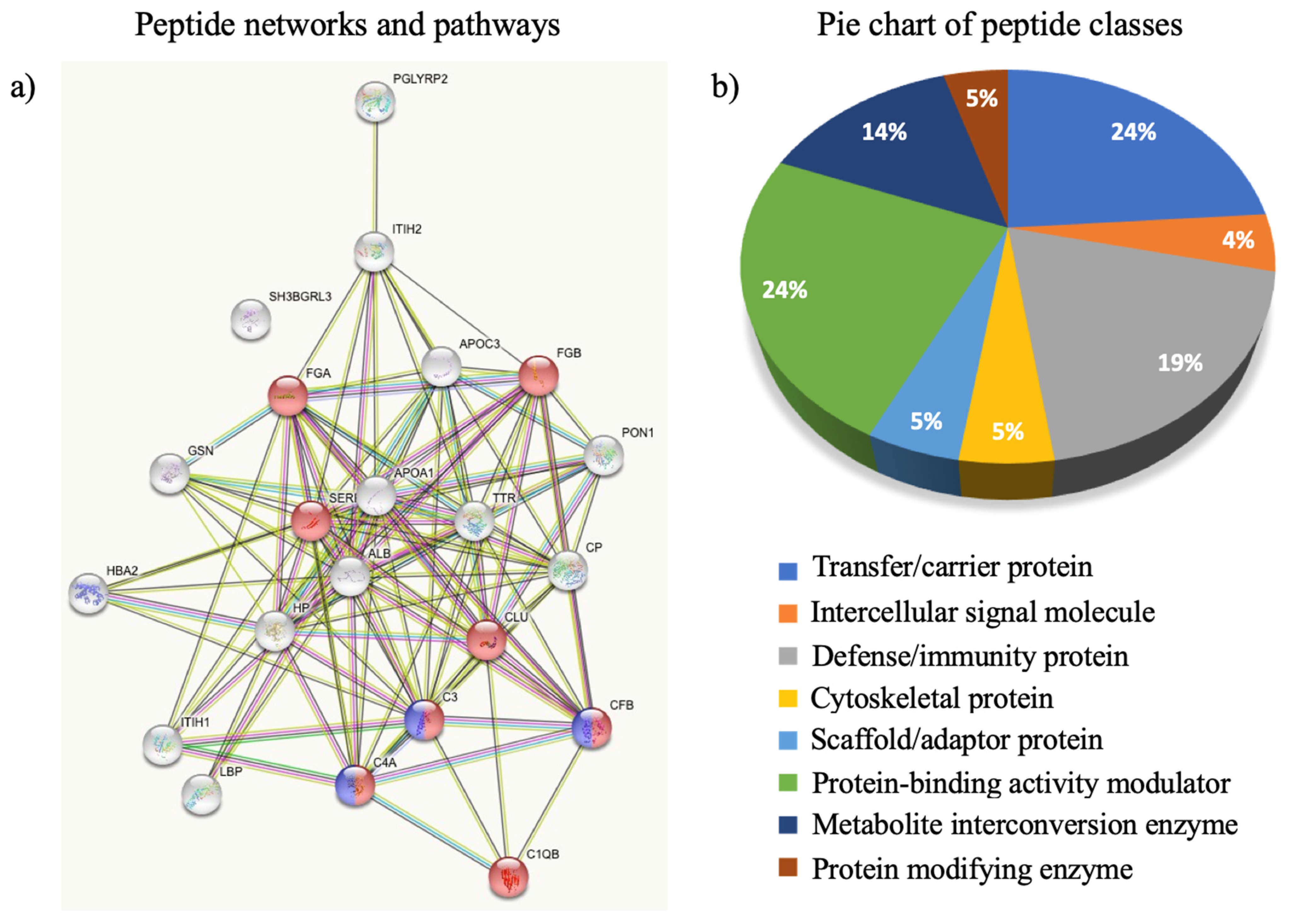
3.2. The Circulating Peptidome Reflects the Disease State
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 13 September 2021).
- D’Alessandro, A.; Thomas, T.; Dzieciatkowska, M.; Hill, R.C.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hod, E.A.; Spitalnik, S.L.; Hansen, K.C. Serum Proteomics in COVID-19 Patients: Altered Coagulation and Complement Status as a Function of IL-6 Level. J. Proteome Res. 2020, 19, 4417–4427. [Google Scholar] [CrossRef] [PubMed]
- Battaglini, D.; Lopes-Pacheco, M.; Castro-Faria-Neto, H.C.; Pelosi, P.; Rocco, P.R.M. Laboratory Biomarkers for Diagnosis and Prognosis in COVID-19. Front. Immunol. 2022, 13, 857573. [Google Scholar] [CrossRef]
- Gordon, D.E.; Hiatt, J.; Bouhaddou, M.; Rezelj, V.V.; Ulferts, S.; Braberg, H.; Jureka, A.S.; Obernier, K.; Guo, J.Z.; Batra, J.; et al. Comparative Host-Coronavirus Protein Interaction Networks Reveal Pan-Viral Disease Mechanisms. Science 2020, 370, eabe9403. [Google Scholar] [CrossRef]
- Feng, L.; Yin, Y.-Y.; Liu, C.-H.; Xu, K.-R.; Li, Q.-R.; Wu, J.-R.; Zeng, R. Proteome-Wide Data Analysis Reveals Tissue-Specific Network Associated with SARS-CoV-2 Infection. J. Mol. Cell Biol. 2020, 12, 946–957. [Google Scholar] [CrossRef]
- Yu, S.; Li, X.; Xin, Z.; Sun, L.; Shi, J. Proteomic Insights into SARS-CoV-2 Infection Mechanisms, Diagnosis, Therapies and Prognostic Monitoring Methods. Front. Immunol. 2022, 13, 923387. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guo, M.; Tian, X.; Wang, X.; Yang, X.; Wu, P.; Liu, C.; Xiao, Z.; Qu, Y.; Yin, Y.; et al. Virus-Host Interactome and Proteomic Survey Reveal Potential Virulence Factors Influencing SARS-CoV-2 Pathogenesis. Med 2021, 2, 99–112. [Google Scholar] [CrossRef]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel Proteomics Reveals Host Perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Münch, C. Proteomics of SARS-CoV-2-Infected Host Cells Reveals Therapy Targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef]
- Appelberg, S.; Gupta, S.; Akusjärvi, S.S.; Ambikan, A.T.; Mikaeloff, F.; Saccon, E.; Végvári, Á.; Benfeitas, R.; Sperk, M.; Ståhlberg, M.; et al. Dysregulation in Akt/MTOR/HIF-1 Signaling Identified by Proteo-Transcriptomics of SARS-CoV-2 Infected Cells. Emerg. Microbes Infect. 2020, 9, 1748–1760. [Google Scholar] [CrossRef]
- Captur, G.; Moon, J.C.; Topriceanu, C.-C.; Joy, G.; Swadling, L.; Hallqvist, J.; Doykov, I.; Patel, N.; Spiewak, J.; Baldwin, T.; et al. Plasma Proteomic Signature Predicts Who Will Get Persistent Symptoms Following SARS-CoV-2 Infection. eBioMedicine 2022, 85, 104293. [Google Scholar] [CrossRef]
- Ciccosanti, F.; Antonioli, M.; Sacchi, A.; Notari, S.; Farina, A.; Beccacece, A.; Fusto, M.; Vergori, A.; D’Offizi, G.; Taglietti, F.; et al. Proteomic Analysis Identifies a Signature of Disease Severity in the Plasma of COVID-19 Pneumonia Patients Associated to Neutrophil, Platelet and Complement Activation. Clin. Proteom. 2022, 19, 38. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020, 182, 59–72. [Google Scholar] [CrossRef]
- Messner, C.B.; Demichev, V.; Wendisch, D.; Michalick, L.; White, M.; Freiwald, A.; Textoris-Taube, K.; Vernardis, S.I.; Egger, A.-S.; Kreidl, M.; et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst. 2020, 11, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Barberis, E.; Timo, S.; Amede, E.; Vanella, V.V.; Puricelli, C.; Cappellano, G.; Raineri, D.; Cittone, M.G.; Rizzi, E.; Pedrinelli, A.R.; et al. Large-Scale Plasma Analysis Revealed New Mechanisms and Molecules Associated with the Host Response to SARS-CoV-2. Int. J. Mol. Sci. 2020, 21, 8623. [Google Scholar] [CrossRef]
- Bi, X.; Liu, W.; Ding, X.; Liang, S.; Zheng, Y.; Zhu, X.; Quan, S.; Yi, X.; Xiang, N.; Du, J.; et al. Proteomic and Metabolomic Profiling of Urine Uncovers Immune Responses in Patients with COVID-19. Cell Rep. 2022, 38, 110271. [Google Scholar] [CrossRef]
- He, F.; Zhang, T.; Xue, K.; Fang, Z.; Jiang, G.; Huang, S.; Li, K.; Gu, Z.; Shi, H.; Zhang, Z.; et al. Fecal Multi-Omics Analysis Reveals Diverse Molecular Alterations of Gut Ecosystem in COVID-19 Patients. Anal. Chim. Acta 2021, 1180, 338881. [Google Scholar] [CrossRef]
- Barberis, E.; Amede, E.; Khoso, S.; Castello, L.; Sainaghi, P.P.; Bellan, M.; Balbo, P.E.; Patti, G.; Brustia, D.; Giordano, M.; et al. Metabolomics Diagnosis of COVID-19 from Exhaled Breath Condensate. Metabolites 2021, 11, 847. [Google Scholar] [CrossRef]
- Haas, P.; Muralidharan, M.; Krogan, N.J.; Kaake, R.M.; Hüttenhain, R. Proteomic Approaches to Study SARS-CoV-2 Biology and COVID-19 Pathology. J. Proteome Res. 2021, 20, 1133–1152. [Google Scholar] [CrossRef]
- Hasan, M.R.; Suleiman, M.; Pérez-López, A. Metabolomics in the Diagnosis and Prognosis of COVID-19. Front. Genet. 2021, 12, 1358. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Huang, W.; Li, Y.; Lai, C.; Huang, S.; Wang, G.; He, Y.; Hu, L.; Chen, C. Lipidomic Alteration of Plasma in Cured COVID-19 Patients Using Ultra High-Performance Liquid Chromatography with High-Resolution Mass Spectrometry. Biosci. Rep. 2021, 41, BSR20204305. [Google Scholar] [CrossRef] [PubMed]
- Scrima, M.; Cossu, A.M.; D’Andrea, E.L.; Bocchetti, M.; Abruzzese, Y.; Iannarone, C.; Miarelli, C.; Grisolia, P.; Melisi, F.; Genua, L.; et al. Genomic Characterization of the Emerging SARS-CoV-2 Lineage in Two Districts of Campania (Italy) Using Next-Generation Sequencing. Front. Virol. 2022, 2, 814114. [Google Scholar] [CrossRef]
- Costanzo, M.; Caterino, M.; Fedele, R.; Cevenini, A.; Pontillo, M.; Barra, L.; Ruoppolo, M. COVIDomics: The Proteomic and Metabolomic Signatures of COVID-19. Int. J. Mol. Sci. 2022, 23, 2414. [Google Scholar] [CrossRef] [PubMed]
- Foreman, R.E.; George, A.L.; Reimann, F.; Gribble, F.M.; Kay, R.G. Peptidomics: A Review of Clinical Applications and Methodologies. J. Proteome Res. 2021, 20, 3782–3797. [Google Scholar] [CrossRef] [PubMed]
- Bastos, P.; Trindade, F.; da Costa, J.; Ferreira, R.; Vitorino, R. Human Antimicrobial Peptides in Bodily Fluids: Current Knowledge and Therapeutic Perspectives in the Postantibiotic Era. Med. Res. Rev. 2018, 38, 101–146. [Google Scholar] [CrossRef]
- Meo, A.D.; Pasic, M.D.; Yousef, G.M. Proteomics and Peptidomics: Moving toward Precision Medicine in Urological Malignancies. Oncotarget 2016, 7, 52460–52474. [Google Scholar] [CrossRef]
- Piovesana, S.; Cerrato, A.; Antonelli, M.; Benedetti, B.; Capriotti, A.L.; Cavaliere, C.; Montone, C.M.; Laganà, A. A Clean-up Strategy for Identification of Circulating Endogenous Short Peptides in Human Plasma by Zwitterionic Hydrophilic Liquid Chromatography and Untargeted Peptidomics Identification. J. Chromatogr. A 2020, 1613, 460699. [Google Scholar] [CrossRef]
- Fricker, L.D.; Lim, J.; Pan, H.; Che, F.-Y. Peptidomics: Identification and Quantification of Endogenous Peptides in Neuroendocrine Tissues. Mass Spectrom. Rev. 2006, 25, 327–344. [Google Scholar] [CrossRef]
- Petricoin, E.F.; Belluco, C.; Araujo, R.P.; Liotta, L.A. The Blood Peptidome: A Higher Dimension of Information Content for Cancer Biomarker Discovery. Nat. Rev. Cancer 2006, 6, 961–967. [Google Scholar] [CrossRef]
- Tammen, H.; Hess, R. Collection and Handling of Blood Specimens for Peptidomics. In Serum/Plasma Proteomics: Methods and Protocols; Simpson, R.J., Greening, D.W., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2011; pp. 151–159. ISBN 978-1-61779-068-3. [Google Scholar]
- Vitorino, R. Digging Deep into Peptidomics Applied to Body Fluids. Proteomics 2018, 18, 1700401. [Google Scholar] [CrossRef]
- Zhao, S.; Yin, C.; Zhai, Y.; Jia, Z.; Su, S.; Lu, Y.; Meng, L.; Li, C.; Liu, X.; Cong, Y.; et al. Serum Peptidomic Screening Identified Circulating Peptide Biomarkers Predictive for Preeclampsia. Front. Cardiovasc. Med. 2022, 9, 2988. [Google Scholar] [CrossRef]
- Shender, V.; Arapidi, G.; Butenko, I.; Anikanov, N.; Ivanova, O.; Govorun, V. Peptidome Profiling Dataset of Ovarian Cancer and Non-Cancer Proximal Fluids: Ascites and Blood Sera. Data Br. 2018, 22, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, Z.; Huang, H.; Suzek, B.E.; Wu, C.H. A Fast Peptide Match Service for UniProt Knowledgebase. Bioinformatics 2013, 29, 2808–2809. [Google Scholar] [CrossRef] [PubMed]
- Overmyer, K.A.; Shishkova, E.; Miller, I.J.; Balnis, J.; Bernstein, M.N.; Peters-Clarke, T.M.; Meyer, J.G.; Quan, Q.; Muehlbauer, L.K.; Trujillo, E.A.; et al. Large-Scale Multi-Omic Analysis of COVID-19 Severity. Cell Syst. 2021, 12, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, M.; Brandi, J.; Di Carlo, C.; Vita Vanella, V.; Barberis, E.; Marengo, E.; Patrone, M.; Cecconi, D. Mining Cancer Biology through Bioinformatic Analysis of Proteomic Data. Expert Rev. Proteom. 2019, 16, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards More Transparent and Integrative Metabolomics Analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed]
- Spick, M.; Campbell, A.; Baricevic-Jones, I.; von Gerichten, J.; Lewis, H.-M.; Frampas, C.F.; Longman, K.; Stewart, A.; Dunn-Walters, D.; Skene, D.J.; et al. Multi-Omics Reveals Mechanisms of Partial Modulation of COVID-19 Dysregulation by Glucocorticoid Treatment. Int. J. Mol. Sci. 2022, 23, 12079. [Google Scholar] [CrossRef] [PubMed]
- Henry, B.M.; Oliveira MHS, d.e.; Benoit, S.; Plebani, M.; Lippi, G. Hematologic, Biochemical and Immune Biomarker Abnormalities Associated with Severe Illness and Mortality in Coronavirus Disease 2019 (COVID-19): A Meta-Analysis. Clin. Chem. Lab. Med. 2020, 58, 1021–1028. [Google Scholar] [CrossRef]
- Kimhofer, T.; Lodge, S.; Whiley, L.; Gray, N.; Loo, R.L.; Lawler, N.G.; Nitschke, P.; Bong, S.-H.; Morrison, D.L.; Begum, S.; et al. Integrative Modeling of Quantitative Plasma Lipoprotein, Metabolic, and Amino Acid Data Reveals a Multiorgan Pathological Signature of SARS-CoV-2 Infection. J. Proteome Res. 2020, 19, 4442–4454. [Google Scholar] [CrossRef]
- Demichev, V.; Tober-Lau, P.; Lemke, O.; Nazarenko, T.; Thibeault, C.; Whitwell, H.; Röhl, A.; Freiwald, A.; Szyrwiel, L.; Ludwig, D.; et al. A Time-Resolved Proteomic and Prognostic Map of COVID-19. Cell Syst. 2021, 12, 780–794. [Google Scholar] [CrossRef]
- Hou, X.; Zhang, X.; Wu, X.; Lu, M.; Wang, D.; Xu, M.; Wang, H.; Liang, T.; Dai, J.; Duan, H.; et al. Serum Protein Profiling Reveals a Landscape of Inflammation and Immune Signaling in Early-Stage COVID-19 Infection. Mol. Cell. Proteom. 2020, 19, 1749–1759. [Google Scholar] [CrossRef]
- He, T.; Pejchinovski, M.; Mullen, W.; Beige, J.; Mischak, H.; Jankowski, V. Peptides in Plasma, Urine, and Dialysate: Toward Unravelling Renal Peptide Handling. Proteom.–Clin. Appl. 2021, 15, 2000029. [Google Scholar] [CrossRef] [PubMed]
- Richter, R.; Schulz-Knappe, P.; Schrader, M.; Ständker, L.; Jürgens, M.; Tammen, H.; Forssmann, W.G. Composition of the Peptide Fraction in Human Blood Plasma: Database of Circulating Human Peptides. J. Chromatogr. B Biomed. Sci. Appl. 1999, 726, 25–35. [Google Scholar] [CrossRef]
- Malerba, M.; Ricciardolo, F.; Radaeli, A.; Torregiani, C.; Ceriani, L.; Mori, E.; Bontempelli, M.; Tantucci, C.; Grassi, V. Neutrophilic Inflammation and IL-8 Levels in Induced Sputum of Alpha-1-antitrypsin PiMZ Subjects. Thorax 2006, 61, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Keshavjee, S.; Liu, M. Alpha-1 Antitrypsin for COVID-19 Treatment: Dual Role in Antiviral Infection and Anti-Inflammation. Front. Pharmacol. 2020, 11, 615398. [Google Scholar] [CrossRef] [PubMed]
- Wettstein, L.; Weil, T.; Conzelmann, C.; Müller, J.A.; Groß, R.; Hirschenberger, M.; Seidel, A.; Klute, S.; Zech, F.; Prelli Bozzo, C.; et al. Alpha-1 Antitrypsin Inhibits TMPRSS2 Protease Activity and SARS-CoV-2 Infection. Nat. Commun. 2021, 12, 1726. [Google Scholar] [CrossRef]
- Lim, Y.-P.; Bendelja, K.; Opal, S.M.; Siryaporn, E.; Hixson, D.C.; Palardy, J.E. Correlation between Mortality and the Levels of Inter-Alpha Inhibitors in the Plasma of Patients with Severe Sepsis. J. Infect. Dis. 2003, 188, 919–926. [Google Scholar] [CrossRef]
- Zhuo, L.; Kimata, K. Structure and Function of Inter-α-Trypsin Inhibitor Heavy Chains. Connect. Tissue Res. 2008, 49, 311–320. [Google Scholar] [CrossRef]
- Paces, J.; Strizova, Z.; Smrz, D.; Cerny, J. COVID-19 and the Immune System. Physiol. Res 2020, 379–388. [Google Scholar] [CrossRef]
- Wu, Y.; Huang, X.; Sun, J.; Xie, T.; Lei, Y.; Muhammad, J.; Li, X.; Zeng, X.; Zhou, F.; Qin, H.; et al. Clinical Characteristics and Immune Injury Mechanisms in 71 Patients with COVID-19. mSphere 2020, 5, e00362-20. [Google Scholar] [CrossRef]
- Fang, S.; Wang, H.; Lu, L.; Jia, Y.; Xia, Z. Decreased Complement C3 Levels Are Associated with Poor Prognosis in Patients with COVID-19: A Retrospective Cohort Study. Int. Immunopharmacol. 2020, 89, 107070. [Google Scholar] [CrossRef]
- Zinellu, A.; Mangoni, A.A. Serum Complement C3 and C4 and COVID-19 Severity and Mortality: A Systematic Review and Meta-Analysis with Meta-Regression. Front. Immunol. 2021, 12, 2184. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, X.; Zhang, W.; Shi, Z.-L.; Zheng, Z.; Wang, T. Clinical Features and Treatment of 2019-NCov Pneumonia Patients in Wuhan: Report of A Couple Cases. Virol. Sin. 2020, 35, 330–336. [Google Scholar] [CrossRef]
- Wieczorek, E.; Ożyhar, A. Transthyretin: From Structural Stability to Osteoarticular and Cardiovascular Diseases. Cells 2021, 10, 1768. [Google Scholar] [CrossRef] [PubMed]
- Dellière, S.; Cynober, L. Is Transthyretin a Good Marker of Nutritional Status? Clin. Nutr. 2017, 36, 364–370. [Google Scholar] [CrossRef]
- Wagener, F.A.D.T.G.; Pickkers, P.; Peterson, S.J.; Immenschuh, S.; Abraham, N.G. Targeting the Heme-Heme Oxygenase System to Prevent Severe Complications Following COVID-19 Infections. Antioxidants 2020, 9, 540. [Google Scholar] [CrossRef]
- Linder, M.C. Ceruloplasmin and Other Copper Binding Components of Blood Plasma and Their Functions: An Update. Metallomics 2016, 8, 887–905. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Akassoglou, K. Fibrinogen as a Key Regulator of Inflammation in Disease. Semin. Immunopathol. 2012, 34, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Hardy, E.; Fernandez-Patron, C. Targeting MMP-Regulation of Inflammation to Increase Metabolic Tolerance to COVID-19 Pathologies: A Hypothesis. Biomolecules 2021, 11, 390. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS Database of Proteolytic Enzymes, Their Substrates and Inhibitors in 2017 and a Comparison with Peptidases in the PANTHER Database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and Tissue Factor-Enriched Neutrophil Extracellular Traps Are Key Drivers in COVID-19 Immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Ueland, T.; Holter, J.C.; Holten, A.R.; Müller, K.E.; Lind, A.; Bekken, G.K.; Dudman, S.; Aukrust, P.; Dyrhol-Riise, A.M.; Heggelund, L. Distinct and Early Increase in Circulating MMP-9 in COVID-19 Patients with Respiratory Failure. J. Infect. 2020, 81, e41–e43. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Su, M.; Shen, G.; Hu, Y.; Yi, F.; Zeng, Z.; Zhu, P.; Yang, G.; Zhou, H.; Li, Q.; et al. Matrix Metalloproteinase 3 as a Valuable Marker for Patients with COVID-19. J. Med. Virol. 2021, 93, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Bauzá-Martinez, J.; Aletti, F.; Pinto, B.B.; Ribas, V.; Odena, M.A.; Díaz, R.; Romay, E.; Ferrer, R.; Kistler, E.B.; Tedeschi, G.; et al. Proteolysis in Septic Shock Patients: Plasma Peptidomic Patterns Are Associated with Mortality. Br. J. Anaesth. 2018, 121, 1065–1074. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-COVID-19 Patients | COVID-19 Patients | ||||
|---|---|---|---|---|---|
| Negative Controls (n = 12) | Total (n = 21) | Mildly Symptomatic (n = 11) | Severe (n = 10) | p-Value (COVID-19 vs. Negative) | |
| Demographic Characteristics | |||||
| Female, n (%) | 6 (50%) | 12 (57.1%) | 6 (54.5%) | 6 (60%) | |
| Age (years) | 75 ± 14.9 | 74.9 ± 16.1 | 71.9 ± 21.8 | 77.2 ± 7.7 | 0.9813 |
| Respiratory Support, n (%) | |||||
| Continuous Positive Airway Pressure (CPAP) | n/a | 10 (47.6%) | 0 (0%) | 10 (100%) | |
| Non-Invasive Oxygen Supplementation | n/a | 11 (52.4%) | 11 (100%) | 0 (0%) | |
| Dexamethasone Regime, n (%) | n/a | 4 (19.04%) | 0 (0%) | 4 (40%) | |
| Outcome, n (%) | |||||
| Discharged | 12 (100%) | 16 (76.2%) | 11 (100%) | 5 (50%) | |
| Deceased | 0 (0%) | 5 (23.8%) | 0 (0%) | 5(50%) | |
| Comorbidity, n | |||||
| Hypertension | 3 | 11 | 5 | 6 | |
| Diabetes | 0 | 4 | 1 | 3 | |
| Respiratory System | 1 | 2 | 1 | 1 | |
| Cardiovascular System | 2 | 8 | 4 | 4 | |
| Other Endocrine System | 1 | 6 | 3 | 3 | |
| Chronic Kidney | 1 | 1 | 1 | 0 | |
| Digestive System | 0 | 3 | 0 | 3 | |
| Time from Disease Onset to Sample Collection, Days | |||||
| Mean ± SD | n/a | 6.45 ± 4.8 | 5.2 ± 4.7 | 7.7 ± 3.9 | |
| Range | n/a | 1.0–11.0 | 1.0–11.0 | 2.0–10.0 | |
| Peptide Sequence | Protein | Modulation | p-Value | Fold Change |
|---|---|---|---|---|
| VDSGNDVTDIADD | HPT_HUMAN | Mild/Negative | 0.00254 | 18.8 |
| SEAEDASL | APOC3_HUMAN | Mild/Negative | 0.00321 | 6.5 |
| SEAEDASLLS | APOC3_HUMAN | Mild/Negative | 0.02524 | 6.4 |
| DSGEGDFLAEGGGVR | FIBA_HUMAN | Mild/Negative | 0.04475 | 4.9 |
| LLSPYSYSTTAVVTNPKE | TTHY_HUMAN | Mild/Negative | 7.96 × 10−5 | 0.0147 |
| DAHKSEVAHRFKDLGEENFKALVLIAF | ALBU_HUMAN | Mild/Negative | 0.03752 | 0.0318 |
| FKVSFLSALEEYTKKLNTQ | APOA1_HUMAN | Mild/Negative | 0.00496 | 0.0371 |
| FEIPINGLSE | ITIH2_HUMAN | Mild/Negative | 0.01937 | 0.0474 |
| VDSGNDVTDIADD | HPT_HUMAN | Severe/Negative | 0.00186 | 85.9 |
| SGASGPENFQVG | TLN1_HUMAN | Severe/Negative | 0.02202 | 20.0 |
| TLEIPGNSD | CO4A_HUMAN | Severe/Negative | 0.02502 | 20.0 |
| WVQKTIAEN | HPT_HUMAN | Severe/Negative | 0.00546 | 13.4 |
| DEAGSEADHEGTHST | FIBA_HUMAN | Severe/Negative | 9.03 × 10−5 | 0.0500 |
| DDPDAPLQPVTPLQ | CO4A_HUMAN | Severe/Negative | 0.00078 | 0.0500 |
| HKSEVAHRFKDLGEENFKALVLIA | ALBU_HUMAN | Severe/Negative | 0.00499 | 0.0500 |
| SGFLLFPDMEA | C1QB_HUMAN | Severe/Negative | 0.01355 | 0.0500 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldanzi, G.; Purghè, B.; Ragnoli, B.; Sainaghi, P.P.; Rolla, R.; Chiocchetti, A.; Manfredi, M.; Malerba, M. Circulating Peptidome Is Strongly Altered in COVID-19 Patients. Int. J. Environ. Res. Public Health 2023, 20, 1564. https://doi.org/10.3390/ijerph20021564
Baldanzi G, Purghè B, Ragnoli B, Sainaghi PP, Rolla R, Chiocchetti A, Manfredi M, Malerba M. Circulating Peptidome Is Strongly Altered in COVID-19 Patients. International Journal of Environmental Research and Public Health. 2023; 20(2):1564. https://doi.org/10.3390/ijerph20021564
Chicago/Turabian StyleBaldanzi, Gianluca, Beatrice Purghè, Beatrice Ragnoli, Pier Paolo Sainaghi, Roberta Rolla, Annalisa Chiocchetti, Marcello Manfredi, and Mario Malerba. 2023. "Circulating Peptidome Is Strongly Altered in COVID-19 Patients" International Journal of Environmental Research and Public Health 20, no. 2: 1564. https://doi.org/10.3390/ijerph20021564
APA StyleBaldanzi, G., Purghè, B., Ragnoli, B., Sainaghi, P. P., Rolla, R., Chiocchetti, A., Manfredi, M., & Malerba, M. (2023). Circulating Peptidome Is Strongly Altered in COVID-19 Patients. International Journal of Environmental Research and Public Health, 20(2), 1564. https://doi.org/10.3390/ijerph20021564