
Cannabis- and Substance-Related Epidemiological Patterns of Chromosomal Congenital Anomalies in Europe: Geospatiotemporal and Causal Inferential Study



Abstract
:1. Introduction
2. Methods
2.1. Data
2.2. National Assignment
2.3. Derived Data
2.4. Data Imputation
2.5. Statistics
2.6. Covariate Selection
2.7. Panel and Geospatial Analysis
2.8. Causal Inference
2.9. Data Availability
2.10. Ethics
3. Results
3.1. Data
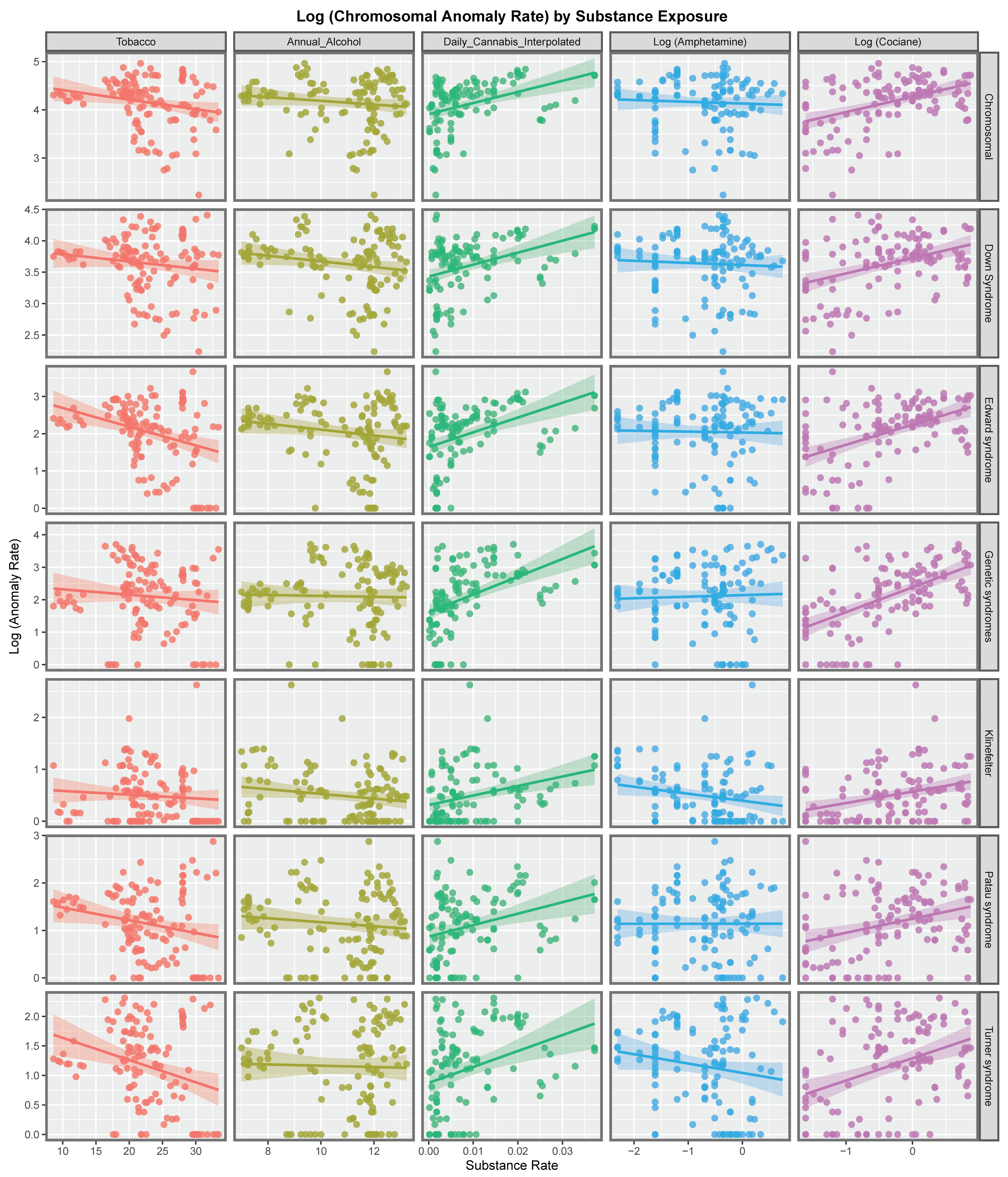
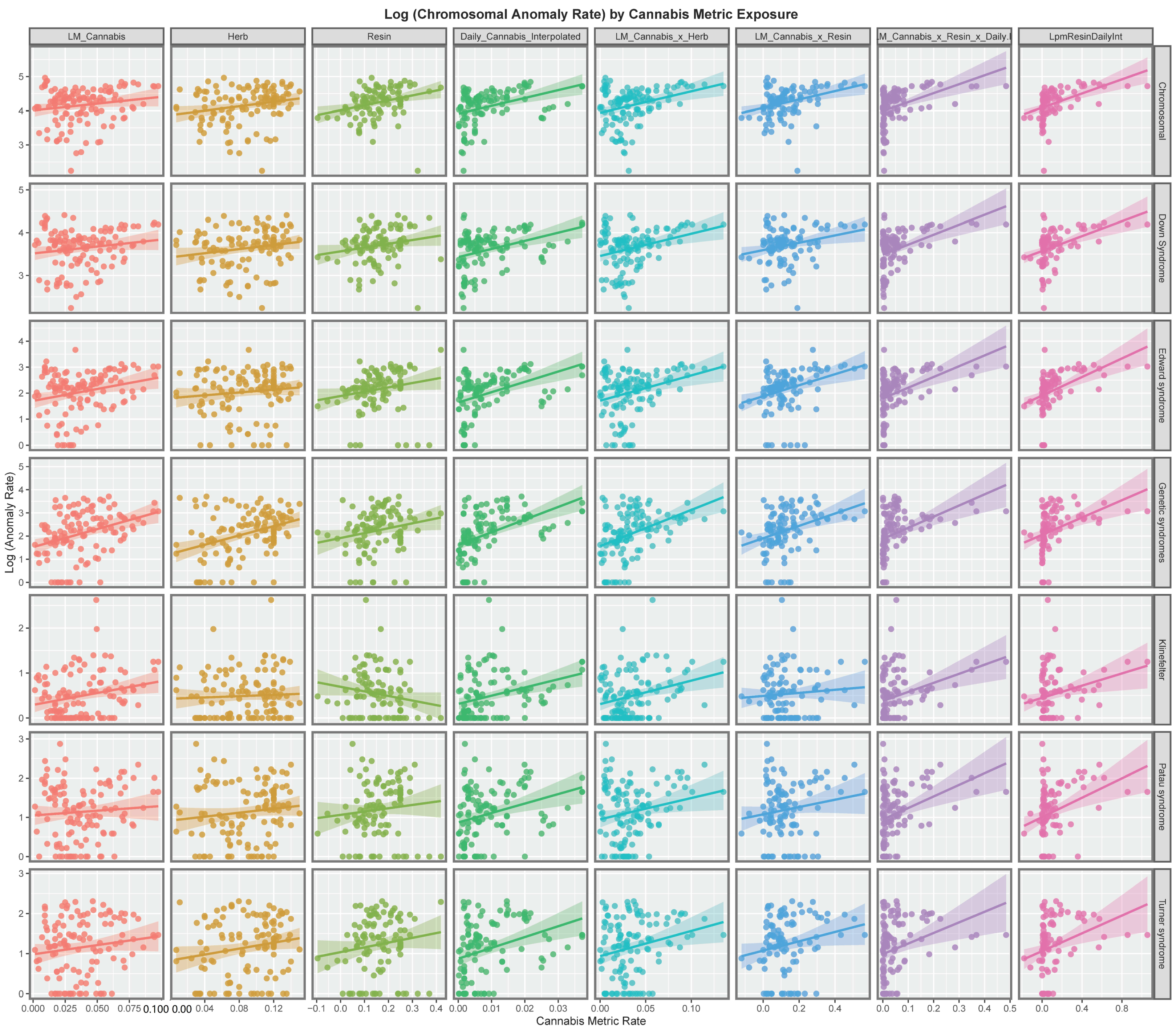
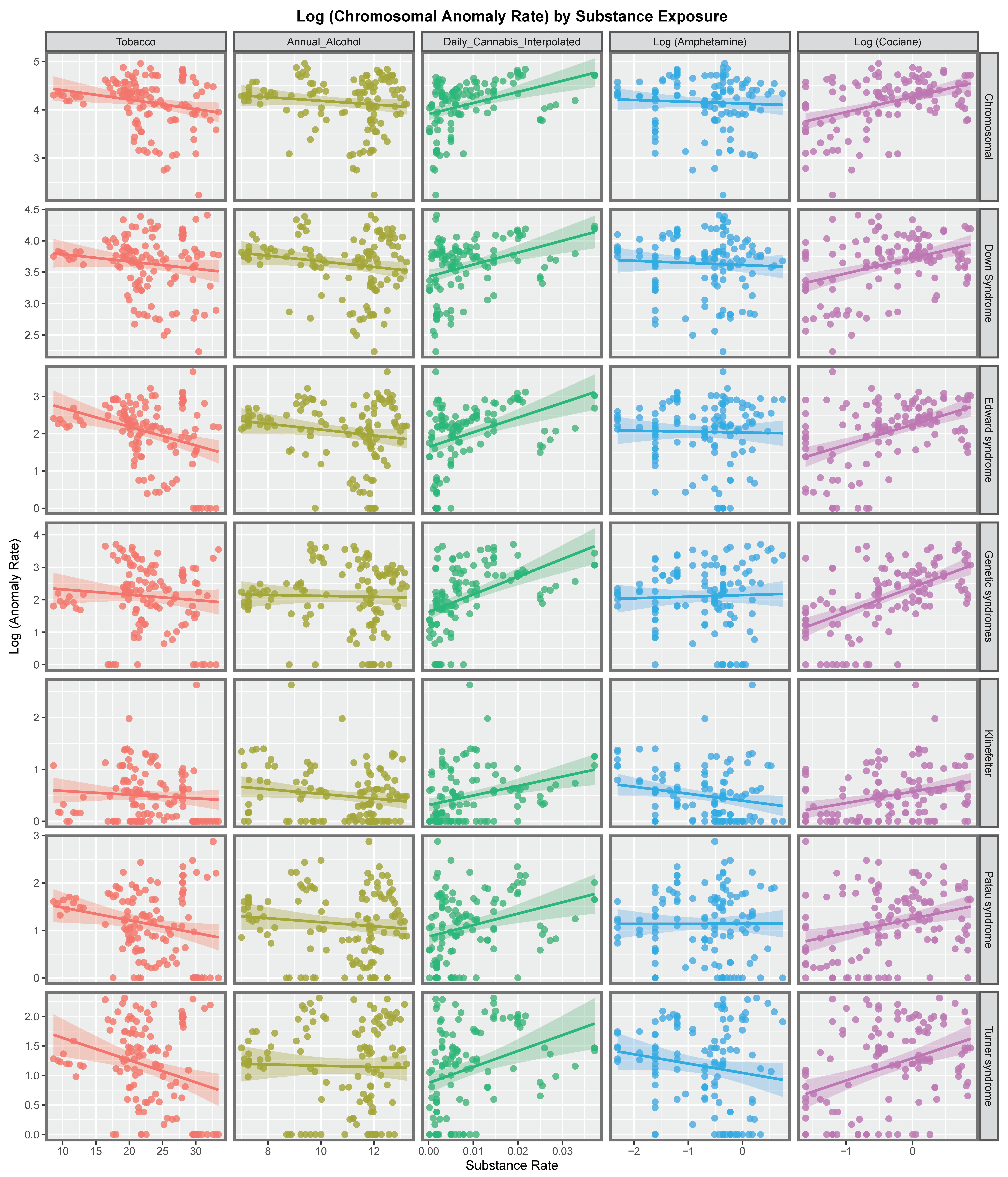
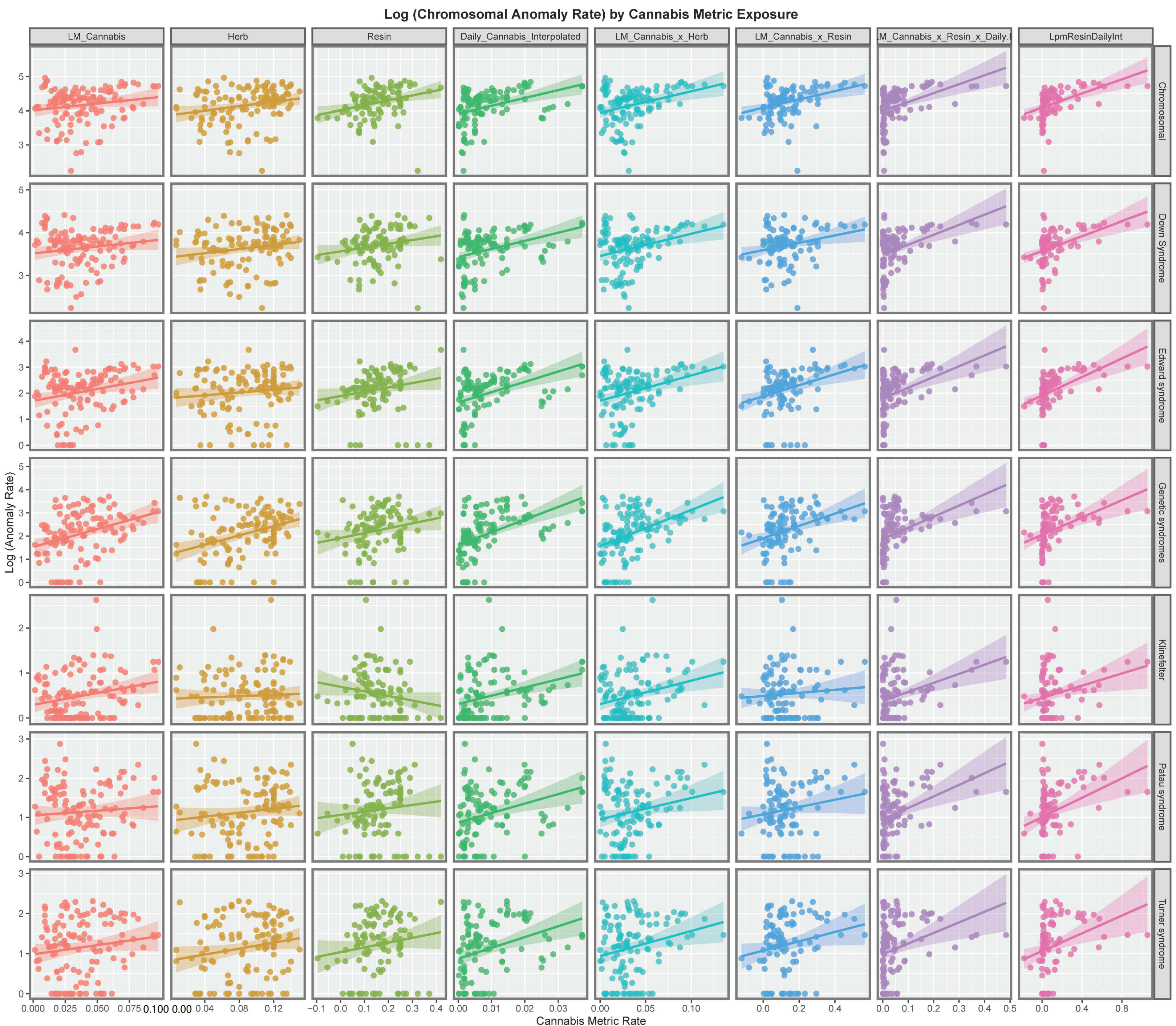
3.2. Bivariate Analysis
3.3. Multivariable Analysis
3.3.1. Random Forest Regression
3.3.2. Inverse Probability Weighted Panel Regression
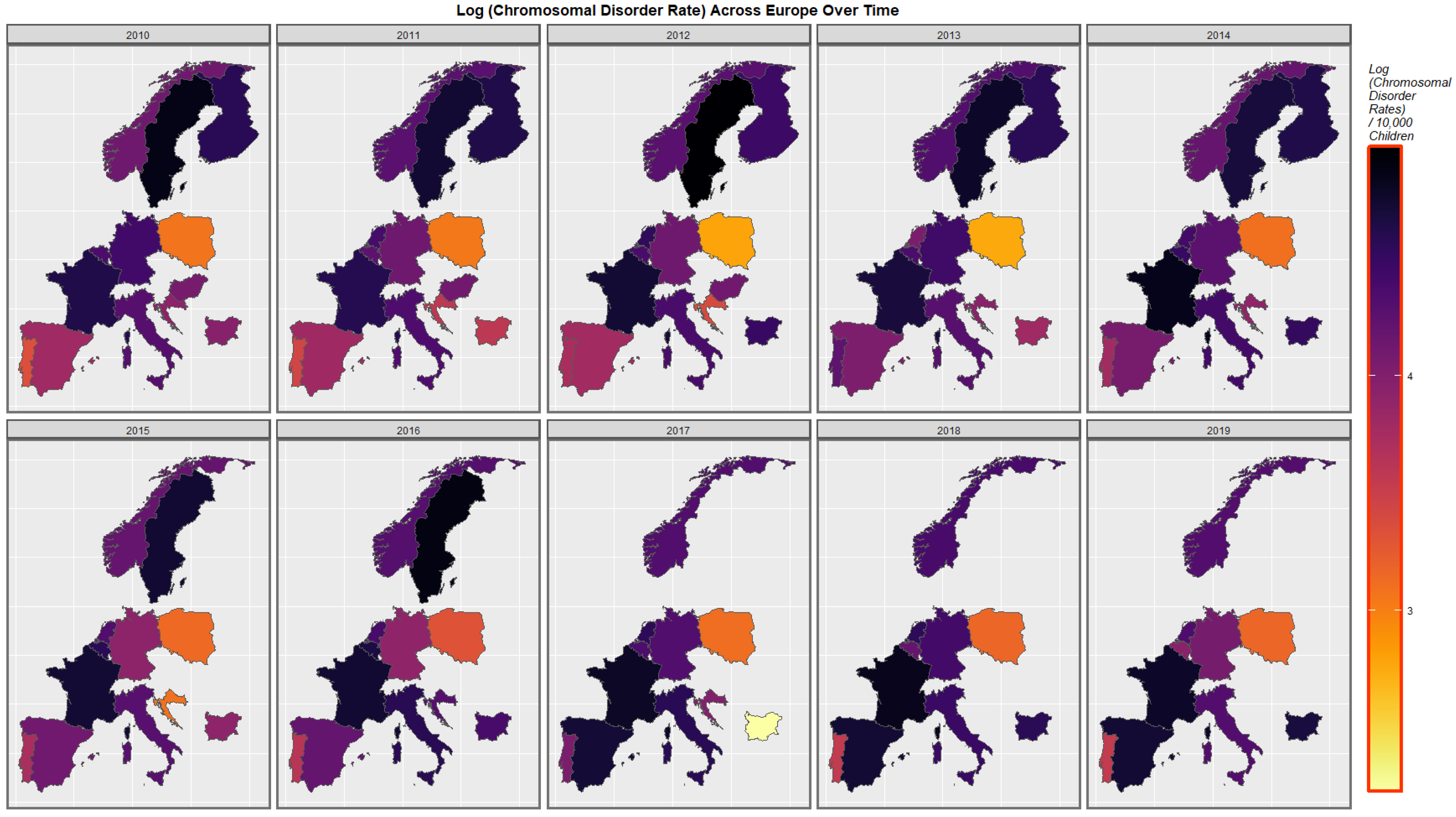
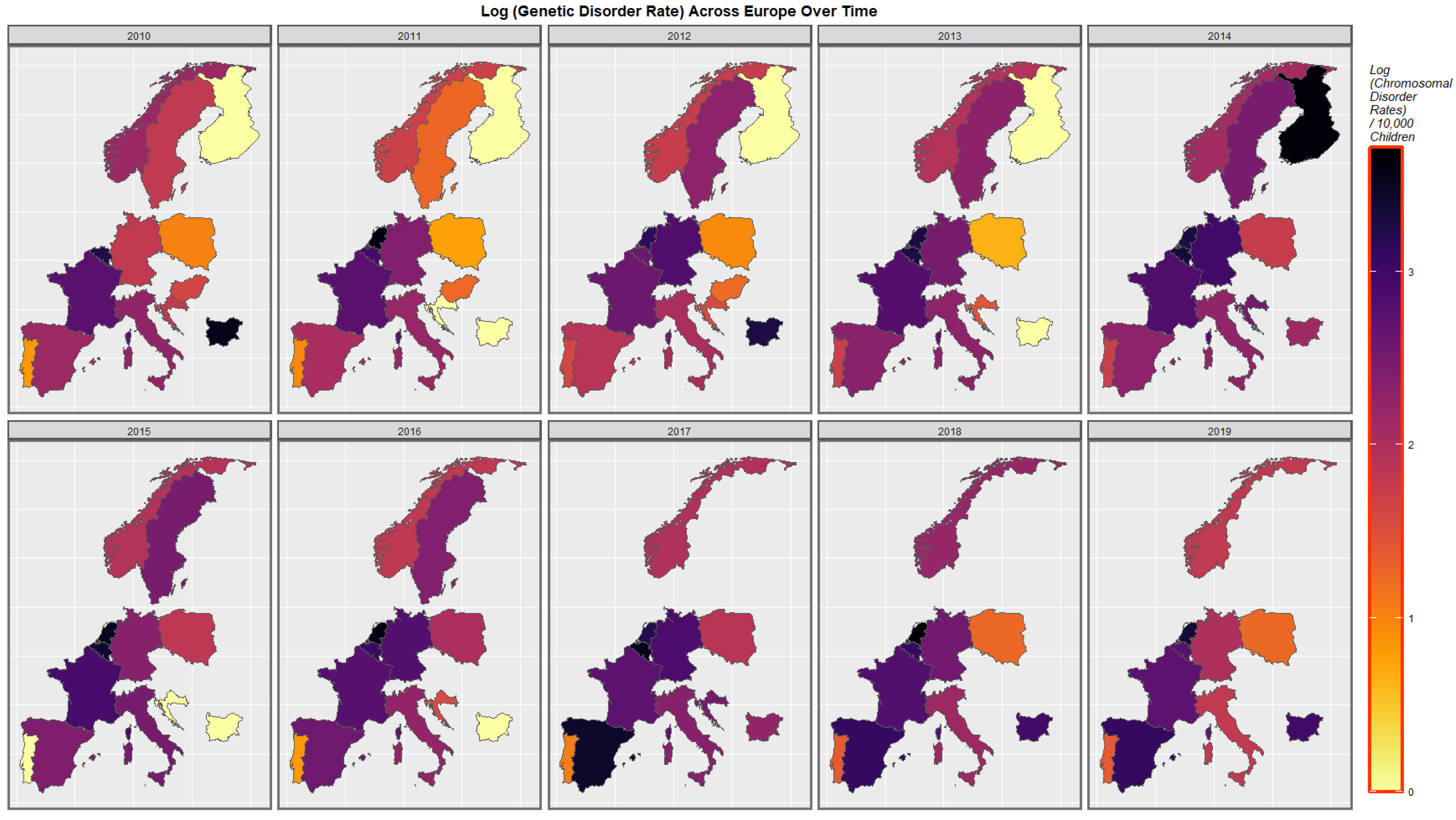
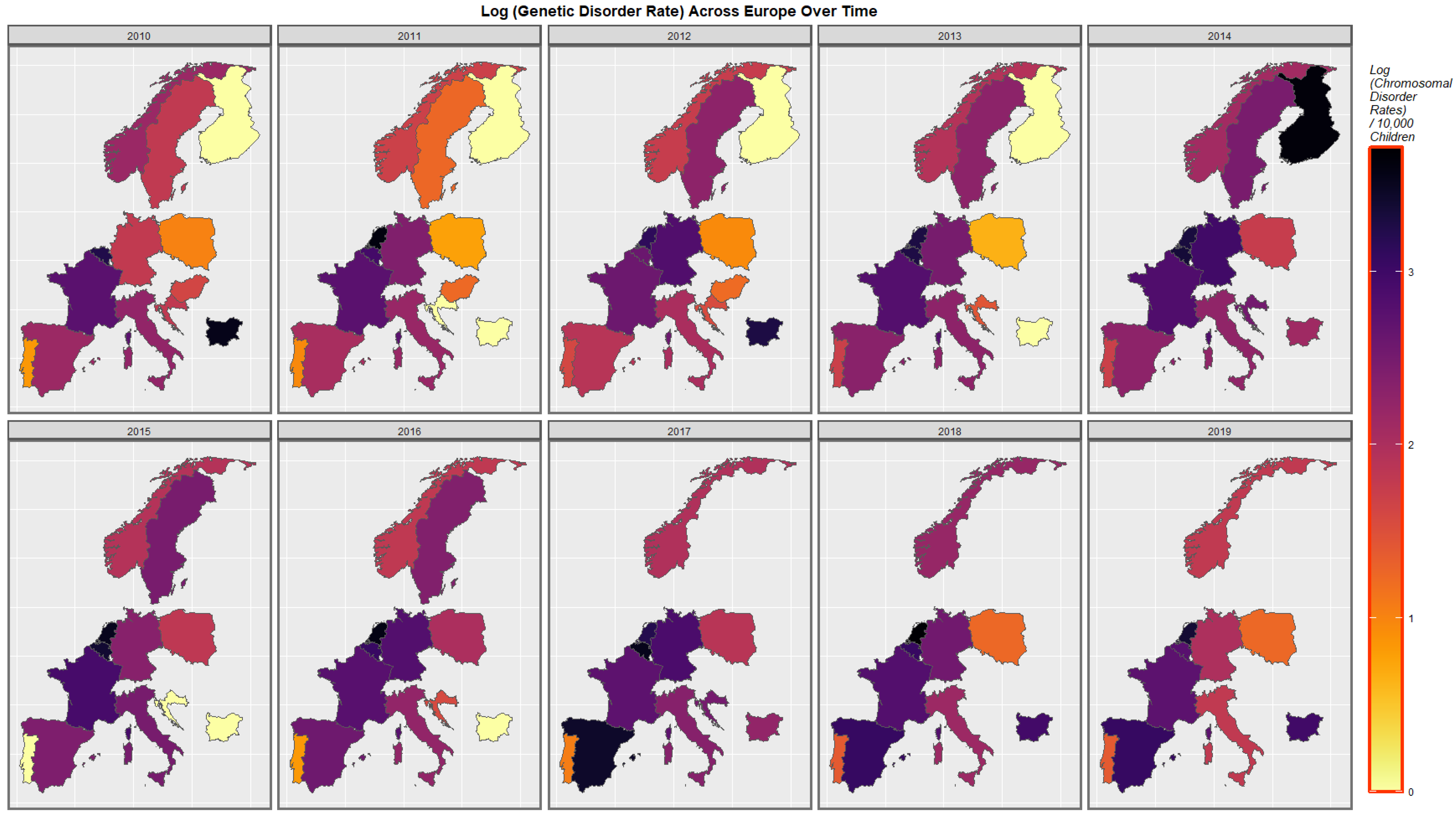
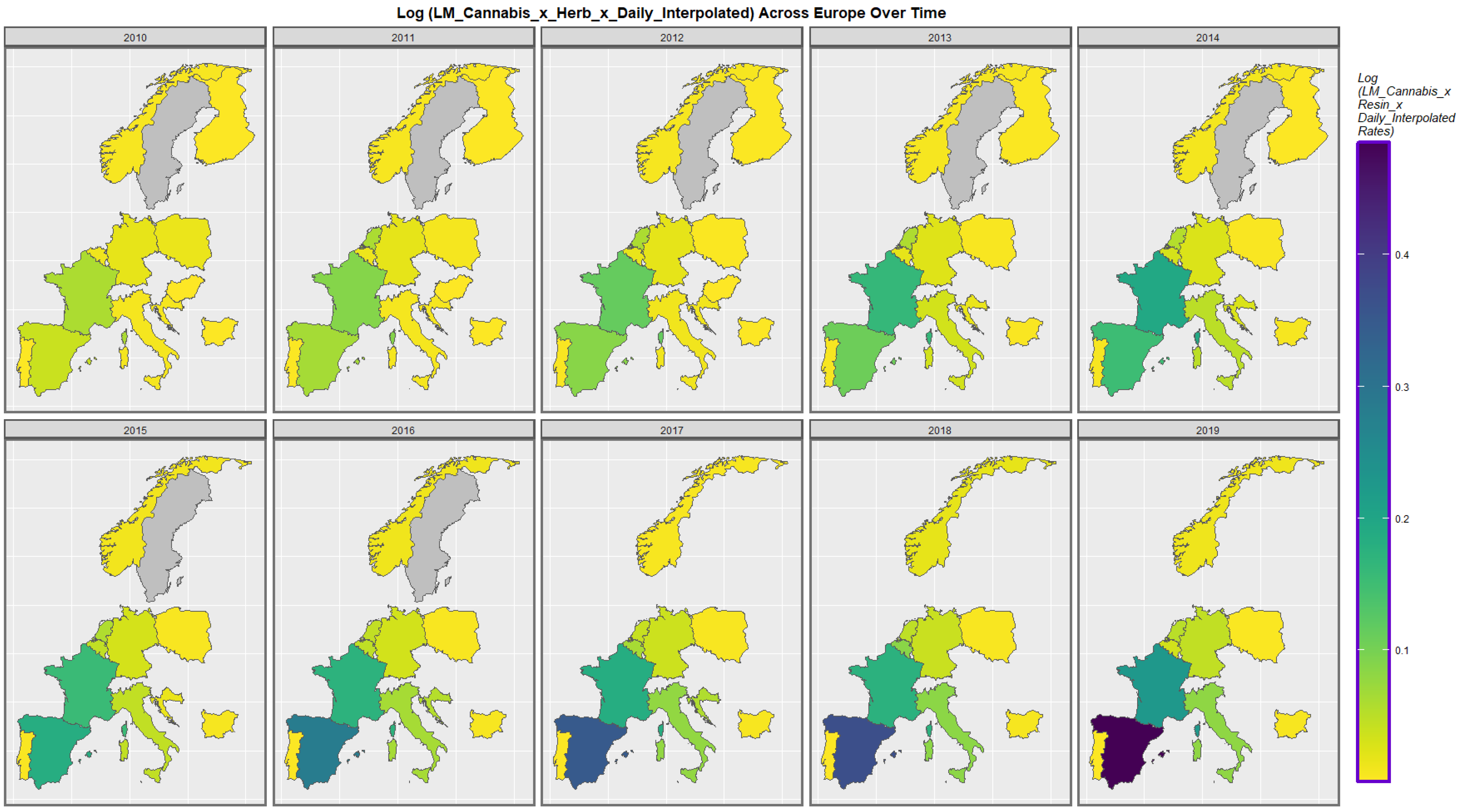
3.3.3. Geospatiotemporal Regression
3.4. Quantitative Causal Inference–E-Values
4. Discussion
4.1. Main Results
4.2. Choice of Anomalies
4.3. Qualitative Causal Inference
4.4. Quantitative Causal Inference
4.5. Mechanisms
4.5.1. Chromosomal Overview
4.5.2. Chromosomal Detachment
4.5.3. Epigenomic Mechanisms
4.5.4. Epigenomic Centrosomal Mechanisms
Epigenomic Gene Activation Post-Human Fertilization
Epigenomic Actions of Extrachromosomal DNA
5. Exponential Genotoxicity
6. Generalizability
7. Strengths and Limitations
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reece, A.S.; Hulse, G.K. Cannabis in Pregnancy—Rejoinder, Exposition and Cautionary Tales. Psychiatr. Times 2020, 37, 1–3. Available online: https://www.psychiatrictimes.com/view/cannabis-pregnancy-rejoinder-exposition-cautionary-tales (accessed on 17 January 2022).
- Reece, A.S.; Hulse, G.K. Epidemiological Overview of Multidimensional Chromosomal and Genome Toxicity of Cannabis Exposure in Congenital Anomalies and Cancer Development. Sci. Rep. 2021, 11, 13892–13912. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Cannabinoid and Substance Relationships of European Congenital Anomaly Patterns: A Space-Time Panel Regression and Causal Inferential Study. Environ. Epigenet. 2022, 8, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Cannabinoid Genotoxicity and Congenital Anomalies: A Convergent Synthesis of European and USA Datasets. In Cannabis, Cannabinoids and Endocannabinoids; Preedy, V., Patel, V., Eds.; Elsevier: London, UK, 2022; Volume 1. [Google Scholar]
- Reece, A.S.; Hulse, G.K. Cannabis Teratology Explains Current Patterns of Coloradan Congenital Defects: The Contribution of Increased Cannabinoid Exposure to Rising Teratological Trends. Clin. Pediatr. 2019, 58, 1085–1123. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Broad Spectrum epidemiological contribution of cannabis and other substances to the teratological profile of northern New South Wales: Geospatial and causal inference analysis. BMC Pharmacol. Toxicol. 2020, 21, 75–103. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Canadian Cannabis Consumption and Patterns of Congenital Anomalies: An Ecological Geospatial Analysis. J. Addict. Med. 2020, 14, e195–e210. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Geotemporospatial and causal inference epidemiological analysis of US survey and overview of cannabis, cannabidiol and cannabinoid genotoxicity in relation to congenital anomalies 2001–2015. BMC Pediatrics 2022, 22, 47–124. [Google Scholar] [CrossRef]
- Forrester, M.B.; Merz, R.D. Risk of selected birth defects with prenatal illicit drug use, Hawaii, 1986–2002. J. Toxicol. Environ. Health 2007, 70, 7–18. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Quadruple convergence—Rising cannabis prevalence, intensity, concentration and use disorder treatment. Lancet Reg. Health—Eur. 2021, 10, 100245–100246. [Google Scholar] [CrossRef]
- Manthey, J.; Freeman, T.P.; Kilian, C.; Lopez-Pelayo, H.; Rehm, J. Public health monitoring of cannabis use in Europe: Prevalence of use, cannabis potency, and treatment rates. Lancet Reg. Health—Eur. 2021, 10, 100227–200237. [Google Scholar] [CrossRef]
- Tahir, S.K.; Trogadis, J.E.; Stevens, J.K.; Zimmerman, A.M. Cytoskeletal organization following cannabinoid treatment in undifferentiated and differentiated PC12 cells. Biochem. Cell Biol. 1992, 70, 1159–1173. [Google Scholar] [CrossRef]
- Vela, G.; Martin, S.; Garcia-Gil, L.; Crespo, J.A.; Ruiz-Gayo, M.; Fernandez-Ruiz, J.J.; Garcia-Lecumberri, C.; Pelaprat, D.; Fuentes, J.A.; Ramos, J.A.; et al. Maternal exposure to delta9-tetrahydrocannabinol facilitates morphine self-administration behavior and changes regional binding to central mu opioid receptors in adult offspring female rats. Brain Res. 1998, 807, 101–109. [Google Scholar] [CrossRef]
- Busch, F.W.; Seid, D.A.; Wei, E.T. Mutagenic activity of marihuana smoke condensates. Cancer Lett. 1979, 6, 319–324. [Google Scholar] [CrossRef]
- Koller, V.J.; Ferk, F.; Al-Serori, H.; Misik, M.; Nersesyan, A.; Auwarter, V.; Grummt, T.; Knasmuller, S. Genotoxic properties of representatives of alkylindazoles and aminoalkyl-indoles which are consumed as synthetic cannabinoids. Food Chem. Toxicol. 2015, 80, 130–136. [Google Scholar] [CrossRef]
- Tahir, S.K.; Zimmerman, A.M. Influence of marihuana on cellular structures and biochemical activities. Pharmacol. Biochem. Behav. 1991, 40, 617–623. [Google Scholar] [CrossRef]
- Zimmerman, A.M.; Raj, A.Y. Influence of cannabinoids on somatic cells in vivo. Pharmacology 1980, 21, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Koller, V.J.; Auwarter, V.; Grummt, T.; Moosmann, B.; Misik, M.; Knasmuller, S. Investigation of the in vitro toxicological properties of the synthetic cannabimimetic drug CP-47,497-C8. Toxicol. Appl. Pharmacol. 2014, 277, 164–171. [Google Scholar] [CrossRef]
- Gant, J. Scientists are baffled by spatter of babies born without hands or arms in France, as investigation fails to discover a cause. Daily Mail, London, UK, 13 July 2019. [Google Scholar]
- Agence France-Presse in Paris. France to investigate cause of upper limb defects in babies. The Guardian, London, UK, 3 November 2018.
- Willsher, K. Baby arm defects prompt nationwide investigation in France. The Guardian, London, UK, 3 November 2018. [Google Scholar]
- Reece, A.S.; Hulse, G.K. Geotemporospatial and Causal Inferential Epidemiological Overview and Survey of USA Cannabis, Cannabidiol and Cannabinoid Genotoxicity Expressed in Cancer Incidence 2003–2017: Part 1—Continuous Bivariate Analysis. Arch. Public Health 2022, 80, 99–133. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Geotemporospatial and Causal Inferential Epidemiological Overview and Survey of USA Cannabis, Cannabidiol and Cannabinoid Genotoxicity Expressed in Cancer Incidence 2003–2017: Part 2—Categorical Bivariate Analysis and Attributable Fractions. Arch. Public Health 2022, 80, 100–135. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Geotemporospatial and Causal Inferential Epidemiological Overview and Survey of USA Cannabis, Cannabidiol and Cannabinoid Genotoxicity Expressed in Cancer Incidence 2003–2017: Part 3—Spatiotemporal, Multivariable and Causal Inferential Pathfinding and Exploratory Analyses of Prostate and Ovarian Cancers. Arch. Public Health 2022, 80, 100–136. [Google Scholar]
- Reece, A.S.; Hulse, G.K. Cannabis Genotoxicity and Cancer Incidence: A Highly Concordant Synthesis of European and USA Datasets. In Cannabis, Cannabinoids and Endocannabinoids; Preedy, V., Patel, V., Eds.; Elsevier: London, UK, 2022; Volume 1. [Google Scholar]
- Huang, H.F.S.; Nahas, G.G.; Hembree, W.C. Effects of Marijuana Inhalation on Spermatogenesis of the Rat. In Marijuana in Medicine; Nahas, G.G., Sutin, K.M., Harvey, D.J., Totowa, A.S., Eds.; Human Press: New York, NY, USA, 1999; Volume 1, pp. 359–366. [Google Scholar]
- Zimmerman, A.M.; Zimmerman, S.; Raj, A.Y. Effects of Cannabinoids on Spermatogensis in Mice. In Marijuana and Medicine, 1st ed.; Nahas, G.G., Sutin, K.M., Harvey, D.J., Totowa, A.S., Eds.; Humana Press: New York, NY, USA, 1999; Volume 1, pp. 347–358. [Google Scholar]
- Morishima, A.; Henrich, R.T.; Jayaraman, J.; Nahas, G.G. Hypoploid metaphases in cultured lymphocytes of marihuana smokers. Adv. Biosci. 1978, 22–23, 371–376. [Google Scholar]
- Henrich, R.T.; Nogawa, T.; Morishima, A. In vitro induction of segregational errors of chromosomes by natural cannabinoids in normal human lymphocytes. Environ. Mutagenesis 1980, 2, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Nahas, G.G.; Morishima, A.; Desoize, B. Effects of cannabinoids on macromolecular synthesis and replication of cultured lymphocytes. Fed. Proc. 1977, 36, 1748–1752. [Google Scholar] [PubMed]
- Morishima, A. Effects of cannabis and natural cannabinoids on chromosomes and ova. NIDA Res. Monogr. 1984, 44, 25–45. [Google Scholar] [PubMed]
- Leuchtenberger, C.; Leuchtenberger, R. Morphological and cytochemical effects of marijuana cigarette smoke on epithelioid cells of lung explants from mice. Nature 1971, 234, 227–229. [Google Scholar] [CrossRef]
- Stenchever, M.A.; Kunysz, T.J.; Allen, M.A. Chromosome breakage in users of marihuana. Am. J. Obstet. Gynecol. 1974, 118, 106–113. [Google Scholar] [CrossRef]
- Mon, M.J.; Haas, A.E.; Stein, J.L.; Stein, G.S. Influence of psychoactive and nonpsychoactive cannabinoids on cell proliferation and macromolecular biosynthesis in human cells. Biochem. Pharmacol. 1981, 30, 31–43. [Google Scholar] [CrossRef]
- Mon, M.J.; Haas, A.E.; Stein, J.L.; Stein, G.S. Influence of psychoactive and nonpsychoactive cannabinoids on chromatin structure and function in human cells. Biochem. Pharmacol. 1981, 30, 45–58. [Google Scholar] [CrossRef]
- Mon, M.J.; Jansing, R.L.; Doggett, S.; Stein, J.L.; Stein, G.S. Influence of delta9-tetrahydrocannabinol on cell proliferation and macromolecular biosynthesis in human cells. Biochem. Pharmacol. 1978, 27, 1759–1765. [Google Scholar] [CrossRef]
- Yang, X.; Hegde, V.L.; Rao, R.; Zhang, J.; Nagarkatti, P.S.; Nagarkatti, M. Histone modifications are associated with Delta9-tetrahydrocannabinol-mediated alterations in antigen-specific T cell responses. J. Biol. Chem. 2014, 289, 18707–18718. [Google Scholar] [CrossRef]
- Wang, J.; Yuan, W.; Li, M.D. Genes and pathways co-associated with the exposure to multiple drugs of abuse, including alcohol, amphetamine/methamphetamine, cocaine, marijuana, morphine, and/or nicotine: A review of proteomics analyses. Mol. Neurobiol. 2011, 44, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Chioccarelli, T.; Cacciola, G.; Altucci, L.; Lewis, S.E.; Simon, L.; Ricci, G.; Ledent, C.; Meccariello, R.; Fasano, S.; Pierantoni, R.; et al. Cannabinoid receptor 1 influences chromatin remodeling in mouse spermatids by affecting content of transition protein 2 mRNA and histone displacement. Endocrinology 2010, 151, 5017–5029. [Google Scholar] [CrossRef] [PubMed]
- Russo, C.; Ferk, F.; Mišík, M.; Ropek, N.; Nersesyan, A.; Mejri, D.; Holzmann, K.; Lavorgna, M.; Isidori, M.; Knasmüller, S. Low doses of widely consumed cannabinoids (cannabidiol and cannabidivarin) cause DNA damage and chromosomal aberrations in human-derived cells. Arch. Toxicol. 2019, 93, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiNieri, J.A.; Wang, X.; Szutorisz, H.; Spano, S.M.; Kaur, J.; Casaccia, P.; Dow-Edwards, D.; Hurd, Y.L. Maternal cannabis use alters ventral striatal dopamine D2 gene regulation in the offspring. Biol. Psychiatry 2011, 70, 763–769. [Google Scholar] [CrossRef]
- Ellis, R.J.; Bara, A.; Vargas, C.A.; Frick, A.L.; Loh, E.; Landry, J.; Uzamere, T.O.; Callens, J.E.; Martin, Q.; Rajarajan, P.; et al. Prenatal Δ(9)-Tetrahydrocannabinol Exposure in Males Leads to Motivational Disturbances Related to Striatal Epigenetic Dysregulation. Biol. Psychiatry 2021, 92, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Szutorisz, H.; Hurd, Y.L. Epigenetic Effects of Cannabis Exposure. Biol. Psychiatry 2016, 79, 586–594. [Google Scholar] [CrossRef]
- Szutorisz, H.; DiNieri, J.A.; Sweet, E.; Egervari, G.; Michaelides, M.; Carter, J.M.; Ren, Y.; Miller, M.L.; Blitzer, R.D.; Hurd, Y.L. Parental THC exposure leads to compulsive heroin-seeking and altered striatal synaptic plasticity in the subsequent generation. Neuropsychopharmacology 2014, 39, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.T.; Szutorisz, H.; Garg, P.; Martin, Q.; Landry, J.A.; Sharp, A.J.; Hurd, Y.L. Genome-Wide DNA Methylation Profiling Reveals Epigenetic Changes in the Rat Nucleus Accumbens Associated With Cross-Generational Effects of Adolescent THC Exposure. Neuropsychopharmacology 2015, 40, 2993–3005. [Google Scholar] [CrossRef]
- Szutorisz, H.; Hurd, Y.L. High times for cannabis: Epigenetic imprint and its legacy on brain and behavior. Neurosci. Biobehav. Rev. 2018, 85, 93–101. [Google Scholar] [CrossRef]
- Schrott, R.; Murphy, S.K.; Modliszewski, J.L.; King, D.E.; Hill, B.; Itchon-Ramos, N.; Raburn, D.; Price, T.; Levin, E.D.; Vandrey, R.; et al. Refraining from use diminishes cannabis-associated epigenetic changes in human sperm. Environ. Epigenet. 2021, 7, 1–10. [Google Scholar] [CrossRef]
- Murphy, S.K.; Itchon-Ramos, N.; Visco, Z.; Huang, Z.; Grenier, C.; Schrott, R.; Acharya, K.; Boudreau, M.H.; Price, T.M.; Raburn, D.J.; et al. Cannabinoid exposure and altered DNA methylation in rat and human sperm. Epigenetics 2018, 13, 1208–1221. [Google Scholar] [CrossRef] [PubMed]
- Schrott, R.; Acharya, K.; Itchon-Ramos, N.; Hawkey, A.B.; Pippen, E.; Mitchell, J.T.; Kollins, S.H.; Levin, E.D.; Murphy, S.K. Cannabis use is associated with potentially heritable widespread changes in autism candidate gene DLGAP2 DNA methylation in sperm. Epigenetics 2019, 15, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Rossato, M.; Ion Popa, F.; Ferigo, M.; Clari, G.; Foresta, C. Human sperm express cannabinoid receptor Cb1, the activation of which inhibits motility, acrosome reaction, and mitochondrial function. J. Clin. Endocrinol. Metab. 2005, 90, 984–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossato, M.; Pagano, C.; Vettor, R. The cannabinoid system and male reproductive functions. J. Neuroendocrinol. 2008, 20 (Suppl. S1), 90–93. [Google Scholar] [CrossRef]
- Mendelson, J.H.; Mello, N.K. Effects of marijuana on neuroendocrine hormones in human males and females. NIDA Res. Monogr. 1984, 44, 97–114. [Google Scholar]
- Smith, C.G.; Asch, R.H. Acute, short-term, and chronic effects of marijuana on the female primate reproductive function. NIDA Res. Monogr. 1984, 44, 82–96. [Google Scholar]
- Borowska, M.; Czarnywojtek, A.; Sawicka-Gutaj, N.; Woliński, K.; Płazińska, M.T.; Mikołajczak, P.; Ruchała, M. The effects of cannabinoids on the endocrine system. Endokrynol. Pol. 2018, 69, 705–719. [Google Scholar] [CrossRef]
- Mokoena, R.D.; George, P.B.; Abrahamse, H. Enhancing Breast Cancer Treatment Using a Combination of Cannabidiol and Gold Nanoparticles for Photodynamic Therapy. Int. J. Mol. Sci. 2019, 20, 4771. [Google Scholar] [CrossRef]
- de Nie, I.; Meißner, A.; Kostelijk, E.H.; Soufan, A.T.; Voorn-de Warem, I.A.C.; den Heijer, M.; Huirne, J.; van Mello, N.M. Impaired semen quality in trans women: Prevalence and determinants. Hum. Reprod. 2020, 35, 1529–1536. [Google Scholar] [CrossRef]
- Farokhnia, M.; McDiarmid, G.R.; Newmeyer, M.N.; Munjal, V.; Abulseoud, O.A.; Huestis, M.A.; Leggio, L. Effects of oral, smoked, and vaporized cannabis on endocrine pathways related to appetite and metabolism: A randomized, double-blind, placebo-controlled, human laboratory study. Transl. Psychiatry 2020, 10, 71. [Google Scholar] [CrossRef]
- Gillies, R.; Lee, K.; Vanin, S.; Laviolette, S.R.; Holloway, A.C.; Arany, E.; Hardy, D.B. Maternal exposure to Δ9-tetrahydrocannabinol impairs female offspring glucose homeostasis and endocrine pancreatic development in the rat. Reprod. Toxicol. 2020, 94, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Maia, J.; Almada, M.; Midão, L.; Fonseca, B.M.; Braga, J.; Gonçalves, D.; Teixeira, N.; Correia-da-Silva, G. The Cannabinoid Delta-9-tetrahydrocannabinol Disrupts Estrogen Signaling in Human Placenta. Toxicol. Sci. 2020, 177, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Sempio, C.; Wymore, E.; Palmer, C.; Bunik, M.; Henthorn, T.K.; Christians, U.; Klawitter, J. Detection of Cannabinoids by LC-MS-MS and ELISA in Breast Milk. J. Anal. Toxicol. 2020, 45, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Meah, F.; Lundholm, M.; Emanuele, N.; Amjed, H.; Poku, C.; Agrawal, L.; Emanuele, M.A. The effects of cannabis and cannabinoids on the endocrine system. Rev. Endocr. Metab. Disord. 2022, 23, 401–420. [Google Scholar] [CrossRef]
- Berta, D.G.; Kuisma, H.; Välimäki, N.; Räisänen, M.; Jäntti, M.; Pasanen, A.; Karhu, A.; Kaukomaa, J.; Taira, A.; Cajuso, T.; et al. Deficient H2A.Z deposition is associated with genesis of uterine leiomyoma. Nature 2021, 596, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Winick-Ng, W.; Kukalev, A.; Harabula, I.; Zea-Redondo, L.; Szabó, D.; Meijer, M.; Serebreni, L.; Zhang, Y.; Bianco, S.; Chiariello, A.M.; et al. Cell-type specialization is encoded by specific chromatin topologies. Nature 2021, 599, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Eurocat Data: Prevalence Charts and Tables. Available online: https://eu-rd-platform.jrc.ec.europa.eu/eurocat/eurocat-data/prevalence_en (accessed on 1 September 2021).
- NBDPN. Major Birth Defects Data from Population-Based Birth Defects Surveillance Programs in the United States, 2011–2015; Centers for Disease Control: Atlanta, GA, USA; National Birth Defects Prevention Network: Atlanta, GA, USA, 2018. [Google Scholar]
- McClean, D.K.; Zimmerman, A.M. Action of delta 9-tetrahydrocannabinol on cell division and macromolecular synthesis in division-synchronized protozoa. Pharmacology 1976, 14, 307–321. [Google Scholar] [CrossRef]
- Zimmerman, S.; Zimmerman, A.M. Genetic effects of marijuana. Int. J. Addict. 1990, 25, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Beh, T.T.; Kalitsis, P. Centromeres and Kinetochores. In Centromeres and Kinetochores; Black, B.E., Ed.; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Beh, T.T.; Kalitsis, P. The Role of Centromere Defects in Cancer. In Centromeres and Kinetochores; Black, B.E., Ed.; Springer: Philadelphia, PA, USA, 2017; Volume 1, pp. 1–554. [Google Scholar]
- Black, B.E. Preface to: Centromeres and Kinetochores. In Centromeres and Kinetochores; Black, B.E., Ed.; Springer: Cham, Switzerland, 2017; Volume 1, pp. V–VIII. [Google Scholar]
- Corbett, K.D. Molecular Mechanisms of Spindle Assembly Checkpoint Activation and Silencing. In Centromeres and Kinetochores; Black, B.E., Ed.; Springer: Philadelphia, PA, USA, 2017; Volume 1, pp. 1–554. [Google Scholar]
- Grishchuk, E.L. Biophysics of Microtubule End Coupling at the Kinetochore. In Centromeres and Kinetochores; Black, B.E., Ed.; Springer: Philadelphia, PA, USA, 2017; Volume 1, pp. 1–554. [Google Scholar]
- Hara, M.; Fukagawa, T. Critical Foundation of the Kinetochore: The Constitutive Centromere—Associated Network (CCAN). In Centromeres and Kinetochores; Black, B.E., Ed.; Springer: Philadelphia, PA, USA, 2017; Volume 1, pp. 1–554. [Google Scholar]
- Cavin-Meza, G.; Kwan, M.M.; Wignall, S.M. Multiple motors cooperate to establish and maintain acentrosomal spindle bipolarity in C. elegans oocyte meiosis. Elife 2022, 11, e72872. [Google Scholar] [CrossRef]
- Tischer, T.; Yang, J.; Barford, D. The APC/C targets the Cep152-Cep63 complex at the centrosome to regulate mitotic spindle assembly. J. Cell Sci. 2022, 135, jcs259273. [Google Scholar] [CrossRef]
- Janke, C.; Magiera, M.M. The tubulin code and its role in controlling microtubule properties and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 307–326. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.Y.; Hochstrasser, M. Histone sumoylation and chromatin dynamics. Nucleic Acids Res. 2021, 49, 6043–6052. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Causal inference multiple imputation investigation of the impact of cannabinoids and other substances on ethnic differentials in US testicular cancer incidence. BMC Pharmacol. Toxicol. 2021, 22, 40–71. [Google Scholar] [CrossRef]
- Global Health Observatory. Available online: https://www.who.int/data/gho/data/indicators/indicator-details/GHO/total-(recorded-unrecorded)-alcohol-per-capita-(15-)-consumption (accessed on 1 September 2021).
- European Monitoring Centre for Drugs and Drug Addiction (EMCDDA): Statistical Bulletin 2021—Prevalence of Drug Use. Available online: https://www.emcdda.europa.eu/data/stats2021/gps_en (accessed on 1 September 2021).
- The World Bank: Crude Data: Adjusted Net National Income Per Capita (Current US$). Available online: https://data.worldbank.org/indicator/NY.ADJ.NNTY.PC.CD (accessed on 1 September 2021).
- R: A Language and Environment for Statistical Computing. Available online: https://cran.r-project.org/ (accessed on 1 September 2021).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; Francios, R.; Groelmund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686–1691. [Google Scholar] [CrossRef]
- Pebesma, E. Simple Features for R: Standardized Support for Spatial Vector Data. R J. 2018, 10, 439–446. [Google Scholar] [CrossRef]
- Viridis: Default Color Maps from ‘matplotlib’. Available online: https://CRAN.R-project.org/package=viridis (accessed on 1 September 2021).
- Colorplaner: ggplot2 Extension to Visualize Two Variables Per Color Aesthetic Through Colorspace Projection. Available online: https://github.com/wmurphyrd/colorplaner (accessed on 1 September 2021).
- Pinheiro, J.; Bates, D.; DebRoy, S.; Sarkar, D.; R Core Team. nlme: Linear and Nonlinear Mixed Effects Models; Comprehensive R Archive Network, 2020; Volume 1. [Google Scholar]
- Broom.mixed: Tidying Methods for Mixed Models. Available online: http://github.com/bbolker/broom.mixed (accessed on 1 September 2021).
- Broom: Convert Statistical Objects into Tidy Tibbles. Available online: https://CRAN.R-project.org/package=broom (accessed on 1 September 2021).
- Leeper, T.J. Margins: Marginal Effects for Model Objects; Leeper, T.J., Ed.; R Package Version 0.3.26; 2021; Volume 1, pp. 1–36. Available online: https://rdrr.io/cran/margins/man/margins.html#:~:text=%20In%20margins%3A%20Marginal%20Effects%20for%20Model%20Objects,See%20Also.%20%208%20Examples.%20%20More%20 (accessed on 10 March 2022).
- Wright, M.N.; Ziegler, A. ranger: A Fast Implementation of Random Forests for High Dimensional Data in C++ and R. J. Stat. Softw. 2017, 77, 1–17. [Google Scholar] [CrossRef]
- Greenwell, B.M.; Boehmke, B.C. Variable Importance Plots—An Introduction to the vip Package. R J. 2021, 12, 343–366. [Google Scholar] [CrossRef]
- Package ‘plm’. Available online: https://cran.r-project.org/web/packages/plm/plm.pdf (accessed on 1 September 2021).
- Bivand, R.; Anselin, L.; Berke, O.; Bernat, A.; Carvalho, M.; Chun, Y.; Dormann, C.; Dray, S.; Halbersma, R.; Lewis-Koh, N.; et al. The spdep Package; CRAN: Houston, TX, USA, 2007; pp. 1–143. [Google Scholar]
- Millo, G.; Piras, G. Splm: Spatial Panel Data Models in R. J. Stastistical Softw. 2012, 47, 1–38. [Google Scholar]
- Millo, G.; Piras, G. Package ‘splm’; CRAN (Central R-Archive Network): Trieste, Italy, 2018; pp. 1–27. Available online: https://cran.r-project.org/web/packages/splm/splm.pdf (accessed on 1 September 2021).
- Croissant, Y.; Millo, G. Panel Data Econometrics with R; John Wiley and Sons: Oxford, UK, 2019; Volume 1. [Google Scholar]
- Wal, W.; Geskus, R. Ipw: An R Package for Inverse Probability Weighting. J. Stat. Softw. 2011, 43, 1–23. [Google Scholar] [CrossRef]
- VanderWeele, T.J.; Ding, P. Sensitivity Analysis in Observational Research: Introducing the E-Value. Ann. Intern. Med. 2017, 167, 268–274. [Google Scholar] [CrossRef] [PubMed]
- VanderWeele, T.J.; Martin, J.N.; Mathur, M.B. E-values and incidence density sampling. Epidemiology 2020, 31, e51–e52. [Google Scholar] [CrossRef]
- VanderWeele, T.J.; Mathur, M.B. Commentary: Developing best-practice guidelines for the reporting of E-values. Int. J. Epidemiol. 2020, 49, 1495–1497. [Google Scholar] [CrossRef] [PubMed]
- VanderWeele, T.J.; Ding, P.; Mathur, M. Technical Considerations in the Use of the E-Value. J. Causal Inference 2019, 7, 1–11. [Google Scholar] [CrossRef]
- Pearl, J.; Mackaenzie, D. The Book of Why: The New Science of Cause and Effect; Basic Books: New York, NY, USA, 2019; Volume 1. [Google Scholar]
- Package ‘EValue’. Available online: https://cran.r-project.org/web/packages/EValue/EValue.pdf (accessed on 1 September 2021).
- Hill, A.B. The Environment and Disease: Association or Causation? Proc. R Soc. Med. 1965, 58, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Hernán, M.A. Methods of Public Health Research—Strengthening Causal Inference from Observational Data. N. Engl. J. Med. 2021, 385, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Lea, R.A.; Niakan, K.K. Human germline genome editing. Nat. Cell Biol. 2019, 21, 1479–1489. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Zahn, L.M. The human genome. Science 2021, 373, 1458–1459. [Google Scholar] [CrossRef]
- Malouf, C.; Ottersbach, K. Molecular processes involved in B cell acute lymphoblastic leukaemia. Cell Mol. Life Sci. 2018, 75, 417–446. [Google Scholar] [CrossRef]
- Gurney, J.; Shaw, C.; Stanley, J.; Signal, V.; Sarfati, D. Cannabis exposure and risk of testicular cancer: A systematic review and meta-analysis. BMC Cancer 2015, 15, 897–906. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. The Production of Homozygous Deficient Tissues with Mutant Characteristics by Means of the Aberrant Mitotic Behavior of Ring-Shaped Chromosomes. Genetics 1938, 23, 315–376. [Google Scholar] [CrossRef] [PubMed]
- Hussein, N.A.E.M.; El-Toukhy, M.A.E.-F.; Kazem, A.H.; Ali, M.E.-S.; Ahmad, M.A.E.-R.; Ghazy, H.M.R.; El-Din, A.M.G. Protective and therapeutic effects of cannabis plant extract on liver cancer induced by dimethylnitrosamine in mice. Alex. J. Med. 2014, 50, 241–251. [Google Scholar] [CrossRef]
- Hall, W.; Degenhardt, L. Adverse health effects of non-medical cannabis use. Lancet 2009, 374, 1383–1391. [Google Scholar] [CrossRef]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M.; Knasmueller, S.; Bolognesi, C.; Holland, N.; Bonassi, S.; Kirsch-Volders, M. Micronuclei as biomarkers of DNA damage, aneuploidy, inducers of chromosomal hypermutation and as sources of pro-inflammatory DNA in humans. Mutat. Res. 2020, 786, 108342. [Google Scholar] [CrossRef] [PubMed]
- Hölzel, B.N.; Pfannkuche, K.; Allner, B.; Allner, H.T.; Hescheler, J.; Derichsweiler, D.; Hollert, H.; Schiwy, A.; Brendt, J.; Schaffeld, M.; et al. Following the adverse outcome pathway from micronucleus to cancer using H2B-eGFP transgenic healthy stem cells. Arch. Toxicol. 2020, 94, 3265–3280. [Google Scholar] [CrossRef]
- Knouse, K.A.; Amon, A. Cell biology: The micronucleus gets its big break. Nature 2015, 522, 162–163. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef]
- Terradas, M.; Martín, M.; Genescà, A. Detection of Impaired DNA Replication and Repair in Micronuclei as Indicators of Genomic Instability and Chromothripsis. Methods Mol. Biol. 2018, 1769, 197–208. [Google Scholar]
- Waldron, D. Genome stability: Chromothripsis and micronucleus formation. Nat. Rev. Genet. 2015, 16, 376–377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed]
- de Pagter, M.S.; van Roosmalen, M.J.; Baas, A.F.; Renkens, I.; Duran, K.J.; van Binsbergen, E.; Tavakoli-Yaraki, M.; Hochstenbach, R.; van der Veken, L.T.; Cuppen, E.; et al. Chromothripsis in healthy individuals affects multiple protein-coding genes and can result in severe congenital abnormalities in offspring. Am. J. Hum. Genet. 2015, 96, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef]
- Kloosterman, W.P.; Guryev, V.; van Roosmalen, M.; Duran, K.J.; de Bruijn, E.; Bakker, S.C.; Letteboer, T.; van Nesselrooij, B.; Hochstenbach, R.; Poot, M.; et al. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum. Mol. Genet. 2011, 20, 1916–1924. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, W.P.; Hoogstraat, M.; Paling, O.; Tavakoli-Yaraki, M.; Renkens, I.; Vermaat, J.S.; van Roosmalen, M.J.; van Lieshout, S.; Nijman, I.J.; Roessingh, W.; et al. Chromothripsis is a common mechanism driving genomic rearrangements in primary and metastatic colorectal cancer. Genome Biol 2011, 12, R103. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Chromothripsis and epigenomics complete causality criteria for cannabis- and addiction-connected carcinogenicity, congenital toxicity and heritable genotoxicity. Mutat. Res. 2016, 789, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Gowran, A.; Murphy, C.E.; Campbell, V.A. Delta(9)-tetrahydrocannabinol regulates the p53 post-translational modifiers Murine double minute 2 and the Small Ubiquitin MOdifier protein in the rat brain. FEBS Lett. 2009, 583, 3412–3418. [Google Scholar] [CrossRef] [PubMed]
- GeneCards: MSH5. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=MSH5&keywords=msh5 (accessed on 10 March 2022).
- RAD21L1—RAD21 Cohesin Complex Component Like 1. Available online: https://www.ncbi.nlm.nih.gov/nuccore/?term=rad21L1 (accessed on 10 March 2022).
- SMC1B—Structural Maintenance of Chromosomes 1B. Available online: https://www.ncbi.nlm.nih.gov/nuccore/?term=smc1b (accessed on 10 March 2022).
- SYCP3—Synaptonemal Complex Protein 3. Available online: https://www.ncbi.nlm.nih.gov/nuccore/?term=sycp3 (accessed on 10 March 2022).
- CDK1—Cyclin Dependent Kinase 1. Available online: https://www.ncbi.nlm.nih.gov/nuccore/?term=cdk1 (accessed on 10 March 2022).
- GeneCards: CDK1. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CDK1&keywords=cdk1 (accessed on 10 March 2022).
- GeneCards: CD3D. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CD3D&keywords=cd3d (accessed on 10 March 2022).
- GeneCards: EPHA2. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=EPHA2&keywords=epha2 (accessed on 10 March 2022).
- GeneCards: FYN. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=FYN&keywords=fyn (accessed on 10 March 2022).
- GeneCards: KMT2A. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=KMT2A&keywords=kmt2a (accessed on 10 March 2022).
- Meharena, H.S.; Marco, A.; Dileep, V.; Lockshin, E.R.; Akatsu, G.Y.; Mullahoo, J.; Watson, L.A.; Ko, T.; Guerin, L.N.; Abdurrob, F.; et al. Down-syndrome-induced senescence disrupts the nuclear architecture of neural progenitors. Cell Stem Cell 2022, 29, 116–130.e117. [Google Scholar] [CrossRef]
- Yilmaz, D.; Furst, A.; Meaburn, K.; Lezaja, A.; Wen, Y.; Altmeyer, M.; Reina-San-Martin, B.; Soutoglou, E. Activation of homologous recombination in G1 preserves centromeric integrity. Nature 2021, 600, 748–753. [Google Scholar] [CrossRef]
- Segbert, C.; Barkus, R.; Powers, J.; Strome, S.; Saxton, W.M.; Bossinger, O. KLP-18, a Klp2 kinesin, is required for assembly of acentrosomal meiotic spindles in Caenorhabditis elegans. Mol. Biol. Cell 2003, 14, 4458–4469. [Google Scholar] [CrossRef] [PubMed]
- Janke, C.; Montagnac, G. Causes and Consequences of Microtubule Acetylation. Curr. Biol. 2017, 27, R1287–R1292. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.J.; Feng, Y.H.; Gu, B.H.; Li, Y.M.; Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Blengini, C.S.; Schindler, K. Acentriolar spindle assembly in mammalian female meiosis and the consequences of its perturbations on human reproduction†. Biol. Reprod. 2021, 106, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Cavazza, T.; Takeda, Y.; Politi, A.Z.; Aushev, M.; Aldag, P.; Baker, C.; Choudhary, M.; Bucevičius, J.; Lukinavičius, G.; Elder, K.; et al. Parental genome unification is highly error-prone in mammalian embryos. Cell 2021, 184, 2860–2877.e2822. [Google Scholar] [CrossRef] [PubMed]
- So, C.; Menelaou, K.; Uraji, J.; Harasimov, K.; Steyer, A.M.; Seres, K.B.; Bucevičius, J.; Lukinavičius, G.; Möbius, W.; Sibold, C.; et al. Mechanism of spindle pole organization and instability in human oocytes. Science 2022, 375, eabj3944. [Google Scholar] [CrossRef]
- Asami, M.; Lam, B.Y.H.; Ma, M.K.; Rainbow, K.; Braun, S.; VerMilyea, M.D.; Yeo, G.S.H.; Perry, A.C.F. Human embryonic genome activation initiates at the one-cell stage. Cell Stem Cell 2022, 29, 209–216.e204. [Google Scholar] [CrossRef]
- Hung, K.L.; Yost, K.E.; Xie, L.; Shi, Q.; Helmsauer, K.; Luebeck, J.; Schöpflin, R.; Lange, J.T.; Chamorro González, R.; Weiser, N.E.; et al. ecDNA hubs drive cooperative intermolecular oncogene expression. Nature 2021, 600, 731–736. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. A geospatiotemporal and causal inference epidemiological exploration of substance and cannabinoid exposure as drivers of rising US pediatric cancer rates. BMC Cancer 2021, 21, 197–230. [Google Scholar] [CrossRef]
- Reece, A.S. Rapid Response: Cannabinoid Genotoxic Trifecta—Cancerogenesis, Clinical Teratogenesis and Cellular Ageing. Br. Med. J. 2022, 376, n3114. [Google Scholar]
- Reece, A.S.; Hulse, G.K. Geospatiotemporal and Causal Inference Study of Cannabis and Other Drugs as Risk Factors for Female Breast Cancer USA 2003–2017. Environ. Epigenet. 2022, 2022, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Novel Insights into Potential Cannabis-Related Cancerogenesis from Recent Key Whole Epigenome Screen of Cannabis Dependence and Withdrawal: Epidemiological Comment and Explication of Schrott. J. Oncol. 2022; submitted. [Google Scholar]
- Aldington, S.; Harwood, M.; Cox, B.; Weatherall, M.; Beckert, L.; Hansell, A.; Pritchard, A.; Robinson, G.; Beasley, R. Cannabis use and risk of lung cancer: A case-control study. Eur. Respir. J. 2008, 31, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Voirin, N.; Berthiller, J.; Benhaim-Luzon, V.; Boniol, M.; Straif, K.; Ayoub, W.B.; Ayed, F.B.; Sasco, A.J. Risk of lung cancer and past use of cannabis in Tunisia. J. Thorac. Oncol. 2006, 1, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, J.; Straif, K.; Boniol, M.; Voirin, N.; Benhaim-Luzon, V.; Ayoub, W.B.; Dari, I.; Laouamri, S.; Hamdi-Cherif, M.; Bartal, M.; et al. Cannabis smoking and risk of lung cancer in men: A pooled analysis of three studies in Maghreb. J. Thorac. Oncol. 2008, 3, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.F.; Morgenstern, H.; Spitz, M.R.; Tashkin, D.P.; Yu, G.P.; Marshall, J.R.; Hsu, T.C.; Schantz, S.P. Marijuana use and increased risk of squamous cell carcinoma of the head and neck. Cancer Epidemiol. Biomark. Prev. 1999, 8, 1071–1078. [Google Scholar]
- Hashibe, M.; Ford, D.E.; Zhang, Z.F. Marijuana smoking and head and neck cancer. J. Clin. Pharmacol. 2002, 42 (Suppl. S11), 103S–107S. [Google Scholar] [CrossRef]
- Sidney, S.; Quesenberry, C.P., Jr.; Friedman, G.D.; Tekawa, I.S. Marijuana use and cancer incidence (California, United States). Cancer Causes Control 1997, 8, 722–728. [Google Scholar] [CrossRef]
- Daling, J.R.; Doody, D.R.; Sun, X.; Trabert, B.L.; Weiss, N.S.; Chen, C.; Biggs, M.L.; Starr, J.R.; Dey, S.K.; Schwartz, S.M. Association of marijuana use and the incidence of testicular germ cell tumors. Cancer 2009, 115, 1215–1223. [Google Scholar] [CrossRef]
- Efird, J.T.; Friedman, G.D.; Sidney, S.; Klatsky, A.; Habel, L.A.; Udaltsova, N.V.; Van den Eeden, S.; Nelson, L.M. The risk for malignant primary adult-onset glioma in a large, multiethnic, managed-care cohort: Cigarette smoking and other lifestyle behaviors. J. Neuro-Oncol. 2004, 68, 57–69. [Google Scholar] [CrossRef]
- Moiche Bokobo, P.; Atxa de la Presa, M.A.; Cuesta Angulo, J. Transitional cell carcinoma in a young heavy marihuana smoker. Arch. Esp. Urol. 2001, 54, 165–167. [Google Scholar]
- Chacko, J.A.; Heiner, J.G.; Siu, W.; Macy, M.; Terris, M.K. Association between marijuana use and transitional cell carcinoma. Urology 2006, 67, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Nieder, A.M.; Lipke, M.C.; Madjar, S. Transitional cell carcinoma associated with marijuana: Case report and review of the literature. Urology 2006, 67, 200. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Cannabinoid exposure as a major driver of pediatric acute lymphoid Leukaemia rates across the USA: Combined geospatial, multiple imputation and causal inference study. BMC Cancer 2021, 21, 984–1017. [Google Scholar] [CrossRef] [PubMed]
- United National Office of Drugs and Crime. World Drug Report 2021; United National World Health Organization: Geneva, Switzerland, 2021; Volume 1–5, Available online: https://wdr.unodc.org/wdr2019/index.html (accessed on 1 September 2021).
- United National Office of Drugs and Crime. World Drug Report 2019; United National World Health Organization: Geneva, Switzerland, 2019; Volume 1–5, Available online: https://wdr.unodc.org/wdr2019/ (accessed on 1 September 2021).
- Reece, A.S.; Hulse, G.K. Contemporary epidemiology of rising atrial septal defect trends across USA 1991–2016: A combined ecological geospatiotemporal and causal inferential study. BMC Pediatr. 2020, 20, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Raad, H.; Cornelius, V.; Chan, S.; Williamson, E.; Cro, S. An evaluation of inverse probability weighting using the propensity score for baseline covariate adjustment in smaller population randomised controlled trials with a continuous outcome. BMC Med. Res. Methodol. 2020, 20, 70. [Google Scholar] [CrossRef] [PubMed]
- Seaman, S.R.; White, I.R. Review of inverse probability weighting for dealing with missing data. Stat. Methods Med. Res. 2013, 22, 278–295. [Google Scholar] [CrossRef]
- VanverWeele, T.J.; Mathur, M.; Chen, Y. Outcome-Wide Longitudinal Designs for Causal Inference: A New Template for Empirical Studies. Stat. Sci. 2020, 35, 437–466. [Google Scholar]
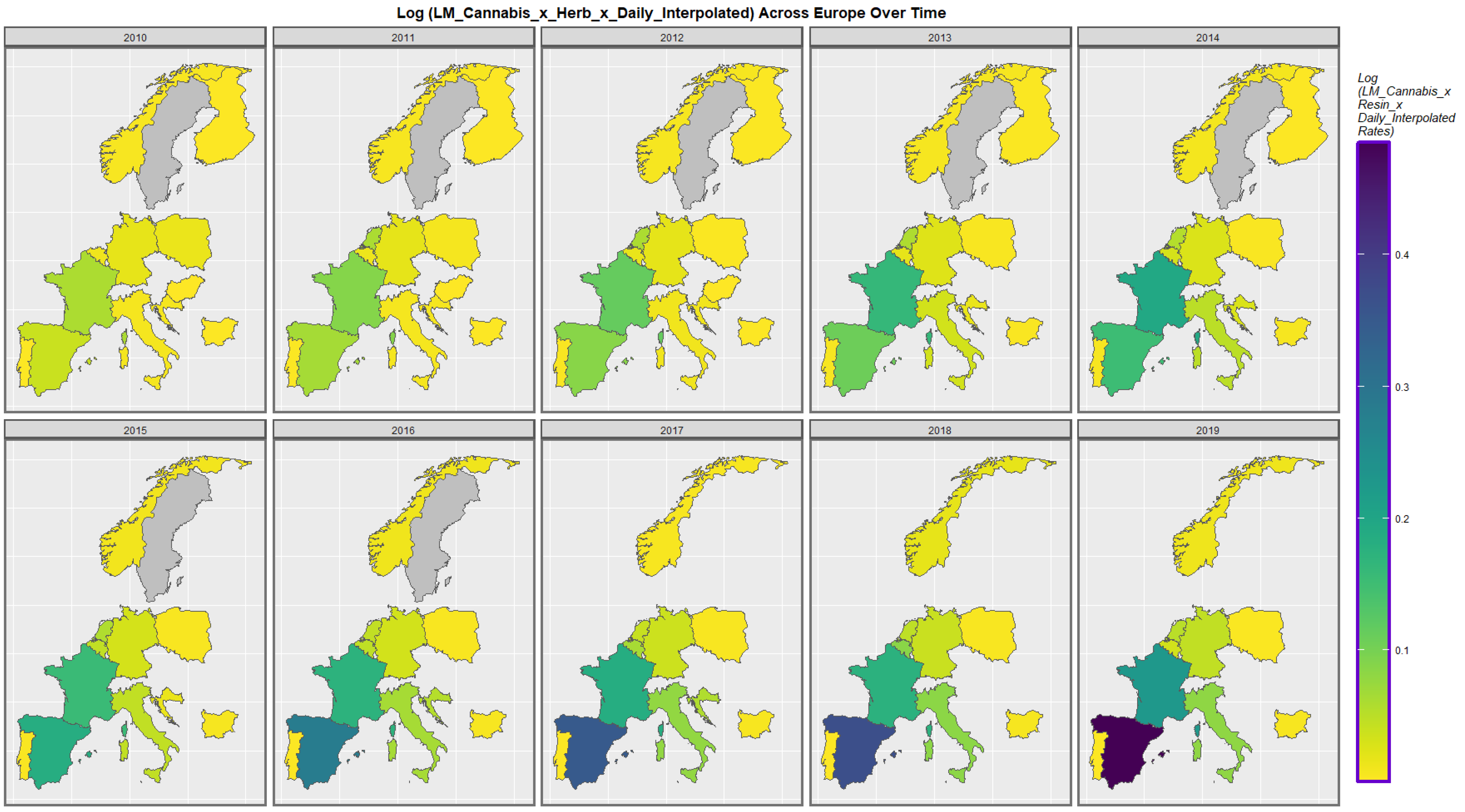
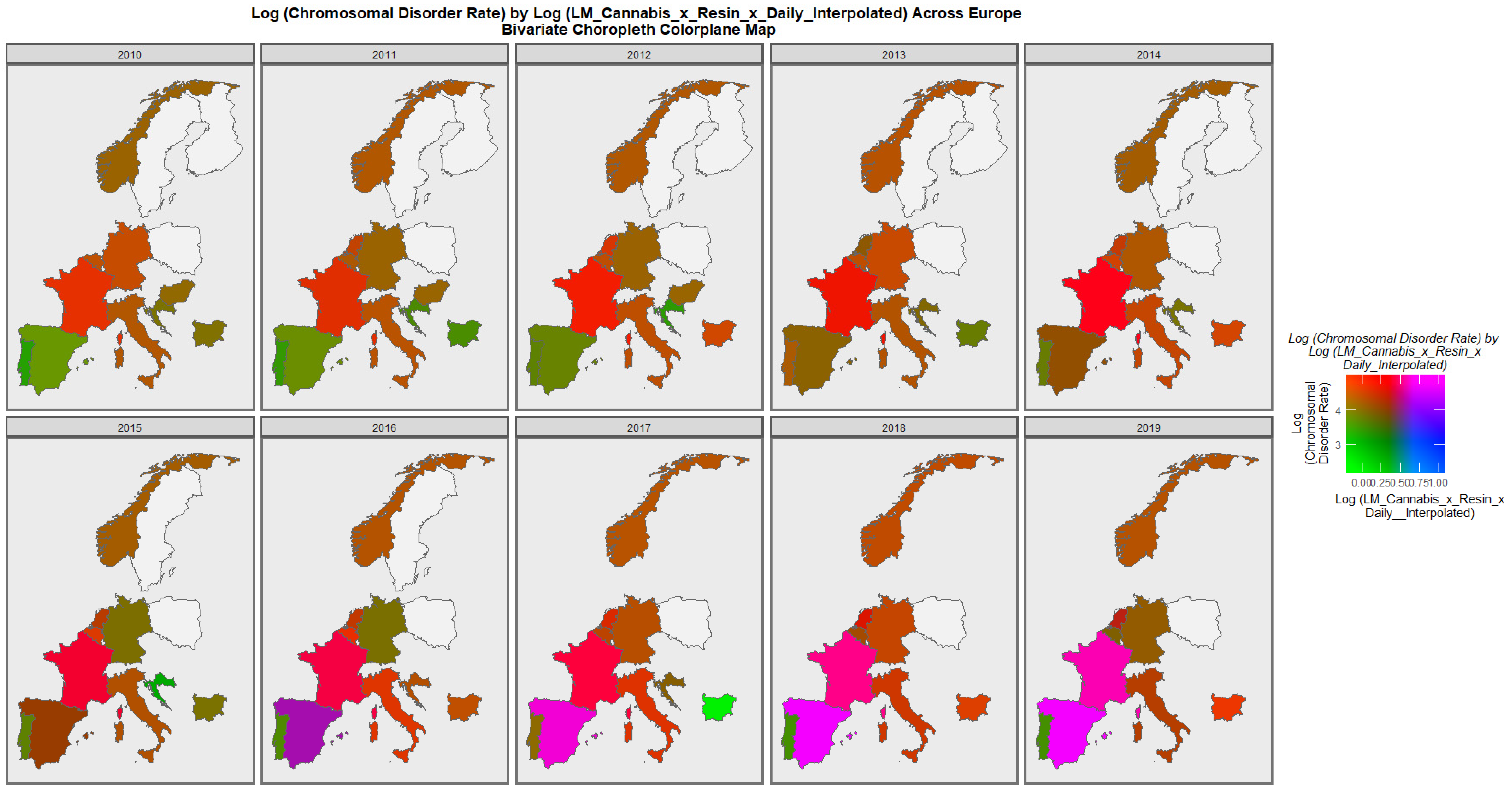

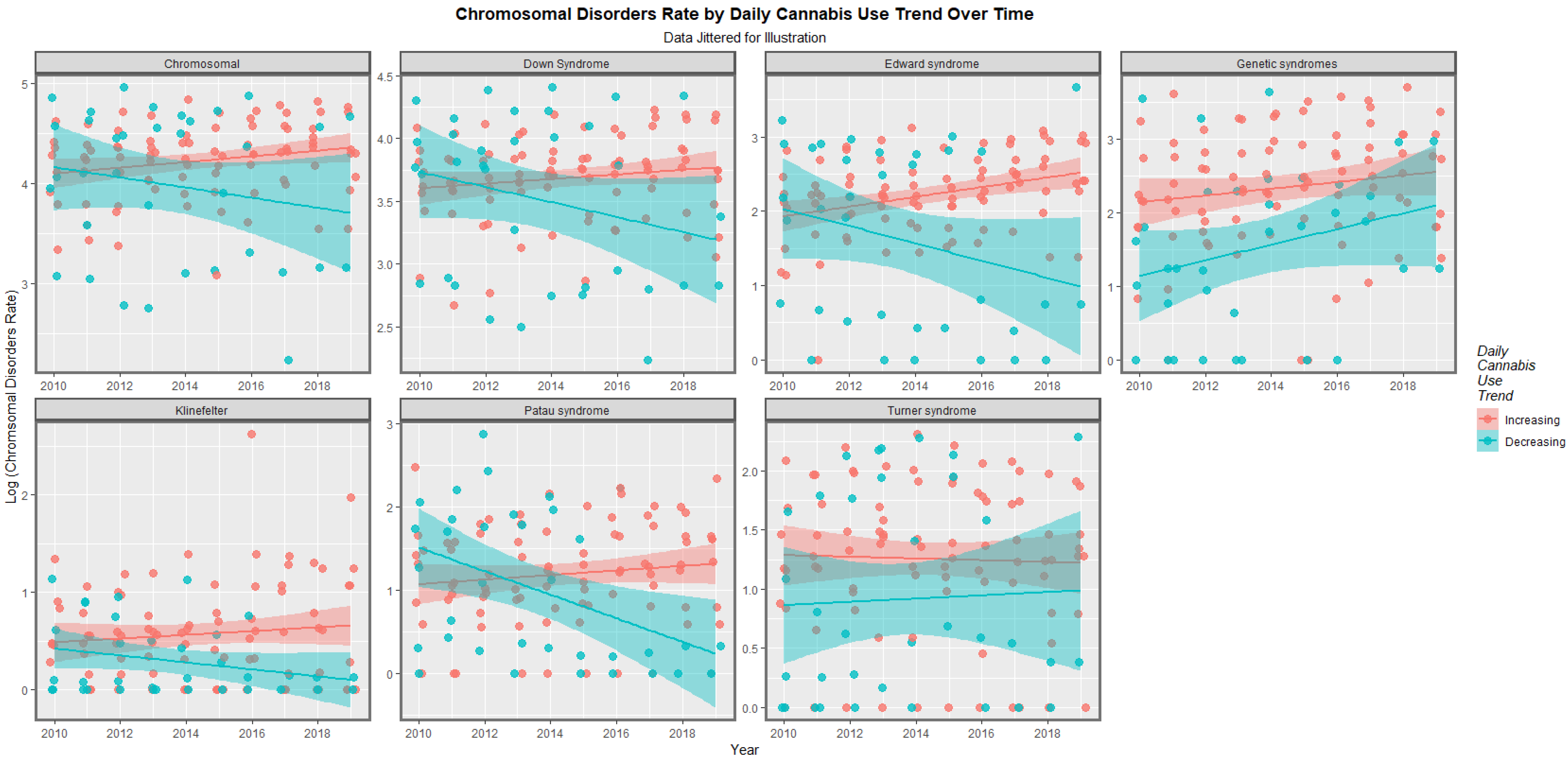
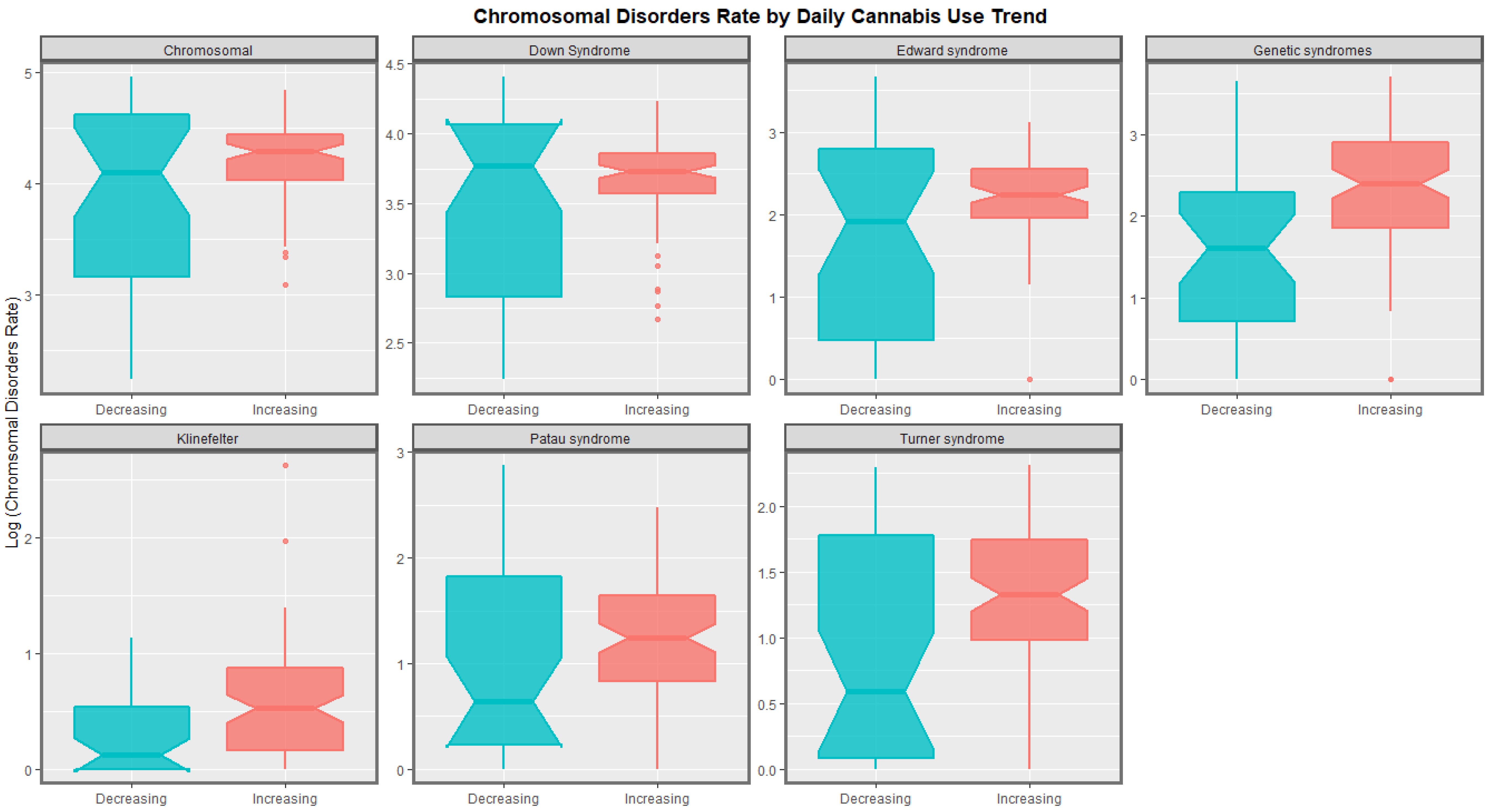

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anomaly | Substance | Mean Anomaly Rate | Estimate | Std. Error | Sigma | t_Statistic | p_Value | E-Value Estimate | E-Value Lower Bound |
|---|---|---|---|---|---|---|---|---|---|
| Genetic syndromes | Daily.Interpol. | 5.9209 | 54.6837 | 9.3918 | 0.8815 | 5.8225 | 5.53 × 10−8 | 6.55 × 1024 | 3.81 × 1016 |
| Edward syndrome | Daily.Interpol. | 4.9688 | 39.6511 | 8.1134 | 0.7616 | 4.8871 | 3.41 × 10−6 | 7.55 × 1020 | 4.39 × 1012 |
| Chromosomal | Daily.Interpol. | 35.8047 | 23.0358 | 5.0643 | 0.4754 | 4.5487 | 1.37 × 10−5 | 2.84 × 1019 | 1.65 × 1011 |
| Down Syndrome | Daily.Interpol. | 20.8129 | 19.0578 | 4.4071 | 0.4137 | 4.3244 | 3.31 × 10−5 | 3.22 × 1018 | 1.87 × 1010 |
| Turner syndrome | Daily.Interpol. | 1.7968 | 27.0000 | 7.2926 | 0.6845 | 3.7024 | 3.32 × 10−4 | 7.75 × 1015 | 4.51 × 107 |
| Klinefelter | Daily.Interpol. | 0.6084 | 18.2674 | 5.0311 | 0.4722 | 3.6309 | 4.26 × 10−4 | 3.87 × 1015 | 2.25 × 107 |
| Patau syndrome | Daily.Interpol. | 1.7793 | 24.1206 | 7.0551 | 0.6622 | 3.4189 | 8.75 × 10−4 | 4.96 × 1014 | 2.89 × 106 |
| Genetic syndromes | LMCannabis_Herb | 5.9209 | 15.6419 | 3.1034 | 0.8908 | 5.0402 | 1.66 × 10−6 | 1.74 × 107 | 3.53 × 104 |
| Genetic syndromes | LM_Cannabis | 5.9209 | 15.5798 | 3.7939 | 0.9182 | 4.1065 | 7.37 × 10−5 | 1.02 × 107 | 6.50 × 103 |
| Genetic syndromes | Herb | 5.9209 | 10.0478 | 2.3492 | 0.9135 | 4.2771 | 3.82 × 10−5 | 4.45 × 104 | 456.72 |
| Chromosomal | LMCannabis_Herb | 35.8047 | 6.2955 | 1.7442 | 0.5007 | 3.6095 | 4.49 × 10−4 | 1.86 × 105 | 377.43 |
| Edward syndrome | LMCannabis_Herb | 4.9688 | 9.7114 | 2.7935 | 0.8019 | 3.4764 | 7.08 × 10−4 | 1.22 × 105 | 247.40 |
| Down Syndrome | LMCannabis_Herb | 20.8129 | 5.2012 | 1.5292 | 0.4390 | 3.4013 | 9.12 × 10−4 | 9.63 × 104 | 194.83 |
| Klinefelter | LMCannabis_Herb | 0.6084 | 5.2172 | 1.6420 | 0.4713 | 3.1774 | 0.0019 | 4.74 × 104 | 95.58 |
| Down Syndrome | LM.Cannabis_x_Herb.THC_x_Daily.Interpol. | 20.8129 | 2.3272 | 0.4751 | 0.4056 | 4.8985 | 3.25 × 10−6 | 369.89 | 45.54 |
| Chromosomal | LM.Cannabis_x_Herb.THC_x_Daily.Interpol. | 35.8047 | 2.6195 | 0.5532 | 0.4723 | 4.7349 | 6.41 × 10−6 | 310.61 | 38.17 |
| Edward syndrome | LM_Cannabis | 4.9688 | 9.1559 | 3.3741 | 0.8166 | 2.7136 | 0.0076 | 5.40 × 104 | 34.01 |
| Edward syndrome | LM.Cannabis_x_Herb.THC_x_Daily.Interpol. | 4.9688 | 4.1359 | 0.9014 | 0.7696 | 4.5883 | 1.16 × 10−5 | 265.61 | 32.57 |
| Klinefelter | LM_Cannabis | 0.6084 | 5.2968 | 1.9692 | 0.4766 | 2.6898 | 0.0082 | 4.94 × 104 | 31.06 |
| Genetic syndromes | LM.Cannabis_x_Herb.THC_x_Daily.Interpol. | 5.9209 | 4.8225 | 1.0864 | 0.9275 | 4.4389 | 2.11 × 10−5 | 226.42 | 27.70 |
| Patau syndrome | LM.Cannabis_x_Herb.THC_x_Daily.Interpol. | 1.7793 | 2.9541 | 0.7660 | 0.6539 | 3.8567 | 1.92 × 10−4 | 121.51 | 14.65 |
| Turner syndrome | LMCannabis_Herb | 1.7968 | 6.2946 | 2.4593 | 0.7060 | 2.5595 | 0.0117 | 6.68 × 103 | 13.03 |
| Klinefelter | LM.Cannabis_x_Herb.THC_x_Daily.Interpol. | 0.6084 | 2.0130 | 0.5530 | 0.4721 | 3.6401 | 4.12 × 10−4 | 96.35 | 11.52 |
| Chromosomal | LM.Cannabis_x_Resin.THC_x_Daily.Interpol. | 35.8047 | 1.0375 | 0.1974 | 0.3694 | 5.2563 | 8.60 × 10−7 | 25.26 | 9.43 |
| Edward syndrome | LM.Cannabis_x_Resin.THC_x_Daily.Interpol. | 4.9688 | 1.7864 | 0.3612 | 0.6758 | 4.9465 | 3.13 × 10−6 | 21.66 | 8.03 |
| Genetic syndromes | LMCannabis_Resin | 5.9209 | 2.6736 | 0.7047 | 0.8438 | 3.7940 | 2.48 × 10−4 | 35.24 | 7.55 |
| Turner syndrome | LM.Cannabis_x_Herb.THC_x_Daily.Interpol. | 1.7968 | 2.6440 | 0.8118 | 0.6930 | 3.2570 | 0.0015 | 63.88 | 7.47 |
| Down Syndrome | LM.Cannabis_x_Resin.THC_x_Daily.Interpol. | 20.8129 | 0.8706 | 0.1835 | 0.3434 | 4.7435 | 7.14 × 10−6 | 19.57 | 7.22 |
| Chromosomal | LMCannabis_Resin | 35.8047 | 1.2280 | 0.3399 | 0.4070 | 3.6129 | 4.66 × 10−4 | 30.64 | 6.50 |
| Edward syndrome | LMCannabis_Resin | 4.9688 | 2.1258 | 0.5980 | 0.7160 | 3.5549 | 5.68 × 10−4 | 29.30 | 6.20 |
| Chromosomal | Resin | 35.8047 | 1.4387 | 0.4591 | 0.4127 | 3.1340 | 0.0022 | 47.23 | 6.05 |
| Chromosomal | Herb | 35.8047 | 3.2385 | 1.3231 | 0.5145 | 2.4478 | 0.0158 | 614.55 | 5.77 |
| Genetic syndromes | LM.Cannabis_x_Resin.THC_x_Daily.Interpol. | 5.9209 | 1.8703 | 0.4563 | 0.8539 | 4.0989 | 8.57 × 10−5 | 14.16 | 5.12 |
| Patau syndrome | LMCannabis_Herb | 1.7793 | 5.4238 | 2.4275 | 0.6968 | 2.2343 | 0.0273 | 2.38 × 103 | 4.27 |
| Patau syndrome | LM.Cannabis_x_Resin.THC_x_Daily.Interpol. | 1.7793 | 1.2368 | 0.3458 | 0.6472 | 3.5761 | 5.44 × 10−4 | 10.86 | 3.82 |
| Genetic syndromes | Cocaine | 5.9209 | 0.7636 | 0.1002 | 0.8051 | 7.6174 | 6.61 × 10−12 | 4.17 | 3.21 |
| Down Syndrome | LMCannabis_Resin | 20.8129 | 0.8596 | 0.3201 | 0.3833 | 2.6853 | 0.0084 | 14.88 | 2.88 |
| Turner syndrome | LM.Cannabis_x_Resin.THC_x_Daily.Interpol. | 1.7968 | 1.1097 | 0.3598 | 0.6732 | 3.0845 | 0.0026 | 8.43 | 2.86 |
| Edward syndrome | Cocaine | 4.9688 | 0.5400 | 0.0924 | 0.7423 | 5.8418 | 4.52 × 10−8 | 3.29 | 2.48 |
| Chromosomal | Cocaine | 35.8047 | 0.3167 | 0.0589 | 0.4732 | 5.3748 | 3.83 × 10−7 | 3.08 | 2.31 |
| Down Syndrome | Cocaine | 20.8129 | 0.2455 | 0.0527 | 0.4229 | 4.6615 | 8.20 × 10−6 | 2.78 | 2.06 |
| Klinefelter | LM.Cannabis_x_Resin.THC_x_Daily.Interpol. | 0.6084 | 0.6802 | 0.2641 | 0.4941 | 2.5759 | 0.0115 | 6.46 | 2.04 |
| Down Syndrome | Herb | 20.8129 | 2.4157 | 1.1613 | 0.4516 | 2.0801 | 0.0396 | 259.68 | 2.01 |
| Turner syndrome | Cocaine | 1.7968 | 0.3768 | 0.0835 | 0.6703 | 4.5142 | 1.49 × 10−5 | 2.72 | 2.01 |
| Klinefelter | Cocaine | 0.6084 | 0.2203 | 0.0577 | 0.4634 | 3.8186 | 2.14 × 10−4 | 2.45 | 1.77 |
| Genetic syndromes | Resin | 5.9209 | 2.1135 | 0.9795 | 0.8805 | 2.1578 | 0.0332 | 17.26 | 1.75 |
| Patau syndrome | Cocaine | 1.7793 | 0.2952 | 0.0844 | 0.6774 | 3.4991 | 6.56 × 10−4 | 2.34 | 1.67 |
| Turner syndrome | Daily.Interpol. | 1.7968 | 3.7174 | 1.8333 | 0.7129 | 2.0277 | 0.0448 | 229.61 | 1.65 |
| Down Syndrome | Resin | 20.8129 | 0.8844 | 0.4322 | 0.3886 | 2.0460 | 0.0433 | 15.35 | 1.42 |
| Edward syndrome | Resin | 4.9688 | 1.6669 | 0.8273 | 0.7437 | 2.0149 | 0.0465 | 14.86 | 1.32 |
| Turner syndrome | LMCannabis_Resin | 1.7968 | 1.1661 | 0.5756 | 0.6892 | 2.0259 | 0.0453 | 8.80 | 1.29 |
| Parameter Values | Model Parameters | ||||
|---|---|---|---|---|---|
| Parameter | Estimate (C.I.) | p-Value | Parameter | Value | Significance |
| Additive | |||||
| Rate ~ Tobacco + Alcohol + LM.Cannabis_x_Herb.THC_x_Daily.Interpol. + LM.Cannabis_x_Resin.THC_x_Daily.Interpol. + Daily.Interpol. + LM.Cannabis_x_Resin.THC + Amphetamines + Cocaine + Income) | |||||
| Alcohol | 0.08 (0.05, 0.12) | 7.28 × 10−7 | rho | 0.5667 | 2.10 × 10−8 |
| LM.Cannabis_x_Resin.THC | 1.47 (0.83, 2.11) | 6.13 × 10−6 | lambda | −0.471 | 1.95 × 10−6 |
| Amphetamines | −0.21 (−0.29, −0.13) | 3.01 × 10−7 | |||
| Cocaine | 0.28 (0.19, 0.37) | 1.27 × 10−9 | |||
| Income | 0 (0, 0) | 1.07 × 10−10 | |||
| Interactive | |||||
| Rate ~ Tobacco * Daily.Interpol. + LM.Cannabis_x_Resin.THC * LM.Cannabis_x_Resin.THC_x_Daily.Interpol. + LM.Cannabis_x_Herb.THC_x_Daily.Interpol. + Alcohol + Amphetamines + Cocaine + Income | |||||
| Tobacco | 0.08 (0.05, 0.11) | 2.11 × 10−7 | rho | 0.2807 | 0.0626 |
| Daily.Interpol. | 81.6 (25.54, 137.66) | 0.004384 | lambda | −0.2844 | 0.0434 |
| LM.Cannabis_x_Resin.THC | 1.61 (0.94, 2.28) | 2.66 × 10−6 | |||
| Amphetamines | −0.28 (−0.37, −0.19) | 1.11 × 10−9 | |||
| Cocaine | 0.28 (0.1, 0.46) | 0.002204 | |||
| Income | 0 (0, 0) | 2.61 × 10−10 | |||
| Tobacco: Daily.Interpol. | −3.7 (−5.88, −1.52) | 0.000847 | |||
| 1 Lag | |||||
| Rate ~ Tobacco * Daily.Interpol. + LM.Cannabis_x_Resin.THC * LM.Cannabis_x_Resin.THC_x_Daily.Interpol. + LM.Cannabis_x_Herb.THC_x_Daily.Interpol. + Alcohol + Amphetamines + Cocaine + Income | |||||
| Tobacco | 0.06 (0.02, 0.09) | 0.003713 | rho | 0.4067 | 0.0137 |
| Daily.Interpol. | 67.7 (3.22, 132.18) | 0.039664 | lambda | −0.3299 | 0.0193 |
| LM.Cannabis_x_Resin.THC | 1.75 (0.94, 2.56) | 2.45 × 10−5 | |||
| Alcohol | 0.05 (0, 0.1) | 0.05342 | |||
| Amphetamines | −0.3 (−0.4, −0.21) | 1.33 × 10−9 | |||
| Cocaine | 0.4 (0.18, 0.61) | 0.000335 | |||
| Income | 0 (0, 0) | 2.12 × 10−6 | |||
| Tobacco: Daily.Interpol. | −3.27 (−5.72, −0.82) | 0.009025 | |||
| 2 Lags | |||||
| Rate ~ Tobacco * LM.Cannabis_x_Herb.THC + LM.Cannabis_x_Resin.THC_x_Daily.Interpol. + LM.Cannabis_x_Herb.THC_x_Daily.Interpol. + Alcohol + Daily.Interpol. + Amphetamines + Cocaine + Income | |||||
| Tobacco | 0.12 (0.09, 0.16) | 1.26 × 10−10 | rho | 0.5497 | 7.96 × 10−6 |
| LM.Cannabis_x_Herb.THC | 40.1 (20.52, 59.68) | 5.89 × 10−5 | lambda | −0.3245 | 0.0144 |
| LM.Cannabis_x_Resin.THC_x_Daily.Interpol. | 3.1 (1.78, 4.42) | 4.28 × 10−6 | |||
| LM.Cannabis_x_Herb.THC_x_Daily.Interpol. | −4 (−7.55, −0.45) | 0.02668 | |||
| Amphetamines | −0.33 (−0.45, −0.21) | 9.89 × 10−8 | |||
| Cocaine | 0.26 (0.08, 0.44) | 0.00442 | |||
| Income | 0 (0, 0) | 3.58 × 10−13 | |||
| Tobacco: LM.Cannabis_x_Herb.THC | −1.79 (−2.54, −1.04) | 3.25 × 10−6 | |||
| Parameter Values | Model Parameters | ||||
|---|---|---|---|---|---|
| Parameter | Estimate (C.I.) | p-Value | Parameter | Value | Significance |
| Additive | |||||
| Rate ~ Tobacco + Alcohol + LM.Cannabis_x_Resin.THC_x_Daily.Interpol. + LM.Cannabis_x_Herb.THC + Daily.Interpol. + LM.Cannabis_x_Herb.THC_x_Daily.Interpol. + Amphetamines + Cocaine + Income | |||||
| Alcohol | 0.08 (0.01, 0.15) | 0.029861 | rho | 0.45269 | 1.85 × 10−7 |
| Daily.Interpol. | 45.7 (32.69, 58.71) | 5.79 × 10−12 | |||
| Income | 0 (0, 0) | 0.000306 | |||
| Interactive | |||||
| Rate ~ Tobacco * Daily.Interpol. + LM.Cannabis_x_Herb.THC + LM.Cannabis_x_Herb.THC_x_Daily.Interpol. + Alcohol + Amphetamines + Cocaine + Income | |||||
| Tobacco | 0.12 (0.06, 0.17) | 3.24 × 10−5 | Least Squares | ||
| Daily.Interpol. | 299 (185.32, 412.68) | 2.58 × 10−7 | S.D. | 0.6888 | |
| Income | 0 (0, 0) | 5.93 × 10−7 | |||
| Tobacco: Daily.Interpol. | −10.6 (−15.23, −5.97) | 7.79 × 10−6 | |||
| 1 Lag | |||||
| Rate ~ Tobacco * LM.Cannabis_x_Herb.THC +Daily.Interpol. + LM.Cannabis_x_Resin.THC_x_Daily.Interpol. + LM.Cannabis_x_Herb.THC_x_Daily.Interpol. + Alcohol + Amphetamines + Cocaine + Income | |||||
| LM.Cannabis_x_Herb.THC | 38.26 (16.43, 60.1) | 0.0006 | rho | 0.4767 | 1.00 × 10−7 |
| LM.Cannabis_x_Resin.THC_x_Daily.Interpol. | 3.53 (1.66, 5.4) | 0.0002 | |||
| LM.Cannabis_x_Herb.THC_x_Daily.Interpol. | −7.65 (−13.21, −2.1) | 0.0069 | |||
| Alcohol | 0.16 (0.09, 0.24) | 3.27 × 10−5 | |||
| Cocaine | 0.79 (0.54, 1.04) | 4.18 × 10−10 | |||
| LM.Cannabis_x_Herb.THC: LM.Cannabis_x_Resin.THC_x_Daily.Interpol. | −1.34 (−2.09, −0.58) | 0.000494 | |||
| 2 Lags | |||||
| Rate ~ Tobacco * Daily.Interpol. + LM.Cannabis_x_Herb.THC + LM.Cannabis_x_Herb.THC_x_Daily.Interpol. + Alcohol + Amphetamines + Cocaine + Income | |||||
| Tobacco | 0.1 (0.02, 0.18) | 0.0111 | psi | 0.3373 | 0.00123 |
| Daily.Interpol. | 278 (111.01, 444.99) | 0.0011 | rho | 0.3269 | 0.00149 |
| Income | 0 (0, 0) | 0.0086 | |||
| Resin | −9.72 (−16.54, −2.9) | 0.0052 | |||
| Parameter Values | Model Parameters | ||||
|---|---|---|---|---|---|
| Parameter | Estimate (C.I.) | p-Value | Parameter | Value | Significance |
| Additive | |||||
| Rate ~ Tobacco + Alcohol + LM.Cannabis_x_Resin.THC_x_Daily.Interpol. + LM.Cannabis_x_Herb.THC_x_Daily.Interpol. + LM.Cannabis_x_Herb.THC + LM.Cannabis_x_Herb.THC + Amphetamines + Cocaine + Income | |||||
| Tobacco | 0.05 (0.03, 0.07) | 1.80 × 10−8 | rho | −0.3628 | 0.0628 |
| Alcohol | −0.07 (−0.1, −0.04) | 2.28 × 10−5 | lambda | 0.2088 | 0.261 |
| LM.Cannabis_x_Herb.THC | 6.89 (4.89, 8.89) | 1.26 × 10−11 | |||
| Amphetamines | −0.09 (−0.17, −0.02) | 0.0138 | |||
| Income | 0 (0, 0) | 1.02 × 10−12 | |||
| Interactive | |||||
| Rate ~ Tobacco * LM.Cannabis_x_Herb.THC + LM.Cannabis_x_Herb.THC_x_Daily.Interpol. * LM.Cannabis_x_Resin.THC_x_Daily.Interpol. + LM.Cannabis_x_Resin.THC + Alcohol + Herb + Amphetamines + Cocaine + Income | |||||
| Tobacco | 0.03 (0.02, 0.05) | 0.0001 | rho | 0.3593 | 0.0319 |
| LM.Cannabis_x_Resin.THC | 1.18 (0.57, 1.79) | 0.0002 | lambda | −0.3577 | 0.0334 |
| Amphetamines | −0.2 (−0.29, −0.12) | 2.24 × 10−6 | |||
| Cocaine | 0.2 (0.11, 0.29) | 2.29 × 10−5 | |||
| Income | 0 (0, 0) | 1.43 × 10−6 | |||
| 2 Lags | |||||
| Rate ~ Tobacco * LM.Cannabis_x_Herb.THC_x_Daily.Interpol. + LM.Cannabis_x_Herb.THC * LM.Cannabis_x_Resin.THC_x_Daily.Interpol. + Alcohol + Amphetamines + Cocaine + Income | |||||
| Tobacco | 0.05 (0.03, 0.08) | 1.86 × 10−5 | rho | −0.1896 | 0.435 |
| LM.Cannabis_x_Resin.THC_x_Daily.Interpol. | 1.24 (0.7, 1.78) | 6.56 × 10−6 | lambda | 0.1934 | 0.402 |
| Alcohol | −0.06 (−0.1, −0.02) | 0.0052 | |||
| Amphetamines | −0.17 (−0.27, −0.07) | 0.0008 | |||
| Income | 0 (0, 0) | 1.64 × 10−7 | |||
| Parameter Values | Model Parameters | ||||
|---|---|---|---|---|---|
| Parameter | Estimate (C.I.) | p-Value | Parameter | Value | Significance |
| Additive | |||||
| Rate ~ Tobacco + Alcohol + LM.Cannabis_x_Resin.THC + Daily.Interpol. + LM.Cannabis_x_Herb.THC_x_Daily.Interpol. + Herb + Amphetamines + Cocaine + Income | |||||
| Tobacco | 0.05 (0.02, 0.08) | 0.0012 | rho | −0.5792 | 1.18 × 10−5 |
| Alcohol | −0.13 (−0.18, −0.07) | 2.96 × 10−6 | lambda | 0.5089 | 4.74 × 10−5 |
| LM.Cannabis_x_Resin.THC | 1.02 (0.29, 1.75) | 0.0058 | |||
| Daily.Interpol. | 33.8 (20.82, 46.78) | 3.35 × 10−7 | |||
| Income | 0 (0, 0) | 1.24 × 10−10 | |||
| Interactive | |||||
| Rate ~ Tobacco * LM.Cannabis_x_Resin.THC_x_Daily.Interpol. * LM.Cannabis_x_Resin.THC + Daily.Interpol. + LM.Cannabis_x_Herb.THC_x_Daily.Interpol. + Alcohol + Amphetamines + Cocaine + Income | |||||
| Tobacco | 0.04 (0.01, 0.07) | 0.0112 | rho | −0.5461 | 3.84 × 10−5 |
| Alcohol | −0.07 (−0.12, −0.02) | 0.0093 | lambda | 0.4785 | 8.76 × 10−5 |
| Amphetamines | −0.17 (−0.29, −0.05) | 0.0067 | |||
| Cocaine | 0.37 (0.18, 0.55) | 0.0001 | |||
| Income | 0 (0, 0) | 4.26 × 10−6 | |||
| Tobacco: LM.Cannabis_x_Resin.THC_x_Daily.Interpol. | 0.09 (0.03, 0.14) | 0.0020 | |||
| Tobacco: LM.Cannabis_x_Resin.THC_x_Daily.Interpol.: LM.Cannabis_x_Resin.THC | −0.12 (−0.23, 0) | 0.049823 | |||
| 2 Lags | |||||
| Rate ~ Tobacco + Daily.Interpol. + LM.Cannabis_x_Resin.THC + LM.Cannabis_x_Resin.THC_x_Daily.Interpol. + LM.Cannabis_x_Herb.THC_x_Daily.Interpol. + Alcohol + Amphetamines + Cocaine + Income | |||||
| Tobacco | 0.04 (0.01, 0.07) | 0.0185 | rho | −0.6449 | 6.04 × 10−8 |
| Daily.Interpol. | 67.2 (42.31, 92.09) | 1.12 × 10−7 | lambda | 0.4422 | 0.0014 |
| LM.Cannabis_x_Resin.THC | 3.86 (2.27, 5.45) | 1.90 × 10−6 | |||
| LM.Cannabis_x_Herb.THC_x_Daily.Interpol. | −6.68 (−10.85, −2.51) | 0.0017 | |||
| Alcohol | −0.11 (−0.17, −0.05) | 0.0004 | |||
| Amphetamines | −0.17 (−0.32, −0.02) | 0.0267 | |||
| Income | 0 (0, 0) | 4.29 × 10−7 | |||
| Parameter Values | Model Parameters | ||||
|---|---|---|---|---|---|
| Parameter | Estimate (C.I.) | p-Value | Parameter | Value | Significance |
| Additive | |||||
| Rate ~ Tobacco + Alcohol + Daily.Interpol. + LM.Cannabis_x_Resin.THC_x_Daily.Interpol. + LM.Cannabis_x_Herb.THC + LM.Cannabis_x_Herb.THC_x_Daily.Interpol. + Herb + Amphetamines + Cocaine + Income | |||||
| Alcohol | 0.07 (0.02, 0.13) | 0.00819 | rho | 0.5696 | 4.39 × 10−8 |
| LM.Cannabis_x_Herb.THC | 14.6 (9.64, 19.56) | 7.52 × 10−9 | lambda | −0.6085 | 1.48 × 10−9 |
| Herb | −5.15 (−9.44, −0.86) | 0.0189 | |||
| Amphetamines | −0.14 (−0.26, −0.02) | 0.0174 | |||
| Income | 0 (0, 0) | 2.94 × 10−12 | |||
| Interactive | |||||
| Rate ~ Tobacco * Daily.Interpol. + LM.Cannabis_x_Herb.THC * LM.Cannabis_x_Resin.THC_x_Daily.Interpol. + LM.Cannabis_x_Herb.THC_x_Daily.Interpol. + Alcohol + Amphetamines + Cocaine + Income | |||||
| Tobacco | 0.04 (0.01, 0.07) | 0.0226 | rho | 0.5546 | 3.44 × 10−6 |
| LM.Cannabis_x_Herb.THC | 9.82 (5.04, 14.6) | 5.79 × 10−5 | lambda | −0.589 | 4.27 × 10−7 |
| Amphetamines | −0.16 (−0.28, −0.03) | 0.0169 | |||
| Income | 0 (0, 0) | 4.81 × 10−9 | |||
| 2 Lags | |||||
| Rate ~ Tobacco * LM.Cannabis_x_Herb.THC + Daily.Interpol. * LM.Cannabis_x_Resin.THC_x_Daily.Interpol. + Alcohol + Daily.Interpol. + Amphetamines + Cocaine + Income | |||||
| Tobacco | 0.05 (0.01, 0.09) | 0.0105 | rho | 0.1822 | 0.459 |
| LM.Cannabis_x_Resin.THC_x_Daily.Interpol. | 1.74 (0.8, 2.68) | 0.0003 | lambda | −0.1076 | 0.644 |
| Amphetamines | −0.23 (−0.4, −0.07) | 0.0060 | |||
| Income | 0 (0, 0) | 1.64 × 10−7 | |||
| No. | E-Value Estimate | Lower Bound E-Value |
|---|---|---|
| 1 | Infinity | Infinity |
| 2 | Infinity | Infinity |
| 3 | Infinity | Infinity |
| 4 | Infinity | Infinity |
| 5 | 2.17 × 10129 | 2.02 × 1071 |
| 6 | 2.17 × 10129 | 2.02 × 1071 |
| 7 | 1.15 × 1099 | 2.21 × 1054 |
| 8 | 1.15 × 1099 | 2.21 × 1054 |
| 9 | 2.20 × 1040 | 2.16 × 1034 |
| 10 | 2.20 × 1040 | 2.16 × 1034 |
| 11 | 1.81 × 1031 | 2.97 × 1025 |
| 12 | 1.81 × 1031 | 2.97 × 1025 |
| 13 | 6.49 × 10115 | 2.92 × 1025 |
| 14 | 6.49 × 10115 | 2.92 × 1025 |
| 15 | 5.48 × 1032 | 1.91 × 1025 |
| 16 | 5.48 × 1032 | 1.91 × 1025 |
| 17 | 2.82 × 1045 | 2.23 × 1020 |
| 18 | 2.82 × 1045 | 2.23 × 1020 |
| 19 | 9.90 × 1018 | 4.11 × 1015 |
| 20 | 9.90 × 1018 | 4.11 × 1015 |
| 21 | 3.98 × 1026 | 2.11 × 1014 |
| 22 | 3.98 × 1026 | 2.11 × 1014 |
| 23 | 2.54 × 1022 | 1.13 × 1014 |
| 24 | 2.53 × 1022 | 1.13 × 1014 |
| 25 | 2.54 × 1022 | 1.13 × 1014 |
| 26 | 2.53 × 1022 | 1.13 × 1014 |
| 27 | 2.46 × 1024 | 2.67 × 1013 |
| 28 | 2.46 × 1024 | 2.67 × 1013 |
| 29 | 2.24 × 1018 | 2.18 × 1013 |
| 30 | 2.24 × 1018 | 2.18 × 1013 |
| 31 | 3.59 × 1010 | 5.29 × 108 |
| 32 | 3.59 × 1010 | 5.29 × 108 |
| 33 | 3.36 × 107 | 2.59 × 105 |
| 34 | 3.36 × 107 | 2.59 × 105 |
| 35 | 2.26 × 106 | 1.07 × 105 |
| 36 | 2.26 × 106 | 1.07 × 105 |
| 37 | 1.19 × 109 | 1.06 × 105 |
| 38 | 1.19 × 109 | 1.06 × 105 |
| 39 | 1.18 × 105 | 1.49 × 103 |
| 40 | 1.18 × 105 | 1.49 × 103 |
| 41 | 1.84 × 103 | 510.05 |
| 42 | 1.84 × 103 | 510.05 |
| 43 | 3.38 × 108 | 194.54 |
| 44 | 3.38 × 108 | 194.54 |
| 45 | 3.38 × 108 | 194.54 |
| 46 | 3.38 × 108 | 194.54 |
| 47 | 927.29 | 176.39 |
| 48 | 927.29 | 176.39 |
| 49 | 4.13 × 105 | 61.12 |
| 50 | 4.13 × 105 | 61.12 |
| 51 | 101.56 | 59.31 |
| 52 | 101.56 | 59.31 |
| 53 | 2.76 × 104 | 58.55 |
| 54 | 2.76 × 104 | 58.55 |
| 55 | 86.25 | 29.18 |
| 56 | 86.25 | 29.18 |
| 57 | 2.78 × 108 | 25.51 |
| 58 | 2.78 × 108 | 25.51 |
| 59 | 104.48 | 21.84 |
| 60 | 104.48 | 21.84 |
| 61 | 2.24 × 105 | 17.73 |
| 62 | 2.24 × 105 | 17.73 |
| 63 | 23.77 | 13.76 |
| 64 | 23.77 | 13.76 |
| 65 | 27.40 | 8.77 |
| 66 | 27.40 | 8.77 |
| 67 | 18.49 | 8.15 |
| 68 | 18.49 | 8.15 |
| 69 | 74.39 | 4.94 |
| 70 | 74.39 | 4.94 |
| 71 | 14.37 | 4.18 |
| 72 | 14.37 | 4.18 |
| 73 | 8.08 | 3.58 |
| 74 | 8.08 | 3.58 |
| 75 | 4.11 × 1012 | 3.51 |
| 76 | 4.11 × 1012 | 3.51 |
| 77 | 7.89 | 2.56 |
| 78 | 7.89 | 2.56 |
| 79 | 18.43 | 2.31 |
| 80 | 18.43 | 2.31 |
| 81 | 3.40 | 1.91 |
| 82 | 3.40 | 1.91 |
| 83 | 2.18 | 1.90 |
| 84 | 2.18 | 1.90 |
| 85 | 3.14 | 1.79 |
| 86 | 3.14 | 1.79 |
| Anomaly | Number | Mean Minimum E-Value | Median Minimum E-Value | Min Minimum E-Value | Max Minimum E-Value | Mean E-Value Estimate | Median E-Value Estimate | Min E-Value Estimate | Max E-Value Estimate |
|---|---|---|---|---|---|---|---|---|---|
| Trisomy 13 | 10 | 4.04 × 1070 | 1.91 × 1025 | 4.18 | 2.02 × 1071 | 4.34 × 10128 | 5.48 × 1032 | 14.37 | 2.17 × 10129 |
| Trisomy 21 | 12 | 3.72 × 1019 | 2.65 × 108 | 1.9 | 2.23 × 1020 | 4.70 × 1044 | 1.80 × 1010 | 2.18 | 2.82 × 1045 |
| Klinefelters | 10 | 8.446 × 1014 | 2.59 × 105 | 3.51 | 4.11 × 1015 | 5.06 × 1021 | 4.11 × 1012 | 3.36 × 107 | 2.53 × 1022 |
| Chromosomes | 20 | 1.10 × 10306 | 5.38 × 104 | 2.31 | 1.10 × 10307 | 1.10 × 10306 | 1,130,920 | 8.08 | 1.10 × 10307 |
| Turners | 6 | 7.37 × 1053 | 1.49 × 103 | 1.79 | 2.21 × 1054 | 3.83 × 1098 | 118,000 | 3.14 | 1.15 × 1099 |
| Genetic | 18 | 1.22 × 10306 | 61.12 | 1.91 | 1.10 × 10307 | 1.22 × 10306 | 413,000 | 3.4 | 1.10 × 10307 |
| Trisomy 18 | 10 | 21,254.994 | 59.31 | 13.76 | 106,000 | 2.94 × 108 | 927.29 | 23.77 | 1.19 × 109 |
| Nearest Gene Name | Chromosome Number | Nearest Gene Number | Dependency Status | Functional Annotation | Page | Distance from Nearest Gene | Relative Position | p-Value | Bonferroni Adjusted p-Value |
|---|---|---|---|---|---|---|---|---|---|
| Centrosomal Organizers | |||||||||
| NUMA1 | 11 | ENSG00000137497 | Withdrawal | Nuclear Mitotic Apparatuis Protein 1 | 212 | 0 | Exon | 1.13 × 10−05 | 0.024154 |
| CEP152 | 15 | ENSG00000103995 | Withdrawal | Centrosomal protein 152 | 195 | 0 | Intron | 7.64 × 10−6 | 0.020250 |
| CEP55 | 10 | ENSG00000138180 | Dependence | Centrosomal protein 55 | 27 | 13,562 | Downstream | 6.71 × 10−7 | 0.005734 |
| Motor Proteins | |||||||||
| Dynein–Dynactin | |||||||||
| DYNC1H1 | 14 | ENSG00000197102 | Withdrawal | Dynein cytoplasmic 1 heavy chain 1 | 135 | 0 | Exon | 2.69 × 10−7 | 0.003989 |
| DYNC1H1 | 14 | ENSG00000197102 | Withdrawal | Dynein cytoplasmic 1 heavy chain 1 | 150 | 0 | Intron | 1.33 × 10−6 | 0.008933 |
| DYNC1H1 | 14 | ENSG00000197102 | Withdrawal | Dynein cytoplasmic 1 heavy chain 1 | 186 | 0 | Intron | 6.04 × 10−6 | 0.018039 |
| DCTN4 | 5 | ENSG00000132912 | Dependence | Dynactin subunit 4 | 37 | 0 | Intron | 1.38 × 10−6 | 0.008195 |
| DCTN4 | 5 | ENSG00000132912 | Withdrawal | Dynactin subunit 4 | 131 | 327 | Downstream | 1.25 × 10−7 | 0.002634 |
| DCTN4 | 5 | ENSG00000132912 | Withdrawal | Dynactin subunit 4 | 151 | 0 | Intron | 1.40 × 10−6 | 0.009142 |
| DCTN4 | 5 | ENSG00000132912 | Withdrawal | Dynactin subunit 4 | 232 | 0 | Intron | 1.63 × 10−5 | 0.028618 |
| Kinesin | |||||||||
| KIFAP3 | 1 | ENSG00000075945 | Dependence | Kinesin Associated protein 3 | 21 | 0 | Intron | 3.63 × 10−7 | 0.004266 |
| KIF26B | 1 | ENSG00000162849 | Dependence | Kinesin family member 26B | 41 | 0 | Intron | 1.69 × 10−6 | 0.009091 |
| KIF2A | 5 | ENSG00000068796 | Dependence | Kinesin family member 2A | 46 | 0 | Intron | 2.20 × 10−6 | 0.010274 |
| KIF26B | 1 | ENSG00000162849 | Dependence | Kinesin family member 26B | 47 | 0 | Intron | 2.26 × 10−6 | 0.010440 |
| KIF27 | 1 | ENSG00000165115 | Dependence | Kinesin family member 27 | 56 | 0 | Exon | 3.24 × 10−6 | 0.012349 |
| KIFC3 | 16 | ENSG00000140859 | Withdrawal | Kinesin family member C3 | 173 | 0 | 3 Untranslated region | 3.87 × 10−6 | 0.014555 |
| KIF14 | 1 | ENSG00000118193 | Withdrawal | Kinesin family member 14 | 200 | 33,155 | Downstream | 8.63 × 10−6 | 0.021429 |
| KIF13B | 1 | ENSG00000197892 | Dependence | Kinesin family member 13B | 60 | 0 | Intron | 3.76 × 10−6 | 0.013269 |
| KIF13B | 6 | ENSG00000197892 | Dependence | Kinesin family member 13B | 77 | 0 | Intron | 6.16 × 10−6 | 0.016839 |
| KIF3C | 2 | ENSG00000137497 | Dependence | Kinesin family member 3C | 79 | 0 | Intron | 6.44 × 10−6 | 0.017205 |
| KIF26B | 1 | ENSG00000162849 | Dependence | Kinesin family member 26B | 83 | 16,530 | Downstream | 7.21 × 10−6 | 0.018129 |
| KIFC2 | 8 | ENSG00000167702 | Withdrawal | Kinesin family member C2 | 221 | 0 | Exon | 1.35 × 10−5 | 0.026224 |
| Nearest Gene Name | Page | Functional Annotation | Number Genes Identified | p-Value |
|---|---|---|---|---|
| KIF13A | 239 | Head and neck cancer | 356 | 7.73 × 10−20 |
| KIF26A | 239 | Head and neck cancer | 356 | 7.73 × 10−20 |
| KIF13A | 239 | Head and neck cancer | 342 | 7.74 × 10−20 |
| KIF26A | 239 | Head and neck cancer | 342 | 7.74 × 10−20 |
| KIF13A | 240 | Head and neck cancer | 333 | 3.34 × 10−19 |
| KIF26A | 240 | Head and neck cancer | 333 | 3.34 × 10−19 |
| KIF13A | 240 | Abdominal carcinoma | 362 | 3.64 × 10−19 |
| KIF26A | 240 | Abdominal carcinoma | 362 | 3.64 × 10−19 |
| KIF13A | 241 | Solid cancer | 381 | 6.93 × 10−18 |
| KIF26A | 241 | Solid cancer | 381 | 6.93 × 10−18 |
| KIF13A | 241 | Thyroid cancer | 318 | 1.21 × 10−17 |
| KIF26A | 241 | Thyroid cancer | 318 | 1.21 × 10−17 |
| KIF13A | 242 | Thyroid cancer | 319 | 1.26 × 10−17 |
| KIF26A | 242 | Thyroid cancer | 319 | 1.26 × 10−17 |
| KIF13A | 242 | Endocrine tumour | 321 | 1.44 × 10−17 |
| KIF26A | 242 | Endocrine tumour | 321 | 1.44 × 10−17 |
| KIF13A | 243 | Adenocarcinoma | 351 | 1.91 × 10−17 |
| KIF26A | 243 | Adenocarcinoma | 351 | 1.91 × 10−17 |
| KIF13A | 243 | Secretory Structure carcinoma | 339 | 1.92 × 10−17 |
| KIF26A | 243 | Secretory Structure carcinoma | 339 | 1.92 × 10−17 |
| KIF13A | 244 | Abdominal carcinoma | 346 | 3.72 × 10−17 |
| KIF26A | 244 | Abdominal carcinoma | 346 | 3.72 × 10−17 |
| KIF13A | 244 | Malignant cancer | 313 | 3.76 × 10−17 |
| KIF26A | 244 | Malignant cancer | 313 | 3.76 × 10−17 |
| KIF13A | 245 | Carcinoma | 376 | 6.96 × 10−17 |
| KIF26A | 245 | Carcinoma | 376 | 6.96 × 10−17 |
| KIF13A | 245 | Non-melanoma solid carcinoma | 379 | 7.21 × 10−17 |
| KIF26A | 245 | Non-melanoma solid carcinoma | 379 | 7.21 × 10−17 |
| Nearest Gene Name | Chromosome | Nearest Gene | Distance to Nearest Gene | Number of Annotations | Relative Location | p-Value | p-Adjust |
|---|---|---|---|---|---|---|---|
| CENPIP1 | 13 | ENSG00000224778 | 1100 | 1 | Upstream | 2.38 × 10−9 | 0.000279 |
| CENPF | 1 | ENSG00000117724 | 72,569 | 3 | Downstream | 2.98 × 10−8 | 0.001109 |
| CENPVL3 | X | ENSG00000224109 | 2146 | 1 | Downstream | 2.80 × 10−6 | 0.001153 |
| CENPK | 5 | ENSG00000123219 | 0 | 1 | Intron | 8.01 × 10−6 | 0.019098 |
| CENPP | 9 | ENSG00000188312 | 0 | 2 | Intron | 8.26 × 10−6 | 0.019330 |
| CENPJ | 13 | ENSG00000151849 | 0 | 1 | Exon | 4.66 × 10−7 | 0.005279 |
| CENPUP1 | 11 | ENSG00000255075 | 8401 | 1 | Upstream | 2.81 × 10−6 | 0.012567 |
| INCENP | 11 | ENSG00000149503 | 0 | 1 | Intron | 3.07 × 10−6 | 0.013077 |
| CENPO | 2 | ENSG00000138092 | 0 | 1 | Exon | 6.25 × 10−6 | 0.018393 |
| CENPI | X | ENSG00000102384 | 0 | 2 | Intron | 7.54 × 10−6 | 0.020123 |
| CENPL | 1 | ENSG00000120334 | 0 | 1 | Intron | 8.22 × 10−6 | 0.020943 |
| CENPX | 17 | ENSG00000169689 | 0 | 1 | Exon | 9.35 × 10−6 | 0.022176 |
| CENPC | 4 | ENSG00000145241 | 0 | 1 | Intron | 9.60 × 10−6 | 0.002248 |
| CENPV | 17 | ENSG00000166582 | 13,237 | 2 | Upstream | 1.63 × 10−5 | 0.002861 |
| CENPN | 16 | ENSG00000166451 | 86 | 7.73 × 10−20 |
| Nearest Gene Name | Chromosome Number | Nearest Gene Number | Dependency Status | Functional Annotation | Page | Number Genes Identified | p-Value |
|---|---|---|---|---|---|---|---|
| CENPN | 16 | ENSG00000166451 | Dependence | Head and Neck Cancer | 239 | 356 | 7.73 × 10−20 |
| CENPN | 16 | ENSG00000166451 | Dependence | Head and Neck Cancer | 239 | 342 | 7.74 × 10−20 |
| CENPN | 16 | ENSG00000166451 | Dependence | Head and Neck Cancer | 240 | 333 | 3.34 × 10−19 |
| CENPN | 16 | ENSG00000166451 | Dependence | Abdominal carcinoma | 240 | 362 | 3.64 × 10−19 |
| CENPN | 16 | ENSG00000166451 | Dependence | Cancer | 241 | 381 | 6.93 × 10−18 |
| CENPN | 16 | ENSG00000166451 | Dependence | Thyroid carcinoma | 241 | 318 | 1.21 × 10−17 |
| CENPN | 16 | ENSG00000166451 | Dependence | Secretory structure cancer | 243 | 339 | 1.92 × 10−17 |
| CENPN | 16 | ENSG00000166451 | Dependence | Malignant Cancer | 244 | 313 | 5.67 × 10−17 |
| CENPN | 16 | ENSG00000166451 | Dependence | Carcinoma | 245 | 376 | 6.96 × 10−17 |
| CENPN | 16 | ENSG00000166451 | Dependence | Non-melanoma solid cancer | 245 | 379 | 7.21 × 10−17 |
| CENPN | 16 | ENSG00000166451 | Dependence | Frequency of Tumour | 246 | 315 | 7.73 × 10−17 |
| CENPN | 16 | ENSG00000166451 | Dependence | Abdominal cancer | 246 | 364 | 8.91 × 10−17 |
| CENPN | 16 | ENSG00000166451 | Dependence | Endocrine gland cancer | 247 | 327 | 1.38 × 10−16 |
| CENPN | 16 | ENSG00000166451 | Dependence | Cancer development | 247 | 310 | 1.72 × 10−16 |
| CENPN | 16 | ENSG00000166451 | Dependence | Epithelial Tumourigenesis | 248 | 312 | 1.74 × 10−16 |
| CENPN | 16 | ENSG00000166451 | Dependence | Endocrine cancer | 249 | 323 | 3.74 × 10−16 |
| CENPN | 16 | ENSG00000166451 | Dependence | Gastrointestinal cancer | 249 | 325 | 4.60 × 10−16 |
| CENPN | 16 | ENSG00000166451 | Dependence | Abdominal cancer | 250 | 365 | 5.17 × 10−16 |
| CENPN | 16 | ENSG00000166451 | Dependence | Digestive system cancer | 250 | 349 | 7.78 × 10−16 |
| CENPN | 16 | ENSG00000166451 | Dependence | Digestive system cancer | 251 | 382 | 8.26 × 10−16 |
| Nearest Gene Name | Page | Chromosome | Status | Nearest Gene | Distance to Nearest Gene | Relative Location | p-Value | p-Adjust |
|---|---|---|---|---|---|---|---|---|
| RAD51–Homologous Recombination | ||||||||
| RAD51B | 28 | 14 | Dependence | ENSG00000182185 | 0 | Intron | 7.33 × 10−7 | 0.006027 |
| RAD51B | 94 | 14 | Dependence | ENSG00000182185 | 0 | Intron | 9.09 × 10−6 | 0.020192 |
| RAD51B | 131 | 14 | Withdrawal | ENSG00000182185 | 0 | Intron | 1.33 × 10−7 | 0.002722 |
| RAD51B | 135 | 14 | Withdrawal | ENSG00000182185 | 0 | Intron | 2.98 × 10−7 | 0.004182 |
| RAD51B | 154 | 14 | Withdrawal | ENSG00000182185 | 0 | Intron | 1.68 × 10−6 | 0.009923 |
| RAD51B | 158 | 14 | Withdrawal | ENSG00000182185 | 0 | Intron | 2.13 × 10−6 | 0.000899 |
| RAD51B | 174 | 14 | Withdrawal | ENSG00000182185 | 0 | Intron | 4.02 × 10−6 | 0.014760 |
| RAD51B | 188 | 14 | Withdrawal | ENSG00000182185 | 0 | Intron | 6.38 × 10−6 | 0.018632 |
| RAD51B | 222 | 14 | Withdrawal | ENSG00000182185 | 0 | Intron | 1.36 × 10−5 | 0.026286 |
| RAD52–Microhomology-Mediated End Joining | ||||||||
| RAD52 | 102 | 12 | Dependence | ENSG00000002016 | 0 | Intron | 1.07 × 10−5 | 0.021847 |
| Nearest Gene Name | Page | Chromosome | Status | Nearest Gene | Distance to Nearest Gene | Relative Location | p-Value | p-Adjust |
|---|---|---|---|---|---|---|---|---|
| AURKAIP1 | 29 | 1 | Dependence | ENSG00000175756 | 84 | Downstream | 8.14 × 10−7 | 0.006318 |
| CCNB3 | 110 | X | Dependence | ENSG00000175756 | 0 | Intron | 1.23 × 10−5 | 0.023286 |
| CCNB2 | 154 | 15 | Withdrawal | ENSG00000175756 | 0 | Intron | 1.60 × 10−6 | 0.009713 |
| CCNB1IP1 | 159 | 14 | Withdrawal | ENSG00000175756 | 0 | Intron | 2.13 × 10−6 | 0.011189 |
| CDC20B | 46 | 5 | Dependence | ENSG00000164287 | 0 | Intron | 2.14 × 10−6 | 0.010145 |
| CDC200 | 70 | 17 | Dependence | ENSG00000236383 | 0 | Intron | 5.13 × 10−6 | 0.015449 |
| CDC200 | 123 | 17 | Dependence | ENSG00000236383 | 0 | Intron | 1.12 × 10−9 | 0.000216 |
| CDC201 | 136 | 7 | Withdrawal | ENSG00000283247 | 0 | Intron | 3.47 × 10−7 | 0.004589 |
| CDC20B | 213 | 5 | Withdrawal | ENSG00000164287 | 18781 | Upstream | 1.14 × 10−5 | 0.024227 |
| CDC200 | 213 | 17 | Withdrawal | ENSG00000236383 | 0 | Intron | 1.38 × 10−5 | 0.026395 |
| CDK1 | 169 | 10 | Withdrawal | ENSG00000170312 | 7683 | Downstream | 3.41 × 10−6 | 0.013821 |
| PPP1CC | 30 | 12 | Dependence | ENSG00000186298 | 0 | Intron | 8.18 × 10−7 | 0.006609 |
| Nearest Gene Name | Page | Functional Annotation | Number Genes Identified | p-Value |
|---|---|---|---|---|
| CDK1 | 169 | Skin lesion | 115 | 1.65 × 10−6 |
| CDK1 | 239 | Head and neck cancer | 356 | 7.73 × 10−20 |
| CDK1 | 325 | Skin cancer | 113 | 4.79 × 10−6 |
| CDK1 | 325 | Lung adenocarcinoma | 42 | 5.84 × 10−6 |
| CDK1 | 325 | Tumourigenesis | 149 | 7.17 × 10−6 |
| CDK1 | 326 | Large bowel cancer | 120 | 7.45 × 10−6 |
| CDK1 | 326 | Cutaneous melanoma | 110 | 7.71 × 10−6 |
| CDK1 | 326 | Abdominal adenocarcinoma | 135 | 8.46 × 10−6 |
| CDK1 | 327 | Solid organ cancer | 150 | 9.16 × 10−6 |
| CDK1 | 327 | Head and neck tumour | 137 | 9.54 × 10−6 |
| CDK1 | 327 | Carcinoma | 148 | 1.38 × 10−5 |
| CDK1 | 328 | CNS solid cancer | 101 | 2.06 × 10−5 |
| CDK1 | 328 | Cancer | 119 | 2.47 × 10−5 |
| CDK1 | 328 | Gastrointestinal adenocarcinoma | 121 | 3.56 × 10−5 |
| Nearest Gene Name | Page | Chromosome | Status | Nearest Gene | Distance to Nearest Gene | Relative Location | p-Value | p-Adjust |
|---|---|---|---|---|---|---|---|---|
| CDK1 | 169 | 10 | Withdrawal | ENSG00000170312 | 7683 | Downstream | 3.41 × 10−6 | 0.013821 |
| CDK1 | See also Table 15 | |||||||
| MCM6 | 166 | 2 | Withdrawal | ENSG00000076003 | 0 | Intron | 2.91 × 10−6 | 0.012759 |
| PCNA | 46 | 20 | Dependence | ENSG00000132646 | 0 | Intron | 2.11 × 10−6 | 0.012759 |
| POLA1 | 121 | x | Dependence | ENSG00000101868 | 0 | Intron | 1.51 × 10−5 | 0.010067 |
| POLA1 | 174 | x | Withdrawal | ENSG00000101868 | 0 | Intron | 3.92 × 10−6 | 0.025791 |
| POLA1 | 190 | x | Withdrawal | ENSG00000101868 | 0 | Intron | 6.68 × 10−6 | 0.019066 |
| TOP2A | 189 | 17 | Withdrawal | ENSG00000131747 | 0 | Intron | 6.53 × 10−6 | 0.018855 |
| ORC2 | 137 | 2 | Withdrawal | ENSG00000170312 | 0 | Intron | 3.82 × 10−7 | 0.004841 |
| POLA1 | 174 | X | Withdrawal | ENSG00000170312 | 0 | Intron | 3.92 × 10−6 | 0.014634 |
| POLA1 | 190 | X | Withdrawal | ENSG00000170312 | 0 | Intron | 6.68 × 10−6 | 0.019066 |
| POLA1 | 121 | X | Dependence | ENSG00000170312 | 0 | Intron | 1.15 × 10−4 | 0.025791 |
| TOP2A | 189 | 17 | Withdrawal | ENSG00000170312 | 6115 | Upstream | 6.53 × 10−6 | 0.018855 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reece, A.S.; Hulse, G.K. Cannabis- and Substance-Related Epidemiological Patterns of Chromosomal Congenital Anomalies in Europe: Geospatiotemporal and Causal Inferential Study. Int. J. Environ. Res. Public Health 2022, 19, 11208. https://doi.org/10.3390/ijerph191811208
Reece AS, Hulse GK. Cannabis- and Substance-Related Epidemiological Patterns of Chromosomal Congenital Anomalies in Europe: Geospatiotemporal and Causal Inferential Study. International Journal of Environmental Research and Public Health. 2022; 19(18):11208. https://doi.org/10.3390/ijerph191811208
Chicago/Turabian StyleReece, Albert Stuart, and Gary Kenneth Hulse. 2022. "Cannabis- and Substance-Related Epidemiological Patterns of Chromosomal Congenital Anomalies in Europe: Geospatiotemporal and Causal Inferential Study" International Journal of Environmental Research and Public Health 19, no. 18: 11208. https://doi.org/10.3390/ijerph191811208
APA StyleReece, A. S., & Hulse, G. K. (2022). Cannabis- and Substance-Related Epidemiological Patterns of Chromosomal Congenital Anomalies in Europe: Geospatiotemporal and Causal Inferential Study. International Journal of Environmental Research and Public Health, 19(18), 11208. https://doi.org/10.3390/ijerph191811208