
Epithelial Ovarian Cancer: Providing Evidence of Predisposition Genes





Abstract
1. Introduction
2. Molecular Landscape
3. Susceptibility to EOC
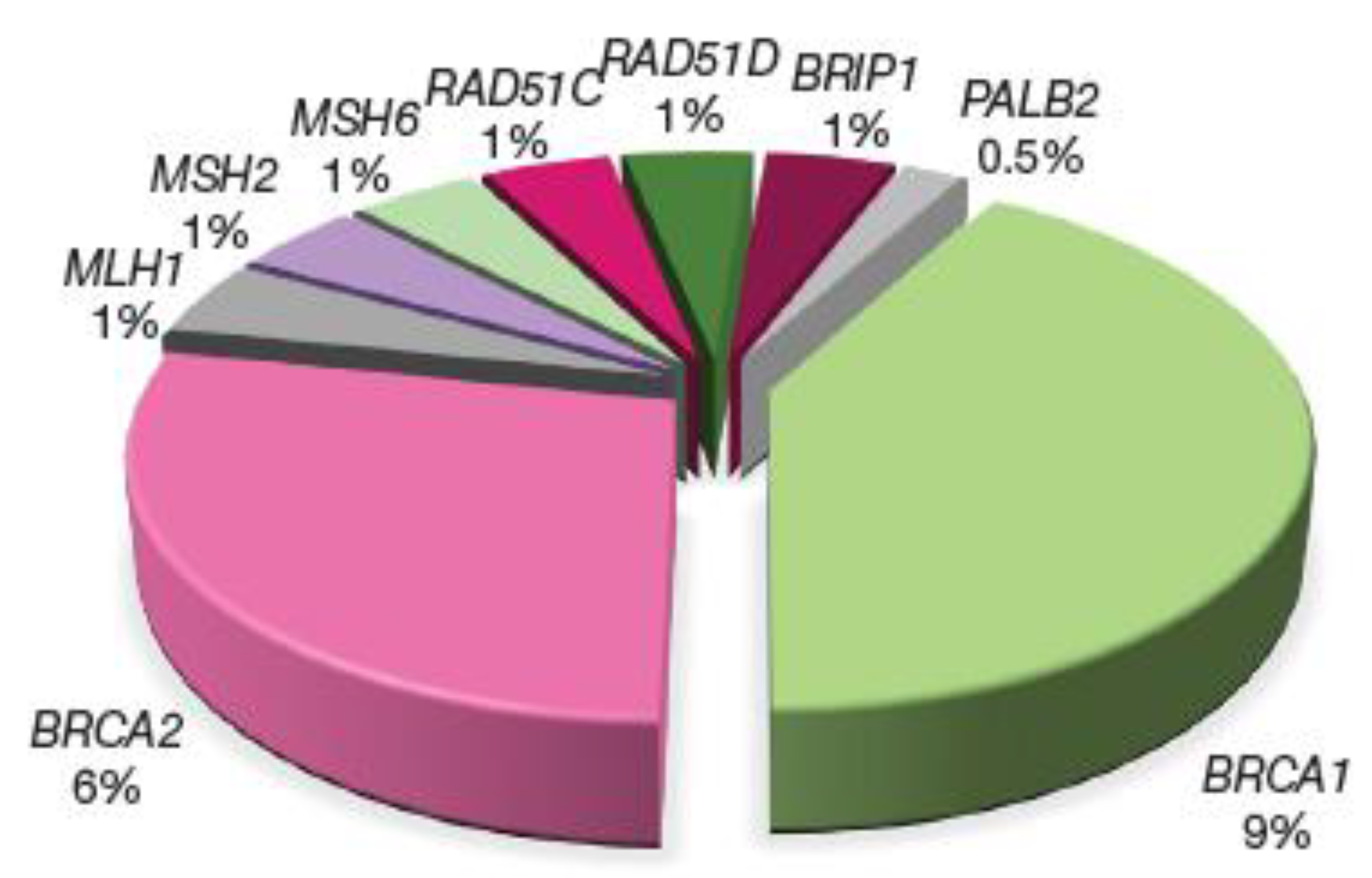
3.1. Germline Predisposing Variants
3.1.1. BRCA1 and BRCA2 Genes
3.1.2. RAD51C, RAD51D, and BRIP1 Genes
3.1.3. PALB2 Gene
3.1.4. MLH1, MSH2, MSH6, and PMS2 Genes
4. Tumour Testing in EOC
4.1. NGS for Cancer Risk Assessments
4.2. Upfront Tumour Testing
4.3. T Cell Repertoire Sequencing
4.4. Pre- and Post-Test Counselling
4.5. Treatment and Recurrent Ovarian Cancer

{kind=link}
{kind=link}
| Study/Reference | Population | Treatment Plan | Median PFS |
|---|---|---|---|
| SOLO 1/[127] | Stage III or IV high-grade serous or endometroid cancer with BRCA1/2 mutation and complete/partial response to CTH | 300 mg olaparib b.d. or placebo | 49.9 vs. 13.8 m (p < 0.0001) |
| SOLO 2/[126] | Relapsed, platinum-sensitive EOC with a BRCA1/2 mutation | 300 mg olaparib b.d. or placebo | 19.9 vs. 5.5 m (p < 0.0001) |
| SOLO 3/[128] | Germline BRCA-mutated ovarian cancer with relapse ≤ 12 m | 300 mg olaparib b.d. or non-platinum CTH of the choice of the physician | 13.4 vs. 9.2 m (p = 0.013) |
| NOVA/[129] | Platinum-sensitive recurrent EOC | Niraparib 300 mg o.d. or placebo | Germline BRCA cohort: 21.0 vs. 5.5 m; non-germline BRCA cohort: 9.3 vs. 3.9 m (p < 0.001) |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Chaudhry, S.R.; Lopes, J.; Levin, N.K.; Kalpage, H.; Tainsky, M.A. Germline mutations in apoptosis pathway genes in ovarian cancer; the functional role of a TP53I3 (PIG3) variant in ROS production and DNA repair. Cell Death Discov. 2021, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Moschetta, M.; Karihtala, P.; Samartzis, E.P.; Sheriff, M.; Pappas-Gogos, G.; Ozturk, M.A.; Uccello, M.; Karathanasi, A.; Tringos, M.; et al. Development of new poly(ADP-ribose) polymerase (PARP) inhibitors in ovarian cancer: Quo Vadis? Ann. Transl. Med. 2020, 8, 1706. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Kamara, D.; Karlan, B.Y.; Pharoah, P.D.P.; Gayther, S.A. Genetic epidemiology of ovarian cancer and prospects for polygenic risk prediction. Gynecol. Oncol. 2017, 147, 705–713. [Google Scholar] [CrossRef] [PubMed]
- The World Ovarian Cancer Coalition Atlas. Global Trends in Incidence, Mortality and Survival. In World Ovarian Cancer Coalition; The World Ovarian Cancer Coalition: Toronto, ON, Canada, 2018; pp. 1–39. Available online: https://worldovariancancercoalition.org/wp-content/uploads/2018/10/THE-WORLD-OVARIAN-CANCER-COALITION-ATLAS-2018.pdf (accessed on 23 April 2022).
- Cancer of the Ovary—Cancer Stat Facts. In Surveillance, Epidemiology, and End Results (SEER) Program; The National Cancer Institute: Bethesda, MD, USA, 2018. Available online: https://seer.cancer.gov/statfacts/html/ovary.html (accessed on 23 April 2022).
- Flaum, N.; Crosbie, E.J.; Edmondson, R.J.; Smith, M.J.; Evans, D.G. Epithelial ovarian cancer risk: A review of the current genetic landscape. Clin. Genet. 2020, 97, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar]
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef]
- Pavlidis, N.; Rassy, E.; Vermorken, J.B.; Assi, T.; Kattan, J.; Boussios, S.; Smith-Gagen, J. The outcome of patients with serous papillary peritoneal cancer, fallopian tube cancer, and epithelial ovarian cancer by treatment eras: 27 years data from the SEER registry. Cancer Epidemiol. 2021, 75, 102045. [Google Scholar] [CrossRef]
- Boussios, S.; Zarkavelis, G.; Seraj, E.; Zerdes, I.; Tatsi, K.; Pentheroudakis, G. Non-epithelial Ovarian Cancer: Elucidating Uncommon Gynaecological Malignancies. Anticancer Res. 2016, 36, 5031–5042. [Google Scholar] [CrossRef]
- Cheung, A.; Shah, S.; Parker, J.; Soor, P.; Limbu, A.; Sheriff, M.; Boussios, S. Non-Epithelial Ovarian Cancers: How Much Do We Really Know? Int. J. Environ. Res. Public Health 2022, 19, 1106. [Google Scholar] [CrossRef]
- Boussios, S.; Moschetta, M.; Zarkavelis, G.; Papadaki, A.; Kefas, A.; Tatsi, K. Ovarian sex-cord stromal tumours and small cell tumours: Pathological, genetic and management aspects. Crit. Rev. Oncol. Hematol. 2017, 120, 43–51. [Google Scholar] [CrossRef]
- Boussios, S.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Tsiouris, A.K.; Chatziantoniou, A.A.; Kanellos, F.S.; Tatsi, K. Ovarian carcinosarcoma: Current developments and future perspectives. Crit. Rev. Oncol. Hematol. 2019, 134, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; Boussios, S.; Rassy, E.; Samartzis, E.P.; Funingana, G.; Uccello, M. Neoadjuvant treatment for newly diagnosed advanced ovarian cancer: Where do we stand and where are we going? Ann. Transl. Med. 2020, 8, 1710. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Moschetta, M.; Tatsi, K.; Tsiouris, A.K.; Pavlidis, N. A review on pregnancy complicated by ovarian epithelial and non-epithelial malignant tumors: Diagnostic and therapeutic perspectives. J. Adv. Res. 2018, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- McCuaig, J.M.; Stockley, T.L.; Shaw, P.; Fung-Kee-Fung, M.; Altman, A.D.; Bentley, J.; Bernardini, M.Q.; Cormier, B.; Hirte, H.; Kieser, K.; et al. Evolution of genetic assessment for BRCA-associated gynaecologic malignancies: A Canadian multisociety roadmap. J. Med. Genet. 2018, 55, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Eoh, K.J.; Kim, J.E.; Park, H.S.; Lee, S.T.; Park, J.S.; Han, J.W.; Lee, J.Y.; Kim, S.; Kim, S.W.; Kim, J.H.; et al. Detection of Germline Mutations in Patients with Epithelial Ovarian Cancer Using Multi-gene Panels: Beyond BRCA1/2. Cancer Res. Treat. 2018, 50, 917–925. [Google Scholar] [CrossRef]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women with Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef]
- Chirasophon, S.; Manchana, T.; Teerapakpinyo, C. High-risk epithelial ovarian cancer patients for hereditary ovarian cancer. J. Obstet. Gynaecol. Res. 2017, 43, 929–934. [Google Scholar] [CrossRef]
- Boussios, S.; Mikropoulos, C.; Samartzis, E.; Karihtala, P.; Moschetta, M.; Sheriff, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Wise Management of Ovarian Cancer: On the Cutting Edge. J. Pers. Med. 2020, 10, 41. [Google Scholar] [CrossRef]
- Santonocito, C.; Rizza, R.; Paris, I.; De Marchis, L.; Paolillo, C.; Tiberi, G.; Scambia, G.; Capoluongo, E. Spectrum of Germline BRCA1 and BRCA2 Variants Identified in 2351 Ovarian and Breast Cancer Patients Referring to a Reference Cancer Hospital of Rome. Cancers 2020, 12, 1286. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182. [Google Scholar] [CrossRef]
- Ford, D.; Easton, D.F.; Stratton, M.; Narod, S.; Goldgar, D.; Devilee, P.; Bishop, D.T.; Weber, B.; Lenoir, G.; Chang-Claude, J.; et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am. J. Hum. Genet. 1998, 62, 676–689. [Google Scholar] [CrossRef] [PubMed]
- King, M.C.; Marks, J.H.; Mandell, J.B. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.; Pharoah, P.D.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, A.; et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 2003, 72, 1117–1130. [Google Scholar] [CrossRef]
- Yang, D.; Khan, S.; Sun, Y.; Hess, K.; Shmulevich, I.; Sood, A.K.; Zhang, W. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA 2011, 306, 1557–1565. [Google Scholar] [CrossRef]
- Vencken, P.M.L.H.; Kriege, M.; Hoogwerf, D.; Beugelink, S.; van der Burg, M.E.L.; Hooning, M.J.; Berns, E.M.; Jager, A.; Collée, M.; Burger, C.W.; et al. Chemosensitivity and outcome of BRCA1- and BRCA2-associated ovarian cancer patients after first-line chemotherapy compared with sporadic ovarian cancer patients. Ann. Oncol. 2011, 22, 1346–1352. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Osher, D.J.; De Leeneer, K.; Michils, G.; Hamel, N.; Tomiak, E.; Poppe, B.; Leunen, K.; Legius, E.; Shuen, A.; Smith, E.; et al. Mutation analysis of RAD51D in non-BRCA1/2 ovarian and breast cancer families. Br. J. Cancer 2012, 106, 1460–1463. [Google Scholar] [CrossRef][Green Version]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef]
- Meindl, A.; Hellebrand, H.; Wiek, C.; Erven, V.; Wappenschmidt, B.; Niederacher, D.; Freund, M.; Lichtner, P.; Hartmann, L.; Schaal, H.; et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010, 42, 410–414. [Google Scholar] [CrossRef]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Da Cunha Colombo Bonadio, R.R.; Fogace, R.N.; Miranda, V.C.; Diz, M.D.P.E. Homologous recombination deficiency in ovarian cancer: A review of its epidemiology and management. Clinics 2018, 73, e450s. [Google Scholar] [CrossRef] [PubMed]
- Angeli, D.; Salvi, S.; Tedaldi, G. Genetic Predisposition to Breast and Ovarian Cancers: How Many and Which Genes to Test? Int. J. Mol. Sci. 2020, 21, 1128. [Google Scholar] [CrossRef]
- de Bono, J.S.; Ashworth, A. Translating cancer research into targeted therapeutics. Nature 2010, 467, 543–549. [Google Scholar] [CrossRef]
- Byrski, T.; Gronwald, J.; Huzarski, T.; Grzybowska, E.; Budryk, M.; Stawicka, M.; Mierzwa, T.; Szwiec, M.; Wisniowski, R.; Siolek, M.; et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J. Clin. Oncol. 2010, 28, 375–379. [Google Scholar] [CrossRef]
- Boussios, S.; Karathanasi, A.; Cooke, D.; Neille, C.; Sadauskaite, A.; Moschetta, M.; Zakynthinakis-Kyriakou, N.; Pavlidis, N. PARP Inhibitors in Ovarian Cancer: The Route to “Ithaca”. Diagnostics 2019, 9, 55. [Google Scholar] [CrossRef]
- Boussios, S.; Abson, C.; Moschetta, M.; Rassy, E.; Karathanasi, A.; Bhat, T.; Ghumman, F.; Sheriff, M.; Pavlidis, N. Poly (ADP-Ribose) Polymerase Inhibitors: Talazoparib in Ovarian Cancer and Beyond. Drugs R D 2020, 20, 55–73. [Google Scholar] [CrossRef]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Combined Strategies with Poly (ADP-Ribose) Polymerase (PARP) Inhibitors for the Treatment of Ovarian Cancer: A Literature Review. Diagnostics 2019, 9, 87. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Abson, C.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Ryan, J.E.; Sheriff, M.; Rassy, E.; Pavlidis, N. Veliparib in ovarian cancer: A new synthetically lethal therapeutic approach. Investig. N. Drugs 2020, 38, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Amé, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, F.; de La Rubia, G.; Ménissier-De Murcia, J.; Hostomsky, Z.; de Murcia, G.; Schreiber, V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry 2000, 39, 7559–7569. [Google Scholar] [CrossRef]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J. Hematol. Oncol. 2020, 13, 118. [Google Scholar] [CrossRef]
- Eskander, R.N.; Tewari, K.S. PARP inhibition and synthetic lethality in ovarian cancer. Expert Rev. Clin. Pharmacol. 2014, 7, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.; Moschetta, M.; Pappas-Gogos, G.; Sheriff, M.; Boussios, S. Genetic Aberrations of DNA Repair Pathways in Prostate Cancer: Translation to the Clinic. Int. J. Mol. Sci. 2021, 22, 9783. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Rachmat, R.; Enyioma, S.; Ghose, A.; Revythis, A.; Boussios, S. BRCA Mutations in Prostate Cancer: Assessment, Implications and Treatment Considerations. Int. J. Mol. Sci. 2021, 22, 12628. [Google Scholar] [CrossRef]
- Boussios, S.; Rassy, E.; Shah, S.; Ioannidou, E.; Sheriff, M.; Pavlidis, N. Aberrations of DNA repair pathways in prostate cancer: A cornerstone of precision oncology. Expert Opin. Ther. Targets 2021, 25, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Pal, T.; Permuth-Wey, J.; Sellers, T.A. A review of the clinical relevance of mismatch-repair deficiency in ovarian cancer. Cancer 2008, 113, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Wajed, S.A.; Laird, P.W.; DeMeester, T.R. DNA methylation: An alternative pathway to cancer. Ann. Surg. 2001, 234, 10–20. [Google Scholar] [CrossRef]
- Cederquist, K.; Emanuelsson, M.; Wiklund, F.; Golovleva, I.; Palmqvist, R.; Grönberg, H. Two Swedish founder MSH6 mutations, one nonsense and one missense, conferring high cumulative risk of Lynch syndrome. Clin. Genet. 2005, 68, 533–541. [Google Scholar] [CrossRef]
- Song, H.; Cicek, M.S.; Dicks, E.; Harrington, P.; Ramus, S.J.; Cunningham, J.M.; Fridley, B.L.; Tyrer, J.P.; Alsop, J.; Jimenez-Linan, M.; et al. The contribution of deleterious germline mutations in BRCA1, BRCA2 and the mismatch repair genes to ovarian cancer in the population. Hum. Mol. Genet. 2014, 23, 4703–4709. [Google Scholar] [CrossRef]
- Jones, M.R.; Peng, P.C.; Coetzee, S.G.; Tyrer, J.; Reyes, A.L.P.; Corona, R.I.; Davis, B.; Chen, S.; Dezem, F.; Seo, J.H.; et al. Ovarian Cancer Risk Variants Are Enriched in Histotype-Specific Enhancers and Disrupt Transcription Factor Binding Sites. Am. J. Hum. Genet. 2020, 107, 622–635. [Google Scholar] [CrossRef]
- Sugino, K.; Tamura, R.; Nakaoka, H.; Yachida, N.; Yamaguchi, M.; Mori, Y.; Yamawaki, K.; Suda, K.; Ishiguro, T.; Adachi, S.; et al. Germline and somatic mutations of homologous recombination-associated genes in Japanese ovarian cancer patients. Sci. Rep. 2019, 9, 17808. [Google Scholar] [CrossRef]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Klotz, D.M.; Wimberger, P. Cells of origin of ovarian cancer: Ovarian surface epithelium or fallopian tube? Arch. Gynecol. Obstet. 2017, 296, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef]
- Kang, S.; Yu, Y.L.; Cho, S.Y.; Park, S.Y. Prevalence of pathogenic variants in actionable genes in advanced ovarian cancer: A next-generation sequencing analysis of a nationwide registry study. Eur. J. Cancer 2020, 141, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Bychkovsky, B.L.; Lo, M.T.; Yussuf, A.; Horton, C.; Richardson, M.; LaDuca, H.; Garber, J.E.; Rana, H.Q. Prevalence and spectrum of pathogenic variants among patients with multiple primary cancers evaluated by clinical characteristics. Cancer 2022, 128, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Silvestri, V.; Leslie, G.; Rebbeck, T.R.; Neuhausen, S.L.; Hopper, J.L.; Nielsen, H.R.; Lee, A.; Yang, X.; McGuffog, L.; et al. Cancer Risks Associated with BRCA1 and BRCA2 Pathogenic Variants. J. Clin. Oncol. 2022, 40, 1529. [Google Scholar] [CrossRef]
- Boni, J.; Idani, A.; Roca, C.; Feliubadaló, L.; Tomiak, E.; Weber, E.; Foulkes, W.D.; Orthwein, A.; El Haffaf, Z.; Lazaro, C.; et al. A decade of RAD51C and RAD51D germline variants in cancer. Hum. Mutat. 2022, 43, 285–298. [Google Scholar] [CrossRef]
- Kotsopoulos, J.; Sopik, V.; Rosen, B.; Fan, I.; McLaughlin, J.R.; Risch, H.; Sun, P.; Narod, S.A.; Akbari, M.R. Frequency of germline PALB2 mutations among women with epithelial ovarian cancer. Fam. Cancer 2017, 16, 29–34. [Google Scholar] [CrossRef]
- Manchana, T.; Phoolcharoen, N.; Tantbirojn, P. BRCA mutation in high grade epithelial ovarian cancers. Gynecol. Oncol. Rep. 2019, 29, 102–105. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Primers 2016, 2, 16061. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Royer, R.; Li, S.; McLaughlin, J.R.; Rosen, B.; Risch, H.A.; Fan, I.; Bradley, L.; Shaw, P.A.; Narod, S.A. Frequencies of BRCA1 and BRCA2 mutations among 1342 unselected patients with invasive ovarian cancer. Gynecol. Oncol. 2011, 121, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Bolton, K.L.; Chenevix-Trench, G.; Goh, C.; Sadetzki, S.; Ramus, S.J.; Karlan, B.Y.; Lambrechts, D.; Despierre, E.; Barrowdale, D.; McGuffog, L.; et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 2012, 307, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Suszynska, M.; Ratajska, M.; Kozlowski, P. BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: Mutation prevalence and precise risk estimates based on a pooled analysis of ~30,000 cases. J. Ovarian Res. 2020, 13, 50. [Google Scholar] [CrossRef]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women with Ovarian Cancer. J. Natl. Cancer Inst. 2015, 107, djv214. [Google Scholar] [CrossRef]
- Lilyquist, J.; LaDuca, H.; Polley, E.; Davis, B.T.; Shimelis, H.; Hu, C.; Hart, S.N.; Dolinsky, J.S.; Couch, F.J.; Goldgar, D.E. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol. Oncol. 2017, 147, 375–380. [Google Scholar] [CrossRef]
- Weber-Lassalle, N.; Hauke, J.; Ramser, J.; Richters, L.; Groß, E.; Blümcke, B.; Gehrig, A.; Kahlert, A.K.; Müller, C.R.; Hackmann, K.; et al. BRIP1 loss-of-function mutations confer high risk for familial ovarian cancer, but not familial breast cancer. Breast Cancer Res. 2018, 20, 7. [Google Scholar] [CrossRef]
- Clague, J.; Wilhoite, G.; Adamson, A.; Bailis, A.; Weitzel, J.N.; Neuhausen, S.L. RAD51C germline mutations in breast and ovarian cancer cases from high-risk families. PLoS ONE 2011, 6, e25632. [Google Scholar] [CrossRef]
- Thompson, D.; Easton, D. Variation in cancer risks, by mutation position, in BRCA2 mutation carriers. Am. J. Hum. Genet. 2001, 68, 410–419. [Google Scholar] [CrossRef]
- Miller, K.A.; Sawicka, D.; Barsky, D.; Albala, J.S. Domain mapping of the Rad51 paralog protein complexes. Nucleic Acids Res. 2004, 32, 169–178. [Google Scholar] [CrossRef]
- Yu, X.; Chini, C.C.; He, M.; Mer, G.; Chen, J. The BRCT domain is a phospho-protein binding domain. Science. 2003, 302, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Bork, P.; Hofmann, K.; Bucher, P.; Neuwald, A.F.; Altschul, S.F.; Koonin, E.V. A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J. 1997, 11, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Seal, S.; Thompson, D.; Renwick, A.; Elliott, A.; Kelly, P.; Barfoot, R.; Chagtai, T.; Jayatilake, H.; Ahmed, M.; Spanova, K.; et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 1239–1241. [Google Scholar] [CrossRef] [PubMed]
- Tyrer, J.P.; Guo, Q.; Easton, D.F.; Pharoah, P.D. The admixture maximum likelihood test to test for association between rare variants and disease phenotypes. BMC Bioinform. 2013, 14, 177. [Google Scholar] [CrossRef][Green Version]
- Helleman, J.; van Staveren, I.L.; Dinjens, W.N.; van Kuijk, P.F.; Ritstier, K.; Ewing, P.C.; van der Burg, M.E.; Stoter, G.; Berns, E.M. Mismatch repair and treatment resistance in ovarian cancer. BMC Cancer 2006, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Banno, K.; Yanokura, M.; Iida, M.; Adachi, M.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Nomura, H.; Hirasawa, A.; et al. Features of ovarian cancer in Lynch syndrome (Review). Mol. Clin. Oncol. 2014, 2, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Bernards, S.S.; Norquist, B.M.; Harrell, M.I.; Agnew, K.J.; Lee, M.K.; Walsh, T.; Swisher, E.M. Genetic characterization of early onset ovarian carcinoma. Gynecol. Oncol. 2016, 140, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Pal, T.; Akbari, M.R.; Sun, P.; Lee, J.H.; Fulp, J.; Thompson, Z.; Coppola, D.; Nicosia, S.; Sellers, T.A.; McLaughlin, J.; et al. Frequency of mutations in mismatch repair genes in a population-based study of women with ovarian cancer. Br. J. Cancer 2012, 107, 1783–1790. [Google Scholar] [CrossRef] [PubMed]
- Crijnen, T.E.; Janssen-Heijnen, M.L.; Gelderblom, H.; Morreau, J.; Nooij, M.A.; Kenter, G.G.; Vasen, H.F. Survival of patients with ovarian cancer due to a mismatch repair defect. Fam. Cancer 2005, 4, 301–305. [Google Scholar] [CrossRef]
- Kamps, R.; Brandão, R.D.; Bosch, B.J.; Paulussen, A.D.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification. Int. J. Mol. Sci. 2017, 18, 308. [Google Scholar] [CrossRef]
- Cobain, E.F.; Milliron, K.J.; Merajver, S.D. Updates on breast cancer genetics: Clinical implications of detecting syndromes of inherited increased susceptibility to breast cancer. Semin. Oncol. 2016, 43, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Lynce, F.; Isaacs, C. How Far Do We Go with Genetic Evaluation? Gene, Panel, and Tumor Testing. Am. Soc. Clin. Oncol. Educ. Book. 2016, 35, e72–e78. [Google Scholar] [CrossRef] [PubMed]
- Andoni, T.; Wiggins, J.; Robinson, R.; Charlton, R.; Sandberg, M.; Eeles, R. Half of germline pathogenic and likely pathogenic variants found on panel tests do not fulfil NHS testing criteria. Sci. Rep. 2022, 12, 2507. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Smith, C.; Salipante, S.J.; Lee, M.K.; Thornton, A.M.; Nord, A.S.; Gulden, C.; Kupfer, S.S.; Swisher, E.M.; Bennett, R.L.; et al. ColoSeq provides comprehensive lynch and polyposis syndrome mutational analysis using massively parallel sequencing. J. Mol. Diagn. 2012, 14, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.K.; Kim, S.H.; Bassett, R.Y.; Martineau, J.; Dalton, E.; Chern, J.Y.; Blank, S.V. Rescreening for genetic mutations using multi-gene panel testing in patients who previously underwent non-informative genetic screening. Gynecol. Oncol. 2015, 139, 211–215. [Google Scholar] [CrossRef]
- Sun, L.; Brentnall, A.; Patel, S.; Buist, D.; Bowles, E.; Evans, D.G.; Eccles, D.; Hopper, J.; Li, S.; Duffy, S.; et al. Should we offer multi-gene testing to all patients with breast cancer: A cost-effectiveness analysis. Int. J. Gynecol. Cancer 2019, 29, A31–A32. [Google Scholar]
- Asphaug, L.; Melberg, H.O. The Cost-Effectiveness of Multigene Panel Testing for Hereditary Breast and Ovarian Cancer in Norway. MDM Policy Pr. 2019, 4, 2381468318821103. [Google Scholar] [CrossRef]
- Bonadio, R.C.; Crespo, J.R.; Estevez-Diz, M.D.P. Ovarian cancer risk assessment in the era of next-generation sequencing. Ann. Transl. Med. 2020, 8, 1704. [Google Scholar] [CrossRef]
- Bekos, C.; Grimm, C.; Kranawetter, M.; Polterauer, S.; Oberndorfer, F.; Tan, Y.; Müllauer, L.; Singer, C.F. Reliability of Tumor Testing Compared to Germline Testing for Detecting BRCA1 and BRCA2 Mutations in Patients with Epithelial Ovarian Cancer. J. Pers. Med. 2021, 11, 593. [Google Scholar] [CrossRef]
- Dougherty, B.A.; Lai, Z.; Hodgson, D.R.; Orr, M.C.M.; Hawryluk, M.; Sun, J.; Yelensky, R.; Spencer, S.K.; Robertson, J.D.; Ho, T.W.; et al. Biological and clinical evidence for somatic mutations in BRCA1 and BRCA2 as predictive markers for olaparib response in high-grade serous ovarian cancers in the maintenance setting. Oncotarget 2017, 8, 43653–43661. [Google Scholar] [CrossRef]
- Fumagalli, C.; Tomao, F.; Betella, I.; Rappa, A.; Calvello, M.; Bonanni, B.; Bernard, L.; Peccatori, F.; Colombo, N.; Viale, G.; et al. Tumor BRCA Test for Patients with Epithelial Ovarian Cancer: The Role of Molecular Pathology in the Era of PARP Inhibitor Therapy. Cancers 2019, 11, 1641. [Google Scholar] [CrossRef] [PubMed]
- Emerson, R.O.; Sherwood, A.M.; Rieder, M.J.; Guenthoer, J.; Williamson, D.W.; Carlson, C.S.; Drescher, C.W.; Tewari, M.; Bielas, J.H.; Robins, H.S. High-throughput sequencing of T-cell receptors reveals a homogeneous repertoire of tumour-infiltrating lymphocytes in ovarian cancer. J. Pathol. 2013, 231, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Ostmeyer, J.; Lucas, E.; Christley, S.; Lea, J.; Monson, N.; Tiro, J.; Cowell, L.G. Biophysicochemical motifs in T cell receptor sequences as a potential biomarker for high-grade serous ovarian carcinoma. PLoS ONE 2020, 15, e0229569. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.T.; Adams, S.F.; Tahirovic, E.; Hagemann, I.S.; Coukos, G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2012, 124, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef]
- Santoiemma, P.P.; Powell, D.J., Jr. Tumor infiltrating lymphocytes in ovarian cancer. Cancer Biol. Ther. 2015, 16, 807–820. [Google Scholar] [CrossRef]
- Aoki, Y.; Takakuwa, K.; Kodama, S.; Tanaka, K.; Takahashi, M.; Tokunaga, A.; Takahashi, T. Use of adoptive transfer of tumor-infiltrating lymphocytes alone or in combination with cisplatin-containing chemotherapy in patients with epithelial ovarian cancer. Cancer Res. 1991, 51, 1934–1939. [Google Scholar]
- Freedman, R.S.; Edwards, C.L.; Kavanagh, J.J.; Kudelka, A.P.; Katz, R.L.; Carrasco, C.H.; Atkinson, E.N.; Scott, W.; Tomasovic, B.; Templin, S.; et al. Intraperitoneal adoptive immunotherapy of ovarian carcinoma with tumor-infiltrating lymphocytes and low-dose recombinant interleukin-2: A pilot trial. J. Immunother. Emphas. Tumor Immunol. 1994, 16, 198–210. [Google Scholar] [CrossRef]
- Wright, S.E.; Rewers-Felkins, K.A.; Quinlin, I.S.; Phillips, C.A.; Townsend, M.; Philip, R.; Dobrzanski, M.J.; Lockwood-Cooke, P.R.; Robinson, W. Cytotoxic T-lymphocyte immunotherapy for ovarian cancer: A pilot study. J. Immunother. 2012, 35, 196–204. [Google Scholar] [CrossRef]
- Owens, D.K.; Davidson, K.W.; Krist, A.H.; Barry, M.J.; Cabana, M.; Caughey, A.B.; Doubeni, C.A.; Epling, J.W., Jr.; Kubik, M.; Landefeld, C.S.; et al. Risk Assessment, Genetic Counseling, and Genetic Testing for BRCA-Related Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2019, 322, 652–665. [Google Scholar]
- Konstantinopoulos, P.A.; Norquist, B.; Lacchetti, C.; Armstrong, D.; Grisham, R.N.; Goodfellow, P.J.; Kohn, E.C.; Levine, D.A.; Liu, J.F.; Lu, K.H.; et al. Germline and Somatic Tumor Testing in Epithelial Ovarian Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 1222–1245. [Google Scholar] [CrossRef] [PubMed]
- Childers, K.K.; Maggard-Gibbons, M.; Macinko, J.; Childers, C.P. National Distribution of Cancer Genetic Testing in the United States: Evidence for a Gender Disparity in Hereditary Breast and Ovarian Cancer. JAMA Oncol. 2018, 4, 876–879. [Google Scholar] [CrossRef] [PubMed]
- McGee, J.; Panabaker, K.; Leonard, S.; Ainsworth, P.; Elit, L.; Shariff, S.Z. Genetics Consultation Rates Following a Diagnosis of High-Grade Serous Ovarian Carcinoma in the Canadian Province of Ontario. Int. J. Gynecol. Cancer 2017, 27, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, K.A.; Fan, I.; McLaughlin, J.; Risch, H.A.; Rosen, B.; Murphy, J.; Bradley, L.; Armel, S.; Sun, P.; Narod, S.A. Uptake of clinical genetic testing for ovarian cancer in Ontario: A population-based study. Gynecol. Oncol. 2009, 112, 68–72. [Google Scholar] [CrossRef]
- Nielsen, S.M.; Eccles, D.M.; Romero, I.L.; Al-Mulla, F.; Balmaña, J.; Biancolella, M.; Bslok, R.; Caligo, M.A.; Calvello, M.; Capone, G.L.; et al. Genetic Testing and Clinical Management Practices for Variants in Non-BRCA1/2 Breast (and Breast/Ovarian) Cancer Susceptibility Genes: An International Survey by the Evidence-Based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) Clinical Working Group. JCO Precis. Oncol. 2018, 2, PO.18.00091. [Google Scholar]
- Mersch, J.; Brown, N.; Pirzadeh-Miller, S.; Mundt, E.; Cox, H.C.; Brown, K.; Aston, M.; Esterling, L.; Manley, S.; Ross, T. Prevalence of Variant Reclassification Following Hereditary Cancer Genetic Testing. JAMA 2018, 320, 1266–1274. [Google Scholar] [CrossRef]
- Fecteau, H.; Vogel, K.J.; Hanson, K.; Morrill-Cornelius, S. The evolution of cancer risk assessment in the era of next generation sequencing. J. Genet. Couns. 2014, 23, 633–639. [Google Scholar] [CrossRef]
- Gilks, C.B.; Prat, J. Ovarian carcinoma pathology and genetics: Recent advances. Hum. Pathol. 2009, 40, 1213–1223. [Google Scholar] [CrossRef]
- Prat, J.; D’Angelo, E.; Espinosa, I. Ovarian carcinomas: At least five different diseases with distinct histological features and molecular genetics. Hum. Pathol. 2018, 80, 11–27. [Google Scholar] [CrossRef]
- Fostira, F.; Papadimitriou, M.; Papadimitriou, C. Current practices on genetic testing in ovarian cancer. Ann. Transl. Med. 2020, 8, 1703. [Google Scholar] [CrossRef]
- Gasparri, M.L.; Taghavi, K.; Fiacco, E.; Zuber, V.; Di Micco, R.; Gazzetta, G.; Valentini, A.; Mueller, M.D.; Papadia, A.; Gentilini, O.D. Risk-Reducing Bilateral Salpingo-Oophorectomy for BRCA Mutation Carriers and Hormonal Replacement Therapy: If It Should Rain, Better a Drizzle than a Storm. Medicina 2019, 55, 415. [Google Scholar] [CrossRef] [PubMed]
- Finch, A.P.; Lubinski, J.; Møller, P.; Singer, C.F.; Karlan, B.; Senter, L.; Rosen, B.; Maehle, L.; Ghadirian, P.; Cybulski, C.; et al. Impact of oophorectomy on cancer incidence and mortality in women with a BRCA1 or BRCA2 mutation. J. Clin. Oncol. 2014, 32, 1547–1553. [Google Scholar] [CrossRef] [PubMed]
- Domchek, S.M.; Rebbeck, T.R. Prophylactic oophorectomy in women at increased cancer risk. Curr. Opin. Obstet. Gynecol. 2007, 19, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Penson, R.T.; Valencia, R.V.; Cibula, D.; Colombo, N.; Leath, C.A., 3rd; Bidziński, M.; Kim, J.W.; Nam, J.H.; Madry, R.; Hernández, C.; et al. Olaparib Versus Nonplatinum Chemotherapy in Patients with Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): A Randomized Phase III Trial. J. Clin. Oncol. 2020, 38, 1164–1174. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Ushijima, K. Treatment for recurrent ovarian cancer-at first relapse. J. Oncol. 2010, 2010, 497429. [Google Scholar] [CrossRef]
- Gadducci, A.; Fuso, L.; Cosio, S.; Landoni, F.; Maggino, T.; Perotto, S.; Sartori, E.; Testa, A.; Galletto, L.; Zola, P. Are surveillance procedures of clinical benefit for patients treated for ovarian cancer?: A retrospective Italian multicentric study. Int. J. Gynecol. Cancer 2009, 19, 367–374. [Google Scholar] [CrossRef]
- Spriggs, D. Optimal sequencing in the treatment of recurrent ovarian cancer. Gynecol. Oncol. 2003, 90, S39–S44. [Google Scholar] [CrossRef]
- Onda, T.; Yoshikawa, H.; Yasugi, T.; Yamada, M.; Matsumoto, K.; Taketani, Y. Secondary cytoreductive surgery for recurrent epithelial ovarian carcinoma: Proposal for patients selection. Br. J. Cancer 2005, 92, 1026–1032. [Google Scholar] [CrossRef] [PubMed]


| Genes Affected (%) | Histology | ||
|---|---|---|---|
| HGSOC | EnOC | CCOC | |
| BRCA1 | 8.0 | - | - |
| BRCA2 | 4.0 | 5.1 | 5.1 |
| ATM | 4.0 | 9.1 | 17.9 |
| BRIP1 | 2.0 | 2.0 | - |
| PALB2 | - | 2.0 | 2.6 |
| RAD50 | - | 1.0 | 2.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, S.; Cheung, A.; Kutka, M.; Sheriff, M.; Boussios, S. Epithelial Ovarian Cancer: Providing Evidence of Predisposition Genes. Int. J. Environ. Res. Public Health 2022, 19, 8113. https://doi.org/10.3390/ijerph19138113
Shah S, Cheung A, Kutka M, Sheriff M, Boussios S. Epithelial Ovarian Cancer: Providing Evidence of Predisposition Genes. International Journal of Environmental Research and Public Health. 2022; 19(13):8113. https://doi.org/10.3390/ijerph19138113
Chicago/Turabian StyleShah, Sidrah, Alison Cheung, Mikolaj Kutka, Matin Sheriff, and Stergios Boussios. 2022. "Epithelial Ovarian Cancer: Providing Evidence of Predisposition Genes" International Journal of Environmental Research and Public Health 19, no. 13: 8113. https://doi.org/10.3390/ijerph19138113
APA StyleShah, S., Cheung, A., Kutka, M., Sheriff, M., & Boussios, S. (2022). Epithelial Ovarian Cancer: Providing Evidence of Predisposition Genes. International Journal of Environmental Research and Public Health, 19(13), 8113. https://doi.org/10.3390/ijerph19138113