Abstract
Although nickel and cobalt compounds have been known to cause induction of the transcription factor hypoxia-inducible factor 1 (HIF-1) and activation of a battery of hypoxia-inducible genes in the cell, the molecular mechanisms of this induction remain unclear. The post-translational modification of HIF-1a, the oxygen-sensitive subunit of HIF-1, regulates stabilization, nuclear translocation, DNA binding activity, and transcriptional activity of the protein. Among the enzymes regulating the post-translational modification of HIF-1a, the factor inhibiting HIF-1 (FIH-1) hydroxylates the protein at asparagine 803, suppressing the interaction of HIF-1a with transcription coactivators p300/CBP and reducing the transcriptional activity of the protein. ARD-1, the acetyltransferase, acetylates HIF-1a at lysine 532, which enhances the interaction of HIF-1a with pVHL. Therefore, FIH-1 and ARD-1 negatively regulate the transcriptional activity and the stability of HIF-1a. We examined the mRNA levels of FIH-1 and ARD-1 genes after exposure nickel (II) or cobalt (II) to the cell and found that both genes were down-regulated by the chemical treatment, which may lead to reduced levels of both proteins and result in increased level of HIF-1a and its transcriptional activity.
Introduction
According to the International Agency for Research on Cancer (IARC 1990), both soluble and insoluble nickel compounds have long been established as human and animal carcinogens [1]. And cobalt compounds are carcinogenic in animals [2]. Epidemiological studies have shown that environmental or occupational exposure to nickel or cobalt compounds could cause lung and nasal cancers, asthma, fibrosis, pneumonitis and some other lung injuries [1–5]. Despite the various differences among the compounds of these two metals regarding the biochemical and molecular mechanisms of their toxicity and carcinogenicity, they mimic hypoxia to induce the HIF-1a transcription factor and hypoxia-inducible genes [5, 6], which are believed to play important roles in carcinogenesis. However, the complete mechanisms by which nickel and cobalt compounds induce HIF-1a are still unknown, although several sites of their impact on HIF-1a have been described [7–9].
The transcription factor hypoxia-inducible factor 1 (HIF-1) plays an essential role in cellular oxygen homeostasis [10, 11]. HIF-1 is a heterodimeric complex composed of alpha and beta two subunits [12]. The beta subunit is also the heterodimerization partner for the aryl hydrocarbon receptor (AhR) and thereby called aryl hydrocarbon receptor nuclear translocator (ARNT) that is constitutively expressed; whereas the alpha subunit (HIF-1a) is highly oxygen-sensitive and is rarely detectable under normal oxygen tension but is dramatically induced with hypoxia [12]. Under reduced oxygen tension, HIF-1a is stabilized and translocates to the nucleus, where it dimerizes with ARNT. Then the active HIF-1 stimulates the transcription of genes involved in angiogenesis, cell survival, glucose transport and metabolism [13, 14].
HIF-1a is regulated by a reduced oxygen level largely at its post-translational modifications, resulting in stabilization, nuclear translocation, DNA binding activity, and transcriptional activity of the protein. The post-translational modifications of HIF-1a include prolyl hydroxylation at proline 402 and 564 within the oxygen-dependent degradation (ODD) domain by HIF-prolyl hydroxylases (HPHs) [9, 15–17], asparaginyl hydroxylation at asparagine 803 in the C-terminal activation domain (C-TAD) by factor inhibiting HIF-1 (FIH-1) [8, 18, 19], acetylation of lysine 532 in the ODD domain by an acetyltransferase ARD-1[20], phosphorylation induced by p42/p44 mitogen-activated protein kinase (MAPK) activity [21], as well as ubiquitination by the von Hippel-lindau (pVHL) complex [22–25].
In normoxia, proline 402 and 564 of HIF-1a are hydroxylated, which is required for the binding of pVHL complex and leads to the ubiquitination of the protein, resulting in targeting of HIF-1a for proteasomal degradation [15–17, 24]. The interaction of HIF-1a with pVHL is enhanced by ARD-1-mediated acetylation at lysine 532 [20]. The acetylation of this lysine residue by ARD-1 is critical to the proteasomal degradation of HIF-1a since a mutant with arginine substituting lysine 532 shown no acetylation by ARD-1 was stabilized and had a decreased interaction with pVHL [20]. At the same time, hydroxylation of asparagine 803 during normoxia suppresses interaction of HIF-1a CAD with transcription coactivators p300/CBP and reduces the transcriptional activity of the protein [18, 26]. In addition to interacting with HIF-1a, the asparaginyl hydroxylase FIH-1 also interacts with pVHL, allowing the formation of complexes containing HIF-1a, FIH-1, and pVHL [27].
In hypoxia, decreased level of prolyl hydroxylation due to the limiting oxygen prevents pVHL binding to HIF-1a, resulting in rapid accumulation of HIF-1a protein [15, 16, 24]. The acetylation level of HIF-1 gradually decreases as the length of hypoxic exposure time increases, which is due to the reduced expression of ARD-1[20]. Meanwhile, stabilized HIF-1a protein is able to bind to p300/CBP to execute its transcriptional activity due to decreased level of hydroxylation at asparagine 803[18, 19]. In addition, during hypoxia, p42/p44 MAPK activity induces phosphorylation of HIF-1a and promotes its transcriptional activity [21]. As a consequence, HIF-1a accumulates and promotes hypoxic tolerance by activating gene transcription.
Among the enzymes that regulate HIF-1a, prolyl hydroxylases (HPHs) and asparaginyl hydroxylase (FIH-1) belong to the family of 2-oxoglutarate-dependent dioxygenases and require Fe2+, 2-oxoglutarate, O2, and ascorbate for their reactions [8, 15, 16]. ARD-1 acetyltransferase acetylates HIF-1a by transferring an acetyl group from acetyl-CoA [20].
Nickel (II) and cobalt (II) exposure in the presence of oxygen causes accumulation of HIF-1a protein and induction of its transcriptional activity [28–30], in part resulting from the inability of HIF-1a binding to pVHL due to the inhibition of HIF-1a hydroxylation by the metals [24, 31]. Furthermore, Cobalt (II) has been shown to inhibit activities of recombinant asparaginyl and prolyl hydroxylase in vitro [8, 9].
In this study, we have examined the effects of nickel (II) and cobalt (II) on the gene expression of FIH-1 and ARD-1 using reverse transcriptase PCR (RT-PCR). Both genes were down-regulated in the metal-exposed cells, which might lead to reduced level of both proteins and result in increased level of HIF-1a and its transcriptional activity.
Materials and Methods
Cell Culture
Human lung adenocarcinoma A549 cells (CCL185) were purchased from American Type Culture Collection (Manassas, VA). Cells were maintained in F-12K medium (Life Technologies, Inc., Gaithersburg, MD) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (equivalent to 100 units/ml and 100 μg/ml, respectively) at 37°C as monolayers in a humidified atmosphere containing 5% CO2.
Chemicals and Cell Exposure
Nickel chloride hexahydrate and cobalt chloride hexahydrate were purchased from Sigma (St. Louis, MO). Cells were seeded into 100-mm dishes and allowed to attach overnight. When cells reached 70–80% confluence, nickel chloride hexahydrate (0.5 mM, 1.0mM), or cobalt chloride hexahydrate (0.2mM, 0.4 mM) was added to the medium for 24h.
RNA Extraction and RT-PCR
At the end of the treatment, total RNA was isolated from the cell using the Trizol reagent (Invitrogen, Carlsbad, CA). The mRNA was isolated using Oligotex kit (Qiagen, Germany). Reverse transcription was carried out with SuperScript first-Strand Synthesis System for RT–PCR (Invitrogen) and 1 μg of mRNA was used for first-strand cDNA synthesis according to the manufacturer’s protocol. The PCR was carried out in a total volume of 50μl and 1μl of first-strand cDNA was used for amplifying genes. The primers used for the PCRs were: FIH-1 forward primer, 5′-GCCAGCACCCACAAGTTCTT-3′; FIH-1 reverse primer, 5′-CCTGTTGGACCTCGGCTTAA-3′ ARD-1 forward primer, 5′-TGGGGTGAGGAGGGGATGG-3′; ARD-1 reverse primer, 5′-GGGAAGATTGTGGGGTATG-3′. Primers for amplifying GAPDH gene were purchased from BD Biosciences Clontech. Amplification conditions for FIH-1 and ARD-1 genes were 95°C for 2 min, 22 cycles at 95°C for 45 s, 57°C for 45 s, 72°C for 1 min, and 72°C for 5 min; the same for GAPDH gene except that 16 cycles were performed. PCR products were then resolved on 1.5% agarose gels containing Ethidium bromide.
Results
Nickel (II) and Cobalt (II) Down-Regulate the Gene Expression of FIH-1and ARD-1 Genes
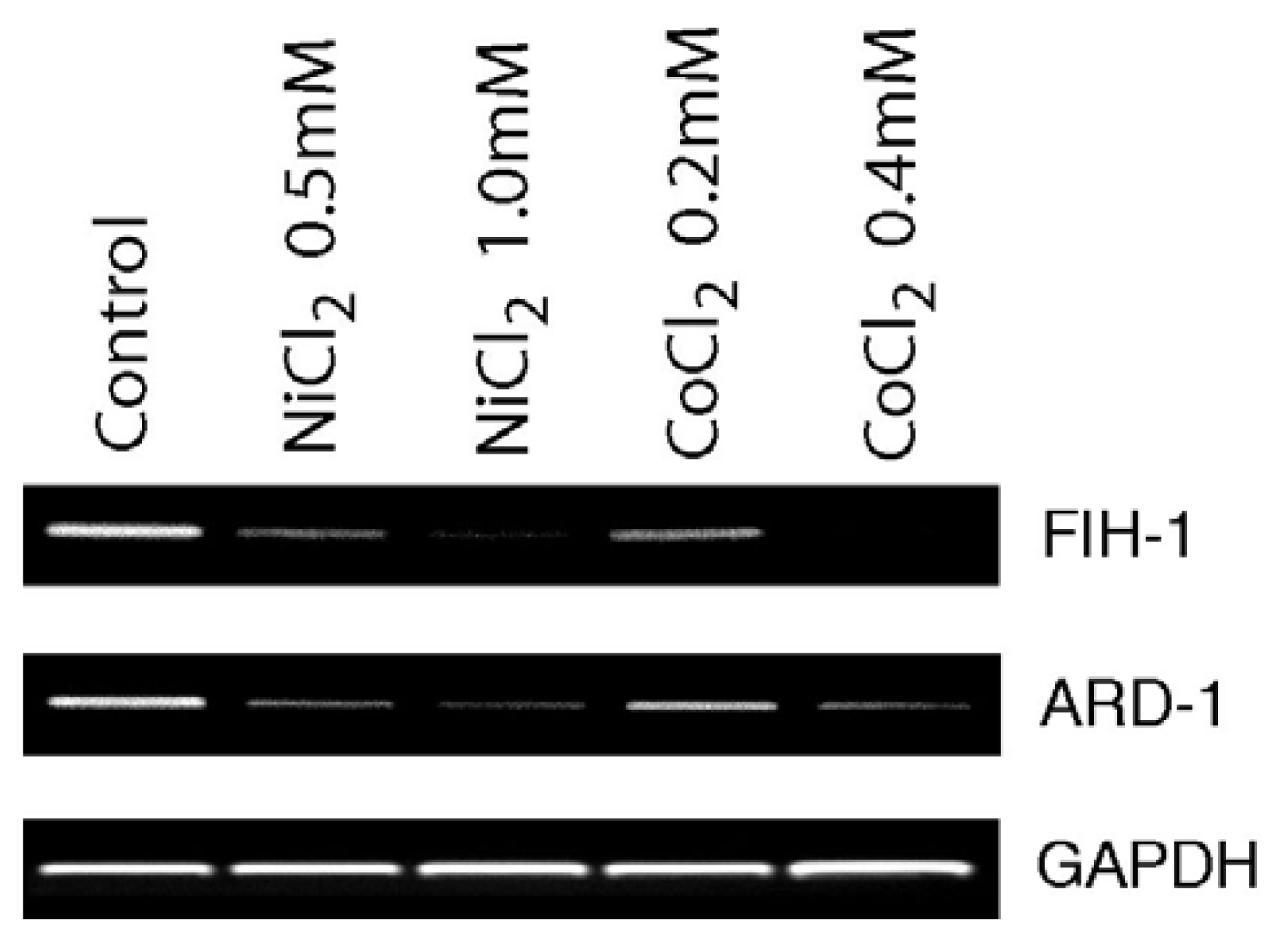
The investigation on the mechanisms of the induction of HIF-1a by nickel (II) and cobalt (II) has been focused on the effects of the metals on the enzyme activities of HPHs and FIH-1. Besides the possible effects of the metals on the enzyme activities, nickel (II) and cobalt (II) might affect the gene expression of the enzymes, so that the levels of the enzymes in the cell could be affected. Therefore, we examined mRNA levels of FIH-1 and ARD-1 by exposure A549 cells to nickel (II) and cobalt (II). As shown in figure 1, nickel (II) and cobalt (II) decreased FIH-1 and ARD-1 mRNA levels in a dose-dependent manner. GAPDH gene served as an internal control. The house keep gene 60S acidic ribosomal protein has also been used for measuring the loading control, which gave the same pattern as that of GAPDH gene (data not shown).
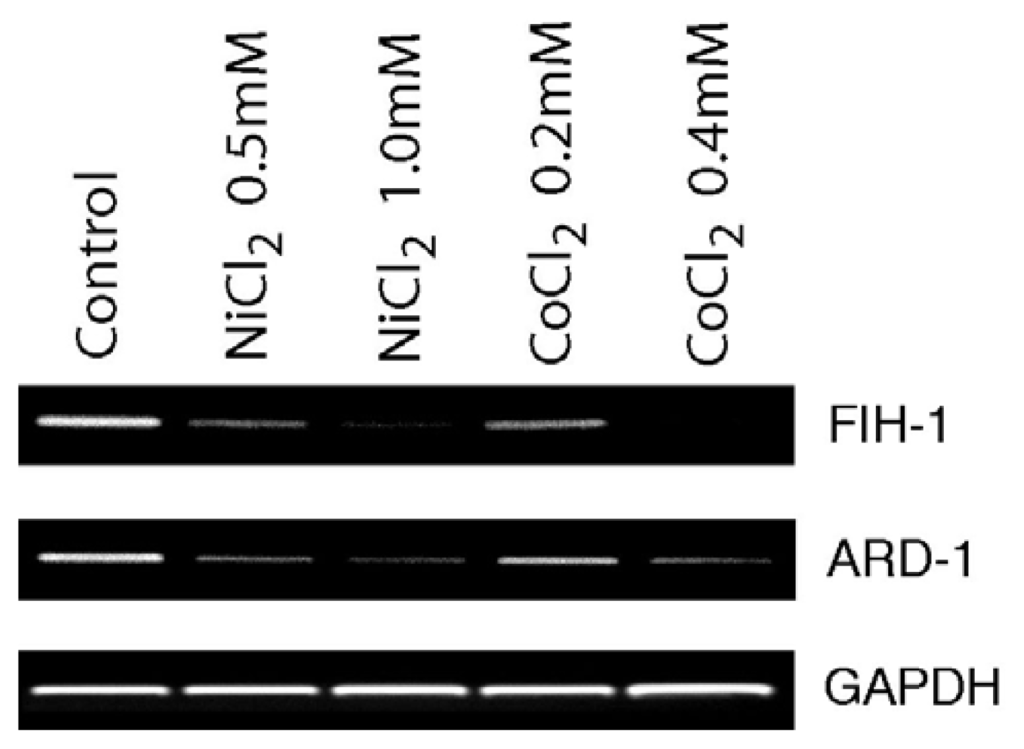
Figure 1.
Nickel (II) and Cobalt (II) down-regulate mRNA levels of FIH-1 and ARD-1genes in A549 cells A549 cells were treated with NiCl2·6H2O (0.5mM and 1 mM) or CoCl2·6H2O (0.2 mM and 0.4mM) for 24 h. The cells were harvested at the end of the treatment and the mRNA was isolated. The FIH-1, ARD-1, and GAPDH genes were amplified by RT-PCR.
Discussion
Nickel (II) and cobalt (II) have long been known to induce hypoxia-like stress by activating HIF-1a [28–30]. However, the mechanisms of this induction are still unclear, although several studies have shown effects at sites of HIF-1a regulation, particularly focusing on the effects of the metals on the hydroxylases activities[7–9]. Since the HIF-1a hydroxylases require iron (II) as a cofactor for their activities and iron, cobalt, and nickel are adjacent in the transition metal group, it has been suggested that nickel (II) and cobalt (II) may substitute for the iron (II) in the hydroxylases, thereby, causing the loss of the enzymatic activity[7]. Unfortunately, there is no direct evidence to support this hypothesis. Recently, the cellular ascorbate, another co-factor required for the hydroxylases activities, has been shown to be depleted by both metals. Since the role of ascorbate is maintaining iron in its reduced state (iron II), this depletion may favor enzyme-bound iron oxidation, which may lead to the inactivation of the hydroxylases[31].
Besides their effects, direct or indirect, on the hydroxylases activities, nickel (II) and cobalt (II) are very likely to induce HIF-1a at additional sites. We demonstrated here that the mRNA levels of FIH-1 and ARD-1 genes were down-regulated by both metals, which could result in reduced levels of the protein products of these genes. Since both FIH-1 and ARD-1 proteins negatively regulate HIF-1a, decreasing of them would lead to accumulation of HIF-1a and increase of its transcriptional activity.
Furthermore, we suspect that nickel (II) or cobalt (II) may affect the acetylation of HIF-1a not only by down-regulating the acetyltransferase ARD-1, but also by decreasing the level of the cellular acetyl-CoA, the supply of the acetyl group, by the way of shutting down the Kreb cycle as well as increasing the ratio of NAD/NADH thereby causing the deacetylation of histones. Since both nickel and cobalt have previously been shown to inhibit histone acetylation and increase the methylation of histone H3 at lysine 9, this effect may play a role in the down-regulation of these two important genes which impact negatively upon HIF-1a activation (Costa et al. Mutation Res. in press).
Our study revealed a possible new mechanism of the nickel- and cobalt- induction of hypoxia-like stress in the cell, contributing insight to better understanding of their carcinogenesis.
References
- IARC, I. A. R. C. Monographs on the Evaluation of Carcinogenic Risks to Humans, Chromium. Nickel and Welding 1990, 49, 677–691.
- Natl. Toxicol. Program. Tech. Rep. Ser 1998, 471, 1–268.
- Costa, M. Mechanism of nickel genotoxicity and carcinogenicity. 1996, 245–251. [Google Scholar]
- Kelleher, P.; Pacheco, K.; Newman, L. S. Inorganic dust pneumonias: the metal-related parenchymal disorders. Environ Health Perspect 2000, 108 Suppl 4, 685–96. [Google Scholar]
- Huang, L. E.; Ho, V.; Arany, Z.; Krainc, D.; Galson, D.; Tendler, D.; Livingston, D. M.; Bunn, H. F. Erythropoietin gene regulation depends on heme-dependent oxygen sensing and assembly of interacting transcription factors. Kidney Int 1997, 51(2), 548–52. [Google Scholar]
- Salnikow, K.; Davidson, T.; Costa, M. The role of hypoxia-inducible signaling pathway in nickel carcinogenesis. Environ Health Perspect 2002, 770 Suppl 5, 831–4. [Google Scholar]
- Goldberg, M. A.; Dunning, S. P.; Bunn, H. F. Regulation of the erythropoietin gene: evidence that the oxygen sensor is a heme protein. Science 1988, 242(4884), 1412–5. [Google Scholar]
- Hewitson, K. S.; McNeill, L. A.; Riordan, M. V.; Tian, Y. M.; Bullock, A. N.; Welford, R. W.; Elkins, J. M.; Oldham, N. J.; Bhattacharya, S.; Gleadle, J. M.; Ratcliffe, P. J.; Pugh, C. W.; Schofield, C. J. Hypoxia-inducible factor (HEF) asparagines hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J. Biol Chem 2002, 277(29), 26351–5. [Google Scholar]
- Epstein, A. C.; Gleadle, J. M.; McNeill, L. A.; Hewitson, K. S.; Oliourke, J.; Mole, D. R.; Mukherji, M.; Metzen, E.; Wilson, M. I.; Dhanda, A.; Tian, Y. M.; Masson, N.; Hamilton, D. L.; Jaakkola, P.; Barstead, R.; Hodgkin, J.; Maxwell, P. H.; Pugh, C. W.; Schofield, C. J.; Ratcliffe, P. J. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107(1), 43–54. [Google Scholar]
- Semenza, G.L. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol 2000, 88(4), 1474–80. [Google Scholar]
- Iyer, N. V.; Kotch, L. E.; Agani, F.; Leung, S. W.; Laughner, E.; Wenger, R. H.; Gassmann, M.; Gearhart, J. D.; Lawler, A. M.; Yu, A. Y.; Semenza, G. L. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev 1998, 12(2), 149–62. [Google Scholar]
- Wang, G. L.; Jiang, B. H.; Rue, E. A.; Semenza, G. L. Hypoxia-inducible factor 1 is a basic-helix-loop- helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995, 92(12), 5510–4. [Google Scholar]
- Semenza, G. L. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003, 3(10), 721–32. [Google Scholar]
- Acker, T.; Plate, K. H. A role for hypoxia and hypoxia-inducible transcription factors in tumor physiology. J Mol Med 2000, 80(9), 562–75. [Google Scholar]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J. M.; Lane, W. S.; Kaelin, W. G., Jr. HJFalpha targeted for VHL- mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001, 292(5516), 464–8. [Google Scholar]
- Jaakkola, P.; Mole, D. R.; Tian, Y. M.; Wilson, M. I.; Gielbert, J.; Gaskell, S. J.; Kriegsheim, A.; Hebestreit, H. F.; Mukherji, M.; Schofield, C. J.; Maxwell, P. H.; Pugh, C. W.; Ratcliffe, P. J. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292(5516), 468–72. [Google Scholar]
- Bruick, R. K.; McKnight, S. L. A conserved family of prolyM-hydroxylases that modify HIF. Science 2001, 294(5545), 1337–10. [Google Scholar]
- Lando, D.; Peet, D. J.; Whelan, D. A.; Gorman, J. J.; Whitelaw, M. L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 295(5556), 858–61. [Google Scholar]
- Lando, D.; Peet, D. J.; Gorman, J. J.; Whelan, D. A.; Whitelaw, M. L.; Bruick, R. K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev 2002, 16(12), 1466–71. [Google Scholar]
- Jeong, J. W.; Bae, M. K.; Ann, M. Y.; Kim, S. H.; Sohn, T. K.; Bae, M. H.; Yoo, M. A.; Song, E. J.; Lee, K. J.; Kim, K. W. Regulation and destabilization of HLF-lalpha by ARD1-mediated acetylation. Cell 2002, 111(5), 709–20. [Google Scholar]
- Richard, D. E.; Berra, E.; Gothie, E.; Roux, D.; Pouyssegur, J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1 alpha (HIF-1 alpha) and enhance the transcriptional activity of HIF-1. J. Biol Chem 1999, 274(46), 32631–7. [Google Scholar]
- Salceda, S.; Caro, J. Hypoxia-inducible factor lalpha (HIF-lalpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 1997, 272(36), 22642–7. [Google Scholar]
- Huang, L. E.; Gu, J.; Schau, M.; Bunn, H. F. Regulation of hypoxia-inducible factor lalpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci. USA 1998, 95(14), 7987–92. [Google Scholar]
- Maxwell, P. H.; Wiesener, M. S.; Chang, G. W.; Clifford, S. C.; Vaux, E. C.; Cockman, M. E.; Wykoff, C. C.; Pugh, C. W.; Maher, E. R.; Ratcliffe, P. J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399(6733), 271–5. [Google Scholar]
- Berra, E.; Roux, D.; Richard, D. E.; Pouyssegur, J. Hypoxia-inducible factor-alpha (HIF-1 alpha) escapes O2-driven proteasomal degradation irrespective of its subcellular localization: nucleus or cytoplasm. EMBO Rep 2001, 2(7), 615–20. [Google Scholar]
- Arany, Z.; Huang, L. E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M. A.; Bunn, H. F.; Livingston, D. M. An essential role or p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci USA 1996, 93(23), 12969–73. [Google Scholar]
- Mahon, P. C.; Hirota, K.; Semenza, G. L. FIH-1: a novel protein that interacts with HIF-lalpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev 2001, 15(20), 2675–86. [Google Scholar]
- Salnikow, K.; Blagosklonny, M. V.; Ryan, H.; Johnson, R.; Costa, M. Carcinogenic nickel induces genes involved with hypoxic stress. Cancer Res 2000, 60(1), 38–41. [Google Scholar]
- Salnikow, K.; An, W. G.; Melillo, G.; Blagosklonny, M. V.; Costa, M. Nickel-induced transformation shifts the balance between HIF-1 and p53 transcription factors. Carcinogenesis 1999, 20(9), 1819–23. [Google Scholar]
- Wang, G. L.; Semenza, G. L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem 1995, 270(3), 1230–7. [Google Scholar]
- Salnikow, K.; Donald, S. P.; Bruick, R. K.; Zhitkovich, A.; Phang, J. M.; Kasprzak, K. S. Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J Biol Chem 2004. [Google Scholar]
© 2005 MDPI. All rights reserved.