Abstract
Arsenic is an environmental toxicant, and one of the major mechanisms by which it exerts its toxic effect is through an impairment of cellular respiration by inhibition of various mitochondrial enzymes, and the uncoupling of oxidative phosphorylation. Most toxicity of arsenic results from its ability to interact with sulfhydryl groups of proteins and enzymes, and to substitute phosphorus in a variety of biochemical reactions. Most toxicity of arsenic results from its ability to interact with sulfhydryl groups of proteins and enzymes, and to substitute phosphorus in a variety of biochemical reactions. Recent studies have pointed out that arsenic toxicity is associated with the formation of reactive oxygen species, which may cause severe injury/damage to the nervous system. The main objective of this study was to conduct biochemical analysis to determine the effect of arsenic trioxide on the activity of acetyl cholinesterase; a critical important nervous system enzyme that hydrolyzes the neurotransmitter acetylcholine. Four groups of six male rats each weighing an average 60 ± 2 g were used in this study. Arsenic trioxide was intraperitoneally administered to the rats at the doses of 5, 10, 15, 20mg/kg body weight (BW), one dose per 24 hour given for five days. A control group was also made of 6 animals injected with distilled water without chemical. Following anaesthesia, blood specimens were immediately collected using heparinized syringes, and acetyl cholinesterase detection and quantification were performed in serum samples by spectrophotometry. Arsenic trioxide exposure significantly decreased the activity of cholinesterase in the Sprague-Dawley rats. Acetyl cholinesterase activities of 6895 ± 822, 5697 ± 468, 5069 ± 624, 4054 ± 980, and 3158 ± 648 U/L were recorded for 0, 5, 10, 15, and 20 mg/kg, respectively; indicating a gradual decrease in acetyl cholinesterase activity with increasing doses of arsenic. These findings indicate that acetyl cholinesterase is a candidate biomarker for arsenic-induced neurotoxicity in Sprague-Dawley rats.
Introduction
Arsenic is widely distributed in nature, being found in food, the soil, water and airborne particles; it derives from both natural and human activities [1]. More than 80% of arsenic compounds are used to manufacture products with agricultural applications such as insecticides, herbicides, fungicides, algicides, sheep dips, wood preservatives, dye-stuffs, and medicines for the eradication of tapeworms in sheep and cattle [2]. Arsenical drugs are still used used in treating certain tropical diseases such as African sleeping sickness and amoebic dysentery, and in veterinary medicine to treat parasitic diseases, including filariasis in dogs and black head in turkeys and chickens [2]. Recently, arsenic has been used as an anticancer agent in the treatment of acute promeylocytic leukemia, and its therapeutic action has been attributed to the induction of programmed cell death (apoptosis) in leukemia cells [3].
A large number of people are exposed to arsenic chroniclly throughout the world. Exposure to arsenic occurs via the oral route (ingestion), inhalation, dermal contact, and the parenteral route to some extent. Humans can be exposed to arsenic through the intake of air, food and water. Although food is usually the major source of arsenic exposure, most adverse effects have been associated with consumption of arsenic-contaminated drinking water. Occupational sources of arsenic to human workers include vineyards, ceramics, glass- making, smelting and refining of metallic ores, during production and use of arsenic containing agricultural products like pesticides and herbicides [4]. The gastrointestinal tract of humans and most experimental animals readily absorbs ingested inorganic arsenic (>90%). Following absorption, arsenic compounds through blood circulation are distributed in various tissues (blood, liver, kidney, lung, skin) [5].
Analyzing the toxic effects of arsenic is complicated because the toxicity varies according to its oxidation state, its solubility and many different inorganic and organic forms. [6]. Several studies have indicated that the toxicity of arsenic depends on the exposure dose, frequency and duration, biological species, age, gender as well as on individual susceptibilities, genetic and nutritional factors [7].
The major metabolic pathway for inorganic arsenic in humans is methylation. Arsenic trioxide is methylated to two major metabolites via a non-enzymatic process to monomethylarsonic acid (MMA), which is further methylated enzymatically to dimethyl arsenic acid (DMA) before excretion in the urine [8]. This methylation mechanism has been widely accepted, and the metabolites MMA (V) and DMA (V) have been consistently observed in human urine [9–11].
Generally, the toxicity of heavy metals is largely due to their reactions with sulfhydryl groups [12]. Lipophilic organometallic compounds easily cross the blood-brain barrier, whereas inorganic metallic compound also reach the brain tissue [12]. Acute and chronic toxic effects of inorganic arsenic involve many organ systems, including the central nervous system (CNS) [13–15]. In experimental animals, arsenic has been shown to affect hepatic mitochondrial enzymes [16]. It can also pass the blood-brain barrier, be accumulated in the brain, and can exert neurochemical effects [17, 18].
The inhibition of acetycholinesterase (AChE) in the nervous tissue and other target organs is generally considered to be the critical effect leading to the acute toxicity of many toxic chemicals [19–22]. Nagaraju and Desiraju [23] further indicated that AChE activity in rats was inhibited in some regions of the brain following inorganic arsenic intake. Despite its deleterious actions on the CNS, there are very few studies of the effects of chronic or acute consumption of arsenic on brain and behaviour. The present study was done to examine the effect of arsenic on the activity of acetyl cholinesterase; a critical important nervous system enzyme that hydrolyzes the neurotransmitter acetylcholine.
Materials and Methods
Chemicals
Arsenic trioxide (As2O3) was purchased from Fischer-Scientific, Houston, TX, USA. Cholinesterase (PTC), and heparin were purchased from Sigma-Aldrich (St.Louis, MO, USA).
Animal Maintenance
Healthy adult male Sprague-Dawley rats (8–10 weeks of age, with average body weight (BW) of 60 ± 2 g) were used in this study. They were obtained from Harlan-Sprague-Dawley Breeding laboratories in Indianpolis, Indiana, USA. The animals were randomly selected and housed in polycarbonate cages (three rats per cage) with steel wire tops and corn-cob bedding. They were maintained in a controlled atmosphere with a 12h:12h dark/light cycle, a temperature of 22 ± 2° C and 50–70% humidity with free access to pelleted feed and fresh tap water. The animals were supplied with commercially available dry food pellets from PMI Feeds Inc. (St. Louis, Missouri). They were allowed to acclimate for 10 days before treatment.
Arsenic Treatment
Groups of six rats each were treated with four different arsenic trioxide dose levels, 5, 10, 15 and 20 mg/kg BW. Arsenic trioxide was diluted with distilled water (as required) and intraperitoneally administered to animals at the doses of 0, 5, 10, 15 and 20 mg/kg BW, one dose per 24h given for 5 days. Each rat received a total of five doses at 24h intervals. The cumulative doses of arsenic trioxide given to rats were thus 25, 50, 75 and 100 mg/kg BW. Distilled water was administered to the 6 animals of control group in the same manner as in the treatment groups. The acute bioassay was performed following standard test protocol.
At the end of the treatment period, rats were anesthetized using 95% CO2 for 70 seconds. Immediately following anaesthetization blood specimens were collected using heparinized syringes to prevent clotting, and the plasma was separated by centrifugation at low speed (2000 g) for 10 min. Plasma samples was evaluated for enzyme identification and quantification.
Enzyme Analysis
Measurement of serum cholinesterase has been used to assess liver function, and monitor exposure to organo-phosphorus insecticides. The enzyme is depressed in acute hepatitis, hepatic metastases and alcoholic cirrhosis.
The reaction for cholinesterase assay is as follows:
Cholinesterase hydrolyzes Propionylthiocholine to form thiocholine that reacts with Dithio bis2-nitrobenzoic acid to yield the yellow 5-thio-2-nitrobenzoate with an absorbance maximum at 405 nm. Therefore the rate of change in absorbance at 405 nm is directly proportional to cholinesterase activity.
Analysis
Cholinesterase reagent (PTC) was prepared by reconstituting with the volume of deionized water indicated on the vial. The temperature of the reaction mixture was maintained at 30° C. Spectrophotometer wavelength was set to 405 nm and absorbance reading to zero with H2O as reference. The reagent was warmed in water bath to assay temperature (30° C). To cuvet labeled TEST, 1.0 ml of cholinesterase (PTC) reagent was added and placed in temperature controlled cuvet compartment. ten μl of serum was added to the above reagent, mixed immediatel by inversion and incubated at 30° C for 15 seconds. The absorbance was read and recorded as (A) of TEST at 405 nm versus water as reference. This is called Initial A. The cuvet labeled TEST was incubated again at 30° C and absorbance was recorded after exactly 30 seconds following the initial absorbance reading. This is called Final A, multiplied by 2 to obtain the change in absorbance per minute at 405nm (Δ A per minute). The cholinesterase activity (U/L) of the sample was determined by using the following formula.
Where:
Δ A per min = change in aborbance per minute at 405 nm.
TV = Total volume (1.01 ml)
SV = Sample volume (0.01 ml)
13.6 = millimolar absorptivity of 5- thio- nitrobenzoic acid at 405 nm.
LP = Lightpath (1-cm)
1000 = conversion of units per ml to units per liter.
Results
Figure 1 show the experimental data obtained from the analysis of acetyl cholinesterase. The results indicate acetyl cholinesterase activities of 6895 ± 822, 5697 ± 468, 5069 ± 624, 4054 ± 980, and 3158 ± 648 U/L for 0, 5, 10, 15, and 20 mg/kg BW respectively. As shown in this figure there was a dose-response relationship with respect to arsenic inhibition of acetyl cholinesterase in the blood.
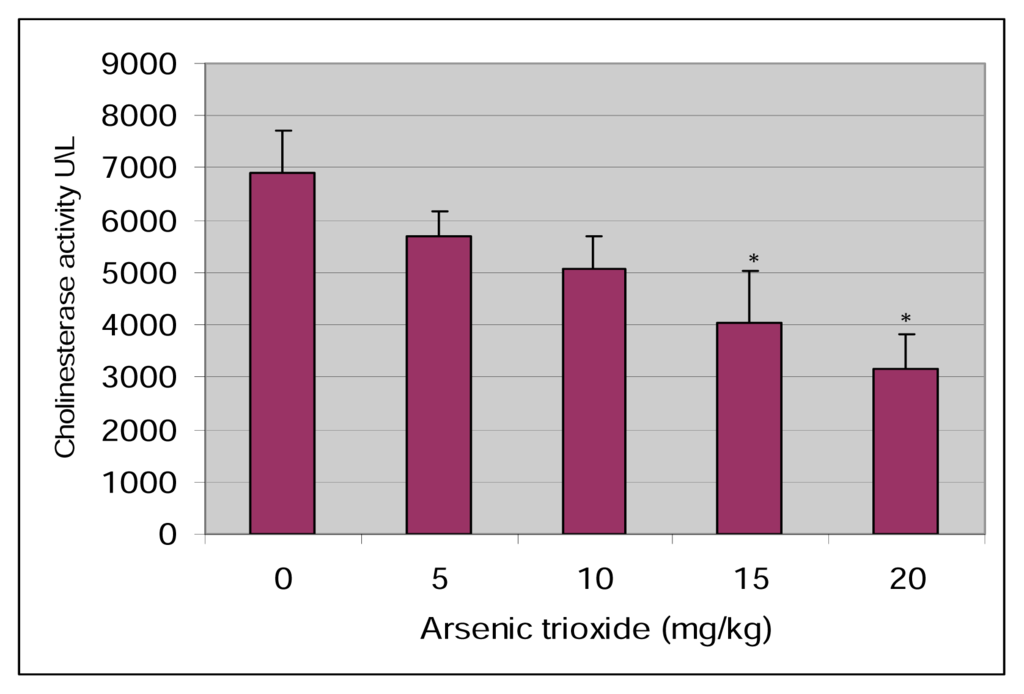
Figure 1.
Effect of Arsenic trioxide on the activity of serum cholinesterase in Sprague-Dawley rats.
Discussion
There is increasing interest in the development of new methods for assessing chemical toxicity, and the use of knowledge of mechanisms of toxic actions to improve this assessment. In order to determine the effect on serum acetyl cholinesterase (AChE), adult male Sprague-Dawley rats were exposed for five days to four different concentrations (5, 10, 15, 20 mg/kg) of arsenic trioxide. The data obtained from this study clearly show that arsenic trioxide significantly decreased the activity of serum acetyl cholinesterase in a dose-dependent manner (Figure 1). The results agree with previous studies that demonstrated a decreased activity of acetyl cholinesterase in neuroblastoma cells of mice [24], in rat whole brain [23, 25] and in two models of fish [26].
In this study, the decrease in the activity of acetyl cholinesterase was positively correlated with the chemical dose. The inhibition of acetyl cholinesterase by arsenic trioxide is a puzzling phenomenon because AChE does not contain the structural features usually associated with the inhibition of enzymes by arsenic. Trivalent arsenic compounds are potent inhibitors of a number of enzymes [27] but the mechanism of this inhibition is the reaction of the arsenical with free sulfhydryl groups, notably those of reduced lipoic acid, to form cyclic thio-arsenite diesters. However, other compounds known to be sulfhydryl reactants such as organomercury compounds or iodoacetamide did not inhibit AChE [28]. The enzyme has been found to contain cysteine only in the form of disulfide bridges and not as free thiol [29].
Acetyl cholinesterase (AChE) is one of many important enzymes needed for the proper functioning of the nervous systems of humans, other vertebrates and insects. Certain chemical classes of pesticides, such as organophosphates (OPs) and carbamates interfere with or inhibit cholinesterase. Acute toxicity manifests as a cholinergic crisis with excessive glandular secretions, altered mental status, and weakness. Several delayed syndromes associated with these pesticides exposure are myasthenic-like syndrome, peripheral neuropathies, neuropsychiatric abnormalities and extrapyramidal disorders [30].
AChE has been shown to be neurotoxic in vivo and in vitro; it accelerates assembly of amyloid peptide in Alzheimer’s fibrils, leading to cell death via apoptosis [31]. Brain AChE has been shown to be toxic to neuronal (Neuro 2a) and glial-like (B12) cells [32]. There are also reports that transgenic mice over-expressing human AChE in brain neurons undergo progressive cognitive deterioration [33]. Organophosphates ability to affect acetylcholine neurotransmission, poisoning commonly manifests with acute dysfunction of the autonomic, central and peripheral nervous system. Failure to recognize these manifestations can result in worsening toxicity, delayed complications, and death [33].
Despite its deleterious actions on the central nervous system, there have been very limited studies of the effects of arsenic on the brain and behaviour. Therefore, the present study provided new insights on the neurotoxicity of arsenic, and indicated that acetyl cholinesterase activity can be used as a biomarker of this neurotoxicity if the specific pre-exposure conditions are characterized.
Acknowledgements
This research was financially supported by NIH-RCMI Grant No. 1G12RR13459. We thank President, Jackson State University, Dr. Ronald Mason Jr. and Dr. Abdul Mohamed, Dean of the College of Science, Engineering and Technology, for their support and advice in this research.
References
- Tchounwou, P. B.; Wilson, B. A.; Ishaque, A. Important considerations in the development of public health advisories for arsenic and arsenic containing compounds in drinking water. Rev. Environ. Hlth 1999, 14, 1–19. [Google Scholar]
- National Academy of Science, Arsenic; Washington, D. C, 1977.
- Rousselot, P.; Laboume, S.; Marolleau, J. P.; Larghero, T.; Noguera, M. L.; Brouet, J. C.; Fermand, J. P. Arsenic trioxide and melarsoprol induce apoptosis in plasma cell lines and in plasma cells from myeloma patients. Cancer Res 1999, 59, 1041–1048. [Google Scholar]
- Tchounwou, P. B.; Centeno, J. A.; Patlolla, A. K. Arsenic toxicity, mutagenesis, and carcinogenesis- A health risk assessment and management approach. Mol. Cell. Biochem 2004, 255, 47–55. [Google Scholar]
- Mckinney, J. D. Metabolism and disposition of inorganic arsenic in laboratory animals and humans. Environ. Oeochem. Health 1992, 14, 43–48. [Google Scholar]
- Tchounwou, P. B.; Patlolla, A.K.; Centeno, J. A. Carcinogenic and systemic health effects associated with arsenic exposure – A Critical Review. Toxicol. Pathol 2003, 31(6), 575–88. [Google Scholar]
- Tchounwou, P. B.; Wilson, B. A.; Ishaque, A.; Schneider, J. Atrazine potentiation of arsenic trioxide induced cytotoxicity and gene expression in HepG2 cells. Mol. Cell. Biochem 2001, 222, 49–59. [Google Scholar]
- Wang, Z.; Rossman, T. G. Cheng, L.W., Ed.; The Toxicology of Metals; Volume 1, CRC Press: Boca Raton, Fl, 1996; pp. 221–243. [Google Scholar]
- Aposhian, H.V.; Gurzau, E. S.; Le, X.C.; Gurzau, A.; Healy, S. M.; Lu, X.; Ma, M.; Yip, L.; Zakharyan, R. A.; Maiorino, R. M.; Dart, R. C.; Tircus, M. G.; Gonzalez-Ramirez, D.; Morgan, D. L.; Avram, D.; Aposhian, M. M. Occurrence of monomethylarsonous acid [MMA (III)], excreted in human urine. Toxicol Appl Pharmacol 2000a, 165, 74–83. [Google Scholar]
- Styblo, M.; Del Razo, L. M.; LeCluyse, E. L.; Hamilton, G. A.; Wang, C.; Cullen, W. R.; Thomas, D. J. Metabolism of arsenic in primary cultures of human and rat hepatocytes. Chem. Res. Toxicol 1999, 12, 560–565. [Google Scholar]
- Styblo, M.; Yamauchi, H.; Thomas, D.J. Comparative in vitro methylation of trivalent and pentavalent arsenicals. Toxicol. Appl. Pharmacol 1995, 135, 172–178. [Google Scholar]
- Bast, A. Anatomy and Toxicological Pathology of the nervous system. In Toxicology: Principles and Applications; Niesink, J. M., de Vries, J., Hollinger, M. A., Eds.; CRC Press: New York, 1996; pp. 974–1001. [Google Scholar]
- Pershagen, G. Sources of exposure and biological effects of arsenic. In Environmental Carcinogens Selected Methods of Analysis, Vol 8, some metals: As, Ni, Oi, Cr, Pb, Se, Zn; O’Neill, J. K., Schuller, P., Fishbein, L., Eds.; Lyon; IARC, 1986; pp. 45–61. [Google Scholar]
- Mazumder, D. N. G.; Chakraborty, A. K.; Ghose, A.; et al. Chronic arsenic toxicity from drinking tubewell water in rural West Bengal. Bulletin of the World Health Organization 1988, 66, 499–506. [Google Scholar]
- Beckett, W. S.; Moore, J. L.; Keogh, J. P.; Blecker, M. L. Acute encephalopathy due to occupational exposure to arsenic. British Journal of Industrial Medicine 1986, 43, 66–7. [Google Scholar]
- Fowler, B. A.; Woods, J. S. The effects of prolonged oral arsenate exposure on liver mitochondria of mice:morphometric and biochemical studies. Toxicol. Appl. Pharmocol 1979, 50, 177–87. [Google Scholar]
- Nagaraju, T. N.; Desiraju, T. Regional alterations in the levels of brain biogenic amines, glutamate, GABA and GAD activity due to chronic consumption of inorganic arsenide in developing and adult rats. Bull. of Environ. Contam. Toxicol 1993, 50, 100–7. [Google Scholar]
- Valkonen, S.; Savolainen, H.; Jarvisalo, J. Arsenic distribution and neurochemical effects in peroral sodium arsenite exposure of rats. Bull. of Environ. Contam. Toxicol 1983, 30, 303–8. [Google Scholar]
- Maxwell, D. M.; Vlahacos, C. P.; Lenz, D. E. A pharmacodynamic model for soman in the rat. Toxicol. lett 1988, 43, 175–188. [Google Scholar]
- Gearhart, J. M.; Jepson, G. W.; Clewell, H. J., III; Anderson, M. E.; Conolly, R. B. Physiologically based pharmacokinetic and pharmacodynamic model for the inhibition of acetylcholinesterase by diisopropylfluorophosphate. Toxicol. Appl. Pharmocol 1990, 106, 295–310. [Google Scholar]
- McCarty, L. S.; Mackay, D. Enhancing Eco-toxicological Modeling and assessment. Environ. Sci. Tech 1993, 27, 1719–1728. [Google Scholar]
- Abbas, R.; Hayton, W. L. A physiologically based pharmacokinetic and pharmacodynamic model for paraoxon in rainbow trout. Toxicol. Appl. Pharmacol 1997, 145(1), 192–201. [Google Scholar]
- Nagaraju, T. N.; Desiraju, T. Effects on operant learning and brain acetylcholine esterase activity in rats following chronic inorganic arsenic intake. Human. Exper. Toxicol 1994, 13, 353–356. [Google Scholar]
- Repetto, G.; Sanz, P.; Repetto, M. Comparative in vitro effects of sodium arsenite and sodium arsenate on neuroblastoma cells. Toxicol 1994, 92, 143–153. [Google Scholar]
- Tripathi, N.; Kannan, G. M.; Pant, B. P.; Jaiswal, D. K.; Malhotra, P. R.; Flora, S. J. S. Arsenic-induced changes in certain neurotransmitter levels and their recoveries following chelation in rat whole brain. Toxicol. Lett 1997, 92, 201–208. [Google Scholar]
- Liao, C. N.; Lin, M. C. Acute toxicity modeling of rainbow trout and silver sea bream exposed to waterborn metals. Environ. Toxicol 2001, 16, 349–360. [Google Scholar]
- Boyer, P. D.; Lardy, H.; Myrback, K. (Eds.) The Enzymes, 2nd ed.; Academic Press, 1963; New York.
- Mounter, L. A.; Whittaker, V. P. The effect of thiol and other group- specific reagents on erythrocyte and plasma cholinesterases. Biochem. J 1953, 53, 167–173. [Google Scholar]
- Rosenberry, T. L. Acetylcholinesterase- A Review. Advan. Enzymol 1975, 43, 103–218. [Google Scholar]
- Rusyniak, D. E.; Nanagas, K. A. Organophosphate Poisoning. Semin. Neurol 2004, 24(2), 197–204. [Google Scholar]
- Yang, L.; Heng-Yi, H.; Zhang, X. J. Increased expression of intranuclear AChE involved in apoptosis of SK-N-SH cells. Neurosci Res 2002, 42, 261–68. [Google Scholar]
- Calderon, F. H.; Von, B. R.; De Ferrari, G.; et al. Toxic effects of acetylcholinesterase on neuronal and glial-like cells in vitro. Mol. Psychiatry 1998, 3, 247–55. [Google Scholar]
- Andres, C.; Seidman, S.; Beeri, R.; et al. Transgenic acetylcholinesterase induces enlargement of murine neuromuscular junctions but leaves spinal cord synapses intact. Neurochem. Int 1998, 32, 449–56. [Google Scholar]
© 2005 MDPI. All rights reserved.