Influence of Second-Hand Smoke and Prenatal Tobacco Smoke Exposure on Biomarkers, Genetics and Physiological Processes in Children—An Overview in Research Insights of the Last Few Years



Abstract
1. Introduction
2. Biomarkers
2.1. Matrix Metalloproteinase-9
2.2. Immune-Regulatory Cytokines
2.3. Cysteinyl Leukotrienes and Urinary Leukotriene E4
2.4. Estimated Glomerular Filtration Rate and Kidney Function
2.5. Cardiovascular Status
2.6. C-Reactive Protein
3. Immune Status
4. Lipid Profile
5. Oxidative Stress
6. Hormone Status
7. Genetic Predisposition
7.1. Glutathione S-Transferase (GST) Genes
7.2. Anti-Inflammatory Cytokine Genes
7.3. CD14 Gene
7.4. Variants at Chromosome 17 Region q21
7.5. ATPase-Related Genes and Bronchial Hyper-Responsiveness
7.6. Mannose-Binding Lectin-2 (MBL2) Gene
7.7. Flavin-Monooxygenase-3 (FMO3) Gene
7.8. O-Sialoglycoprotein Endopeptidase (OSGEP) Gene
7.9. MSX1 Gene
8. Protein Expression in Foetal Liver
9. Dysregulation of Diverse MicroRNAs
10. Leukocyte Telomere Length
11. DNA Methylation
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- NIH National Cancer Institute. Secondhand Smoke and Cancer. 2018. Available online: https://www.cancer.gov/about-cancer/causes-prevention/risk/tobacco/second-hand-smoke-fact-sheet (accessed on 16 September 2019).
- Protano, C.; Vitali, M. The new danger of thirdhand smoke: Why passive smoking does not stop at secondhand smoke. Environ. Health Perspect. 2011, 119, A422. [Google Scholar] [CrossRef]
- ACS American Cancer Society. Health Risks of Secondhand Smoke. 2019. Available online: https://www.cancer.org/cancer/cancer-causes/tobacco-and-cancer/secondhand-smoke.html (accessed on 16 September 2019).
- WHO World Health Organization. Global Health Observatory (GHO) Data. Mortality and Burden of Disease from Second-Hand Smoke. 2004. Available online: https://www.who.int/gho/phe/secondhand_smoke/burden/en/ (accessed on 16 September 2019).
- Tsai, J.; Homa, D.M.; Gentzke, A.S.; Mahoney, M.; Sharapova, S.R.; Sosnoff, C.S.; Caron, K.T.; Wang, L.; Melstrom, P.C.; Trivers, K.F. Exposure to Secondhand Smoke Among Nonsmokers - United States, 1988–2014. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 1342–1346. [Google Scholar] [CrossRef]
- Besaratinia, A.; Pfeifer, G.P. Second-hand smoke and human lung cancer. Lancet Oncol. 2008, 9, 657–666. [Google Scholar] [CrossRef]
- Lee, P.N.; Hamling, J.S. Environmental tobacco smoke exposure and risk of breast cancer in nonsmoking women. An updated review and meta-analysis. Inhal. Toxicol. 2016, 28, 431–454. [Google Scholar] [CrossRef]
- Farber, H.J.; Groner, J.; Walley, S.; Nelson, K. Protecting Children From Tobacco, Nicotine, and Tobacco Smoke. Pediatrics 2015, 136, e1439–e1467. [Google Scholar] [CrossRef]
- DiGiacomo, S.I.; Jazayeri, M.A.; Barua, R.S.; Ambrose, J.A. Environmental Tobacco Smoke and Cardiovascular Disease. Int. J. Environ. Res. Public Health 2018, 16, 96. [Google Scholar] [CrossRef]
- Strzelak, A.; Ratajczak, A.; Adamiec, A.; Feleszko, W. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int. J. Environ. Res. Public Health 2018, 15, 1033. [Google Scholar] [CrossRef]
- Rahman, I.; MacNee, W. Lung glutathione and oxidative stress: Implications in cigarette smoke-induced airway disease. Am. J. Physiol. 1999, 277, L1067–L1088. [Google Scholar] [CrossRef]
- Han, C.; Liu, Y.; Gong, X.; Ye, X.; Zhou, J. Relationship Between Secondhand Smoke Exposure and Depressive Symptoms: A Systematic Review and Dose(-)Response Meta-Analysis. Int. J. Environ. Res. Public Health 2019, 16, 1356. [Google Scholar] [CrossRef]
- Mund, M.; Louwen, F.; Klingelhoefer, D.; Gerber, A. Smoking and pregnancy—A review on the first major environmental risk factor of the unborn. Int. J. Environ. Res. Public Health 2013, 10, 6485–6499. [Google Scholar] [CrossRef]
- Waterland, R.A.; Michels, K.B. Epigenetic epidemiology of the developmental origins hypothesis. Annu. Rev. Nutr. 2007, 27, 363–388. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. The Health Consequences of Smoking—50 Years of Progress; A Report of the Surgeon General; U.S. Department of Health and Human Services, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health: Atlanta, GA, USA, 2014. Available online: https://www.ncbi.nlm.nih.gov/books/NBK179276/pdf/Bookshelf_NBK179276.pdf (accessed on 17 September 2019).
- von Ehrenstein, O.S.; von Mutius, E.; Maier, E.; Hirsch, T.; Carr, D.; Schaal, W.; Roscher, A.A.; Olgemoller, B.; Nicolai, T.; Weiland, S.K. Lung function of school children with low levels of alpha1-antitrypsin and tobacco smoke exposure. Eur. Respir. J. 2002, 19, 1099–1106. [Google Scholar] [CrossRef]
- Gilliland, F.D.; Li, Y.F.; Dubeau, L.; Berhane, K.; Avol, E.; McConnell, R.; Gauderman, W.J.; Peters, J.M. Effects of glutathione S-transferase M1, maternal smoking during pregnancy, and environmental tobacco smoke on asthma and wheezing in children. Am. J. Respir. Crit. Care Med. 2002, 166, 457–463. [Google Scholar] [CrossRef]
- Kabesch, M.; Hoefler, C.; Carr, D.; Leupold, W.; Weiland, S.K.; von Mutius, E. Glutathione S transferase deficiency and passive smoking increase childhood asthma. Thorax 2004, 59, 569–573. [Google Scholar] [CrossRef]
- Chen, Y.; Wong, G.W.; Li, J. Environmental Exposure and Genetic Predisposition as Risk Factors for Asthma in China. Allergy Asthma Immunol. Res. 2016, 8, 92–100. [Google Scholar] [CrossRef]
- Oberg, M.; Jaakkola, M.S.; Woodward, A.; Peruga, A.; Pruss-Ustun, A. Worldwide burden of disease from exposure to second-hand smoke: A retrospective analysis of data from 192 countries. Lancet 2011, 377, 139–146. [Google Scholar] [CrossRef]
- Homa, D.M.; Neff, L.J.; King, B.A.; Caraballo, R.S.; Bunnell, R.E.; Babb, S.D.; Garrett, B.E.; Sosnoff, C.S.; Wang, L.; Centers for Disease, C.; et al. Vital signs: Disparities in nonsmokers’ exposure to secondhand smoke--United States, 1999–2012. MMWR Morb. Mortal. Wkly. Rep. 2015, 64, 103–108. [Google Scholar]
- Lupsa, I.R.; Nunes, B.; Ligocka, D.; Gurzau, A.E.; Jakubowski, M.; Casteleyn, L.; Aerts, D.; Biot, P.; Den Hond, E.; Castano, A.; et al. Urinary cotinine levels and environmental tobacco smoke in mothers and children of Romania, Portugal and Poland within the European human biomonitoring pilot study. Environ. Res. 2015, 141, 106–117. [Google Scholar] [CrossRef]
- WHO World Health Organization. Tobacco. Key Facts. 2019. Available online: https://www.who.int/news-room/fact-sheets/detail/tobacco (accessed on 17 September 2019).
- Lau, E.M.; Celermajer, D.S. Protecting our children from environmental tobacco smoke: One of our great healthcare challenges. Eur. Heart J. 2014, 35, 2452–2453. [Google Scholar] [CrossRef][Green Version]
- Van den Steen, P.E.; Dubois, B.; Nelissen, I.; Rudd, P.M.; Dwek, R.A.; Opdenakker, G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9). Crit. Rev. Biochem. Mol. 2002, 37, 375–536. [Google Scholar] [CrossRef]
- De, S.; Leong, S.C.; Fenton, J.E.; Carter, S.D.; Clarke, R.W.; Jones, A.S. The effect of passive smoking on the levels of matrix metalloproteinase 9 in nasal secretions of children. Am. J. Rhinol. Allergy 2011, 25, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Vignola, A.M.; Riccobono, L.; Mirabella, A.; Profita, M.; Chanez, P.; Bellia, V.; Mautino, G.; D’Accardi, P.; Bousquet, J.; Bonsignore, G. Sputum metalloproteinase-9/tissue inhibitor of metalloproteinase-1 ratio correlates with airflow obstruction in asthma and chronic bronchitis. Am. J. Respir. Crit. Care Med. 1998, 158, 1945–1950. [Google Scholar] [CrossRef] [PubMed]
- Kopp, B.T.; Thompson, R.; Kim, J.; Konstan, R.; Diaz, A.; Smith, B.; Shrestha, C.; Rogers, L.K.; Hayes, D., Jr.; Tumin, D.; et al. Secondhand smoke alters arachidonic acid metabolism and inflammation in infants and children with cystic fibrosis. Thorax 2019, 74, 237–246. [Google Scholar] [CrossRef]
- Yilmaz, O.; Turkeli, A.; Onur, E.; Bilge, S.; Yuksel, H. Secondhand tobacco smoke and severity in wheezing children: Nasal oxidant stress and inflammation. J. Asthma 2018, 55, 477–482. [Google Scholar] [CrossRef]
- Opal, S.M.; DePalo, V.A. Anti-inflammatory cytokines. Chest 2000, 117, 1162–1172. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H.; Fahy, J.V. The Cytokines of Asthma. Immunity 2019, 50, 975–991. [Google Scholar] [CrossRef]
- Berry, M.; Brightling, C.; Pavord, I.; Wardlaw, A. TNF-alpha in asthma. Curr. Opin. Pharmacol. 2007, 7, 279–282. [Google Scholar] [CrossRef]
- Chahal, N.; McLain, A.C.; Ghassabian, A.; Michels, K.A.; Bell, E.M.; Lawrence, D.A.; Yeung, E.H. Maternal Smoking and Newborn Cytokine and Immunoglobulin Levels. Nicotine Tob. Res. 2017, 19, 789–796. [Google Scholar] [CrossRef]
- Wilson, K.M.; Wesgate, S.C.; Pier, J.; Weis, E.; Love, T.; Evans, K.; Chhibber, A. Secondhand smoke exposure and serum cytokine levels in healthy children. Cytokine 2012, 60, 34–37. [Google Scholar] [CrossRef][Green Version]
- Mahabee-Gittens, E.M.; Merianos, A.L.; Fulkerson, P.C.; Stone, L.; Matt, G.E. The Association of Environmental Tobacco Smoke Exposure and Inflammatory Markers in Hospitalized Children. Int. J. Environ. Res. Public Health 2019, 16, 4625. [Google Scholar] [CrossRef]
- Riis, J.L.; Granger, D.A.; DiPietro, J.A.; Bandeen-Roche, K.; Johnson, S.B. Salivary cytokines as a minimally-invasive measure of immune functioning in young children: Correlates of individual differences and sensitivity to laboratory stress. Dev. Psychobiol. 2015, 57, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Feleszko, W.; Zawadzka-Krajewska, A.; Matysiak, K.; Lewandowska, D.; Peradzynska, J.; Dinh, Q.T.; Hamelmann, E.; Groneberg, D.A.; Kulus, M. Parental tobacco smoking is associated with augmented IL-13 secretion in children with allergic asthma. J. Allergy Clin. Immunol. 2006, 117, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, L.A.; Laitinen, A.; Haahtela, T.; Vilkka, V.; Spur, B.W.; Lee, T.H. Leukotriene E4 and granulocytic infiltration into asthmatic airways. Lancet 1993, 341, 989–990. [Google Scholar] [CrossRef]
- Kumlin, M. Measurement of leukotrienes in humans. Am. J. Respir. Crit. Care Med. 2000, 161, S102–S106. [Google Scholar] [CrossRef]
- Lazarus, S.C.; Chinchilli, V.M.; Rollings, N.J.; Boushey, H.A.; Cherniack, R.; Craig, T.J.; Deykin, A.; DiMango, E.; Fish, J.E.; Ford, J.G.; et al. Smoking affects response to inhaled corticosteroids or leukotriene receptor antagonists in asthma. Am. J. Respir. Crit. Care Med. 2007, 175, 783–790. [Google Scholar] [CrossRef]
- Rabinovitch, N.; Strand, M.; Stuhlman, K.; Gelfand, E.W. Exposure to tobacco smoke increases leukotriene E4-related albuterol usage and response to montelukast. J. Allergy Clin. Immunol. 2008, 121, 1365–1371. [Google Scholar] [CrossRef]
- Rabinovitch, N.; Reisdorph, N.; Silveira, L.; Gelfand, E.W. Urinary leukotriene E(4) levels identify children with tobacco smoke exposure at risk for asthma exacerbation. J. Allergy Clin. Immunol. 2011, 128, 323–327. [Google Scholar] [CrossRef]
- Kott, K.S.; Salt, B.H.; McDonald, R.J.; Jhawar, S.; Bric, J.M.; Joad, J.P. Effect of secondhand cigarette smoke, RSV bronchiolitis and parental asthma on urinary cysteinyl LTE4. Pediatr. Pulmonol. 2008, 43, 760–766. [Google Scholar] [CrossRef]
- Chilmonczyk, B.A.; Salmun, L.M.; Megathlin, K.N.; Neveux, L.M.; Palomaki, G.E.; Knight, G.J.; Pulkkinen, A.J.; Haddow, J.E. Association between exposure to environmental tobacco smoke and exacerbations of asthma in children. N. Engl. J. Med. 1993, 328, 1665–1669. [Google Scholar] [CrossRef]
- Lang, J.E.; Dozor, A.J.; Holbrook, J.T.; Mougey, E.; Krishnan, S.; Sweeten, S.; Wise, R.A.; Teague, W.G.; Wei, C.Y.; Shade, D.; et al. Biologic mechanisms of environmental tobacco smoke in children with poorly controlled asthma: Results from a multicenter clinical trial. J. Allergy Clin. Immunol. Pract. 2013, 1, 172–180. [Google Scholar] [CrossRef]
- Hernandez-Alvidrez, E.; Alba-Reyes, G.; Munoz-Cedillo, B.C.; Arreola-Ramirez, J.L.; Furuya, M.E.; Becerril-Angeles, M.; Vargas, M.H. Passive smoking induces leukotriene production in children: Influence of asthma. J. Asthma 2013, 50, 347–353. [Google Scholar] [CrossRef]
- Gill, R.; Krishnan, S.; Dozor, A.J. Low-level environmental tobacco smoke exposure and inflammatory biomarkers in children with asthma. J. Asthma 2014, 51, 355–359. [Google Scholar] [CrossRef]
- Marmarinos, A.; Saxoni-Papageorgiou, P.; Cassimos, D.; Manoussakis, E.; Tsentidis, C.; Doxara, A.; Paraskakis, I.; Gourgiotis, D. Urinary leukotriene E4 levels in atopic and non-atopic preschool children with recurrent episodic (viral) wheezing: A potential marker? J. Asthma 2015, 52, 554–559. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Tsai, M.H.; Yao, T.C.; Tu, Y.L.; Hua, M.C.; Yeh, K.W.; Huang, J.L. Urinary LTE4 levels as a diagnostic marker for IgE-mediated asthma in preschool children: A birth cohort study. PLoS ONE 2014, 9, e115216. [Google Scholar] [CrossRef]
- Hagan, J.B.; Laidlaw, T.M.; Divekar, R.; O’Brien, E.K.; Kita, H.; Volcheck, G.W.; Hagan, C.R.; Lal, D.; Teaford, H.G., 3rd; Erwin, P.J.; et al. Urinary Leukotriene E4 to Determine Aspirin Intolerance in Asthma: A Systematic Review and Meta-Analysis. J. Allergy Clin. Immunol. Pract. 2017, 5, 990–997. [Google Scholar] [CrossRef]
- Hoffman, B.C.; Rabinovitch, N. Urinary Leukotriene E4 as a Biomarker of Exposure, Susceptibility, and Risk in Asthma: An Update. Immunol. Allergy Clin. 2018, 38, 599–610. [Google Scholar] [CrossRef]
- Taal, H.R.; Geelhoed, J.J.; Steegers, E.A.; Hofman, A.; Moll, H.A.; Lequin, M.; van der Heijden, A.J.; Jaddoe, V.W. Maternal smoking during pregnancy and kidney volume in the offspring: The Generation R Study. Pediatr. Nephrol. 2011, 26, 1275–1283. [Google Scholar] [CrossRef]
- Hogan, S.L.; Vupputuri, S.; Guo, X.; Cai, J.; Colindres, R.E.; Heiss, G.; Coresh, J. Association of cigarette smoking with albuminuria in the United States: The third National Health and Nutrition Examination Survey. Ren. Fail. 2007, 29, 133–142. [Google Scholar] [CrossRef]
- Omoloja, A.; Chand, D.; Greenbaum, L.; Wilson, A.; Bastian, V.; Ferris, M.; Bernert, J.; Stolfi, A.; Patel, H. Cigarette smoking and second-hand smoking exposure in adolescents with chronic kidney disease: A study from the Midwest Pediatric Nephrology Consortium. Nephrol. Dial. Transplant. 2011, 26, 908–913. [Google Scholar] [CrossRef][Green Version]
- Garcia-Esquinas, E.; Loeffler, L.F.; Weaver, V.M.; Fadrowski, J.J.; Navas-Acien, A. Kidney function and tobacco smoke exposure in US adolescents. Pediatrics 2013, 131, e1415–e1423. [Google Scholar] [CrossRef]
- Omoloja, A.; Jerry-Fluker, J.; Ng, D.K.; Abraham, A.G.; Furth, S.; Warady, B.A.; Mitsnefes, M. Secondhand smoke exposure is associated with proteinuria in children with chronic kidney disease. Pediatr. Nephrol. 2013, 28, 1243–1251. [Google Scholar] [CrossRef]
- Kooijman, M.N.; Bakker, H.; Franco, O.H.; Hofman, A.; Taal, H.R.; Jaddoe, V.W. Fetal Smoke Exposure and Kidney Outcomes in School-Aged Children. Am. J. Kidney Dis. 2015, 66, 412–420. [Google Scholar] [CrossRef]
- He, J.; Vupputuri, S.; Allen, K.; Prerost, M.R.; Hughes, J.; Whelton, P.K. Passive smoking and the risk of coronary heart disease—A meta-analysis of epidemiologic studies. N. Engl. J. Med. 1999, 340, 920–926. [Google Scholar] [CrossRef]
- Torok, J.; Gvozdjakova, A.; Kucharska, J.; Balazovjech, I.; Kysela, S.; Simko, F.; Gvozdjak, J. Passive smoking impairs endothelium-dependent relaxation of isolated rabbit arteries. Physiol. Res. 2000, 49, 135–141. [Google Scholar]
- Hackshaw, A.; Rodeck, C.; Boniface, S. Maternal smoking in pregnancy and birth defects: A systematic review based on 173 687 malformed cases and 11.7 million controls. Hum. Reprod. Update 2011, 17, 589–604. [Google Scholar] [CrossRef]
- Groner, J.A.; Huang, H.; Nagaraja, H.; Kuck, J.; Bauer, J.A. Secondhand smoke exposure and endothelial stress in children and adolescents. Acad. Pediatr. 2015, 15, 54–60. [Google Scholar] [CrossRef]
- Raitakari, O.T.; Juonala, M.; Kahonen, M.; Taittonen, L.; Laitinen, T.; Maki-Torkko, N.; Jarvisalo, M.J.; Uhari, M.; Jokinen, E.; Ronnemaa, T.; et al. Cardiovascular risk factors in childhood and carotid artery intima-media thickness in adulthood: The Cardiovascular Risk in Young Finns Study. JAMA 2003, 290, 2277–2283. [Google Scholar] [CrossRef]
- Gunes, T.; Koklu, E.; Yikilmaz, A.; Ozturk, M.A.; Akcakus, M.; Kurtoglu, S.; Coskun, A.; Koklu, S. Influence of maternal smoking on neonatal aortic intima-media thickness, serum IGF-I and IGFBP-3 levels. Eur. J. Pediatr. 2007, 166, 1039–1044. [Google Scholar] [CrossRef]
- Geerts, C.C.; Bots, M.L.; van der Ent, C.K.; Grobbee, D.E.; Uiterwaal, C.S. Parental smoking and vascular damage in their 5-year-old children. Pediatrics 2012, 129, 45–54. [Google Scholar] [CrossRef]
- Gall, S.; Huynh, Q.L.; Magnussen, C.G.; Juonala, M.; Viikari, J.S.; Kahonen, M.; Dwyer, T.; Raitakari, O.T.; Venn, A. Exposure to parental smoking in childhood or adolescence is associated with increased carotid intima-media thickness in young adults: Evidence from the Cardiovascular Risk in Young Finns study and the Childhood Determinants of Adult Health Study. Eur. Heart J. 2014, 35, 2484–2491. [Google Scholar] [CrossRef]
- Groh, C.A.; Vittinghoff, E.; Benjamin, E.J.; Dupuis, J.; Marcus, G.M. Childhood Tobacco Smoke Exposure and Risk of Atrial Fibrillation in Adulthood. J. Am. Coll. Cardiol. 2019, 74, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Black, S.; Kushner, I.; Samols, D. C-reactive Protein. J. Biol. Chem. 2004, 279, 48487–48490. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.G.; Mendall, M.A.; Whincup, P.H.; Carey, I.M.; Ballam, L.; Morris, J.E.; Miller, G.J.; Strachan, D.P. C-reactive protein concentration in children: Relationship to adiposity and other cardiovascular risk factors. Atherosclerosis 2000, 149, 139–150. [Google Scholar] [CrossRef]
- Wilkinson, J.D.; Lee, D.J.; Arheart, K.L. Secondhand smoke exposure and C-reactive protein levels in youth. Nicotine Tob. Res. 2007, 9, 305–307. [Google Scholar] [CrossRef]
- Nagel, G.; Arnold, F.J.; Wilhelm, M.; Link, B.; Zoellner, I.; Koenig, W. Environmental tobacco smoke and cardiometabolic risk in young children: Results from a survey in south-west Germany. Eur. Heart J. 2009, 30, 1885–1893. [Google Scholar] [CrossRef]
- Kang, E.; Kim, S.Y.; Chang, S.S.; Lim, S.; Kim, H.C.; Lee, C.G.; Kim, Y.M.; Kim, S.Y.; Lee, K.J.; Kim, S.; et al. Environmental Tobacco Smoke Exposure at Home and High-Sensitivity C-Reactive Protein Levels in Three-to-Five-Year-Old Children. Int. J. Environ. Res. Public Health 2017, 14, 1105. [Google Scholar] [CrossRef]
- Wang, D.; Juonala, M.; Viikari, J.S.A.; Wu, F.; Hutri-Kahonen, N.; Raitakari, O.T.; Magnussen, C.G. Exposure to Parental Smoking in Childhood is Associated with High C-Reactive Protein in Adulthood: The Cardiovascular Risk in Young Finns Study. J. Atheroscler. Thromb. 2017, 24, 1231–1241. [Google Scholar] [CrossRef]
- Palomares, O.; Yaman, G.; Azkur, A.K.; Akkoc, T.; Akdis, M.; Akdis, C.A. Role of Treg in immune regulation of allergic diseases. Eur. J. Immunol. 2010, 40, 1232–1240. [Google Scholar] [CrossRef]
- Hinz, D.; Bauer, M.; Roder, S.; Olek, S.; Huehn, J.; Sack, U.; Borte, M.; Simon, J.C.; Lehmann, I.; Herberth, G.; et al. Cord blood Tregs with stable FOXP3 expression are influenced by prenatal environment and associated with atopic dermatitis at the age of one year. Allergy 2012, 67, 380–389. [Google Scholar] [CrossRef]
- Herberth, G.; Bauer, M.; Gasch, M.; Hinz, D.; Roder, S.; Olek, S.; Kohajda, T.; Rolle-Kampczyk, U.; von Bergen, M.; Sack, U.; et al. Maternal and cord blood miR-223 expression associates with prenatal tobacco smoke exposure and low regulatory T-cell numbers. J. Allergy Clin. Immunol. 2014, 133, 543–550. [Google Scholar] [CrossRef]
- Hedrick, S.M.; Hess Michelini, R.; Doedens, A.L.; Goldrath, A.W.; Stone, E.L. FOXO transcription factors throughout T cell biology. Nat. Rev. Immunol. 2012, 12, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, C.J.; Kim, J.S.; Lee, D.C.; Ahn, S.; Yoon, B.H. Increased miR-223 expression in foetal organs is a signature of acute chorioamnionitis with systemic consequences. J. Cell. Mol. Med. 2018, 22, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Wang, W.; Liu, Q. Passive smoking induces pediatric asthma by affecting the balance of Treg/Th17 cells. Pediatr. Res. 2019, 85, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.H.; Shi, G.C.; Wan, H.Y.; Jiang, L.H.; Ai, X.Y.; Zhu, H.X.; Tang, W.; Ma, J.Y.; Jin, X.Y.; Zhang, B.Y. Coexistence of Th1/Th2 and Th17/Treg imbalances in patients with allergic asthma. Chin. Med. J. 2011, 124, 1951–1956. [Google Scholar] [PubMed]
- Vardavas, C.I.; Plada, M.; Tzatzarakis, M.; Marcos, A.; Warnberg, J.; Gomez-Martinez, S.; Breidenassel, C.; Gonzalez-Gross, M.; Tsatsakis, A.M.; Saris, W.H.; et al. Passive smoking alters circulating naive/memory lymphocyte T-cell subpopulations in children. Pediatr. Allergy Immunol. 2010, 21, 1171–1178. [Google Scholar] [CrossRef]
- Marseglia, G.L.; Avanzini, M.A.; Caimmi, S.; Caimmi, D.; Marseglia, A.; Valsecchi, C.; Poddighe, D.; Ciprandi, G.; Pagella, F.; Klersy, C.; et al. Passive exposure to smoke results in defective interferon-gamma production by adenoids in children with recurrent respiratory infections. J. Interferon Cytokine Res. 2009, 29, 427–432. [Google Scholar] [CrossRef]
- Tagliacarne, S.C.; Valsecchi, C.; Castellazzi, A.M.; Licari, A.; Klersy, C.; Montagna, L.; Castagnoli, R.; Benazzo, M.; Ciprandi, G.; Marseglia, G.L. Impact of passive smoke and/or atopy on adenoid immunoglobulin production in children. Immunol. Lett. 2015, 165, 70–77. [Google Scholar] [CrossRef]
- Yao, T.C.; Chang, S.W.; Hua, M.C.; Liao, S.L.; Tsai, M.H.; Lai, S.H.; Tseng, Y.L.; Yeh, K.W.; Tsai, H.J.; Huang, J.L.; et al. Tobacco smoke exposure and multiplexed immunoglobulin E sensitization in children: A population-based study. Allergy 2016, 71, 90–98. [Google Scholar] [CrossRef]
- Thacher, J.D.; Gruzieva, O.; Pershagen, G.; Neuman, A.; van Hage, M.; Wickman, M.; Kull, I.; Melen, E.; Bergstrom, A. Parental smoking and development of allergic sensitization from birth to adolescence. Allergy 2016, 71, 239–248. [Google Scholar] [CrossRef]
- Feleszko, W.; Ruszczynski, M.; Jaworska, J.; Strzelak, A.; Zalewski, B.M.; Kulus, M. Environmental tobacco smoke exposure and risk of allergic sensitisation in children: A systematic review and meta-analysis. Arch. Dis. Child. 2014, 99, 985–992. [Google Scholar] [CrossRef]
- Hirata, K.; Yamano, Y.; Suzuki, H.; Miyagawa, S.; Nakadate, T. Passive smoking is associated with lower serum HDL-C levels in school children. Pediatr. Int. 2010, 52, 252–256. [Google Scholar] [CrossRef] [PubMed]
- El-Hodhod, M.A.; Hamdy, A.M.; Ahmed, M.B.; Youssef, S.R.; Aly, S.M. Effect of passive smoking on blood lymphocyte apoptosis in children. Eur. J. Clin. Investig. 2011, 41, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Palmer, P.H.; Pang, Z.; Sun, P.; Duan, H.; Johnson, C.A. Environmental tobacco use and indicators of metabolic syndrome in Chinese adults. Nicotine Tob. Res. 2010, 12, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Groner, J.A.; Huang, H.; Joshi, M.S.; Eastman, N.; Nicholson, L.; Bauer, J.A. Secondhand Smoke Exposure and Preclinical Markers of Cardiovascular Risk in Toddlers. J. Pediatr. 2017, 189, 155–161. [Google Scholar] [CrossRef]
- Ayer, J.G.; Belousova, E.; Harmer, J.A.; David, C.; Marks, G.B.; Celermajer, D.S. Maternal cigarette smoking is associated with reduced high-density lipoprotein cholesterol in healthy 8-year-old children. Eur. Heart J. 2011, 32, 2446–2453. [Google Scholar] [CrossRef]
- Le-Ha, C.; Beilin, L.J.; Burrows, S.; Huang, R.C.; Oddy, W.H.; Hands, B.; Mori, T.A. Gender difference in the relationship between passive smoking exposure and HDL-cholesterol levels in late adolescence. J. Clin. Endocrinol. Metab. 2013, 98, 2126–2135. [Google Scholar] [CrossRef]
- Kallio, K.; Jokinen, E.; Saarinen, M.; Hamalainen, M.; Volanen, I.; Kaitosaari, T.; Ronnemaa, T.; Viikari, J.; Raitakari, O.T.; Simell, O. Arterial intima-media thickness, endothelial function, and apolipoproteins in adolescents frequently exposed to tobacco smoke. Circ. Cardiovasc. Qual. Outcomes 2010, 3, 196–203. [Google Scholar] [CrossRef]
- Cupul-Uicab, L.A.; Skjaerven, R.; Haug, K.; Travlos, G.S.; Wilson, R.E.; Eggesbo, M.; Hoppin, J.A.; Whitworth, K.W.; Longnecker, M.P. Exposure to tobacco smoke in utero and subsequent plasma lipids, ApoB, and CRP among adult women in the MoBa cohort. Environ. Health Perspect. 2012, 120, 1532–1537. [Google Scholar] [CrossRef]
- Rolle-Kampczyk, U.E.; Krumsiek, J.; Otto, W.; Roder, S.W.; Kohajda, T.; Borte, M.; Theis, F.; Lehmann, I.; von Bergen, M. Metabolomics reveals effects of maternal smoking on endogenous metabolites from lipid metabolism in cord blood of newborns. Metabolomics 2016, 12, 76. [Google Scholar] [CrossRef]
- Zakhar, J.; Amrock, S.M.; Weitzman, M. Passive and Active Tobacco Exposure and Children’s Lipid Profiles. Nicotine Tob. Res. 2016, 18, 982–987. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Free radicals, antioxidants, and human disease: Curiosity, cause, or consequence? Lancet 1994, 344, 721–724. [Google Scholar] [CrossRef]
- Sahiner, U.M.; Birben, E.; Erzurum, S.; Sackesen, C.; Kalayci, O. Oxidative stress in asthma: Part of the puzzle. Pediatr. Allergy Immunol. 2018, 29, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Raghuveer, G.; White, D.A.; Hayman, L.L.; Woo, J.G.; Villafane, J.; Celermajer, D.; Ward, K.D.; de Ferranti, S.D.; Zachariah, J.; American Heart Association Committee on Atherosclerosis, Hypertension, and Obesity in the Young of the Council on Cardiovascular Disease in the Young; et al. Cardiovascular Consequences of Childhood Secondhand Tobacco Smoke Exposure: Prevailing Evidence, Burden, and Racial and Socioeconomic Disparities: A Scientific Statement From the American Heart Association. Circulation 2016, 134, e336–e359. [Google Scholar] [CrossRef]
- Perrone, S.; Tataranno, M.L.; Stazzoni, G.; Buonocore, G. Biomarkers of oxidative stress in fetal and neonatal diseases. J. Matern. Fetal Neonatal Med. 2012, 25, 2575–2578. [Google Scholar] [CrossRef]
- Bono, R.; Bellisario, V.; Romanazzi, V.; Pirro, V.; Piccioni, P.; Pazzi, M.; Bugiani, M.; Vincenti, M. Oxidative stress in adolescent passive smokers living in urban and rural environments. Int. J. Hyg. Environ. Health 2014, 217, 287–293. [Google Scholar] [CrossRef]
- Squillacioti, G.; Bellisario, V.; Grignani, E.; Mengozzi, G.; Bardaglio, G.; Dalmasso, P.; Bono, R. The Asti Study: The Induction of Oxidative Stress in A Population of Children According to Their Body Composition and Passive Tobacco Smoking Exposure. Int. J. Environ. Res. Public Health 2019, 16, 490. [Google Scholar] [CrossRef]
- Cahill-Smith, S.; Li, J.M. Oxidative stress, redox signalling and endothelial dysfunction in ageing-related neurodegenerative diseases: A role of NADPH oxidase 2. Br. J. Clin. Pharmacol. 2014, 78, 441–453. [Google Scholar] [CrossRef]
- Loffredo, L.; Zicari, A.M.; Occasi, F.; Perri, L.; Carnevale, R.; Angelico, F.; Del Ben, M.; Martino, F.; Nocella, C.; De Castro, G.; et al. Role of NADPH oxidase-2 and oxidative stress in children exposed to passive smoking. Thorax 2018, 73, 986–988. [Google Scholar] [CrossRef]
- Roberts, C.K.; Sindhu, K.K. Oxidative stress and metabolic syndrome. Life Sci. 2009, 84, 705–712. [Google Scholar] [CrossRef]
- Weitzman, M.; Cook, S.; Auinger, P.; Florin, T.A.; Daniels, S.; Nguyen, M.; Winickoff, J.P. Tobacco smoke exposure is associated with the metabolic syndrome in adolescents. Circulation 2005, 112, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.F.; Clark, M.L.; Bachand, A.; Reynolds, S.J.; Nelson, T.L.; Peel, J.L. Interactions Between Diet and Exposure to Secondhand Smoke on Metabolic Syndrome Among Children: NHANES 2007–2010. J. Clin. Endocrinol. Metab. 2016, 101, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Chelchowska, M.; Ambroszkiewicz, J.; Gajewska, J.; Rowicka, G.; Maciejewski, T.M.; Mazur, J. Cord Blood Adiponectin and Visfatin Concentrations in relation to Oxidative Stress Markers in Neonates Exposed and Nonexposed In Utero to Tobacco Smoke. Oxid. Med. Cell. Longev. 2016, 2016, 4569108. [Google Scholar] [CrossRef] [PubMed]
- Bono, R.; Tassinari, R.; Bellisario, V.; Gilli, G.; Pazzi, M.; Pirro, V.; Mengozzi, G.; Bugiani, M.; Piccioni, P. Urban air and tobacco smoke as conditions that increase the risk of oxidative stress and respiratory response in youth. Environ. Res. 2015, 137, 141–146. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Bossley, C.; Gupta, A.; Akashi, K.; Tsartsali, L.; Mercado, N.; Barnes, P.J.; Bush, A.; Ito, K. Passive smoking impairs histone deacetylase-2 in children with severe asthma. Chest 2014, 145, 305–312. [Google Scholar] [CrossRef]
- Rathkopf, M.M. Passive smoking impairs histone deacetylase-2 in children with severe asthma. Pediatrics 2014, 134 (Suppl. 3), S147–S148. [Google Scholar] [CrossRef]
- Cohen, R.T.; Raby, B.A.; Van Steen, K.; Fuhlbrigge, A.L.; Celedon, J.C.; Rosner, B.A.; Strunk, R.C.; Zeiger, R.S.; Weiss, S.T.; Childhood Asthma Management Program Research Group. In utero smoke exposure and impaired response to inhaled corticosteroids in children with asthma. J. Allergy Clin. Immunol. 2010, 126, 491–497. [Google Scholar] [CrossRef]
- Podlecka, D.; Malewska-Kaczmarek, K.; Jerzynska, J.; Stelmach, W.; Stelmach, I. Secondhand smoke exposure increased the need for inhaled corticosteroids in children with asthma. Ann. Allergy Asthma Immunol. 2018, 121, 119–121. [Google Scholar] [CrossRef]
- Lisboa, P.C.; de Oliveira, E.; de Moura, E.G. Obesity and endocrine dysfunction programmed by maternal smoking in pregnancy and lactation. Front. Physiol. 2012, 3, 437. [Google Scholar] [CrossRef]
- Paslakis, G.; Buchmann, A.F.; Westphal, S.; Banaschewski, T.; Hohm, E.; Zimmermann, U.S.; Laucht, M.; Deuschle, M. Intrauterine exposure to cigarette smoke is associated with increased ghrelin concentrations in adulthood. Neuroendocrinology 2014, 99, 123–129. [Google Scholar] [CrossRef]
- Duskova, M.; Hruskovicova, H.; Simunkova, K.; Starka, L.; Parizek, A. The effects of smoking on steroid metabolism and fetal programming. J. Steroid Biochem. Mol. Biol. 2014, 139, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Gollenberg, A.L.; Addo, O.Y.; Zhang, Z.; Hediger, M.L.; Himes, J.H.; Lee, P.A. In utero exposure to cigarette smoking, environmental tobacco smoke and reproductive hormones in US girls approaching puberty. Horm. Res. Paediatr. 2015, 83, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Kristensen, S.L.; Toft, G.; Thulstrup, A.M.; Hakonsen, L.B.; Olsen, S.F.; Ramlau-Hansen, C.H. Maternal smoking during pregnancy and reproductive health of daughters: A follow-up study spanning two decades. Hum. Reprod. 2012, 27, 3593–3600. [Google Scholar] [CrossRef] [PubMed]
- Ittermann, T.; Thamm, M.; Schipf, S.; John, U.; Rettig, R.; Volzke, H. Relationship of smoking and/or passive exposure to tobacco smoke on the association between serum thyrotropin and body mass index in large groups of adolescents and children. Thyroid 2013, 23, 262–268. [Google Scholar] [CrossRef]
- Filis, P.; Hombach-Klonisch, S.; Ayotte, P.; Nagrath, N.; Soffientini, U.; Klonisch, T.; O’Shaughnessy, P.; Fowler, P.A. Maternal smoking and high BMI disrupt thyroid gland development. BMC Med. 2018, 16, 194. [Google Scholar] [CrossRef]
- Singhal, A.; Farooqi, I.S.; Cole, T.J.; O’Rahilly, S.; Fewtrell, M.; Kattenhorn, M.; Lucas, A.; Deanfield, J. Influence of leptin on arterial distensibility: A novel link between obesity and cardiovascular disease? Circulation 2002, 106, 1919–1924. [Google Scholar] [CrossRef]
- Wilce, M.C.; Parker, M.W. Structure and function of glutathione S-transferases. Biochim. Biophys. Acta 1994, 1205, 1–18. [Google Scholar] [CrossRef]
- Panasevich, S.; Lindgren, C.; Kere, J.; Wickman, M.; Pershagen, G.; Nyberg, F.; Melen, E. Interaction between early maternal smoking and variants in TNF and GSTP1 in childhood wheezing. Clin. Exp. Allergy 2010, 40, 458–467. [Google Scholar] [CrossRef]
- Lee, Y.L.; Lee, Y.C.; Guo, Y.L. Associations of glutathione S-transferase P1, M1, and environmental tobacco smoke with wheezing illness in school children. Allergy 2007, 62, 641–647. [Google Scholar] [CrossRef]
- Palmer, C.N.; Doney, A.S.; Lee, S.P.; Murrie, I.; Ismail, T.; Macgregor, D.F.; Mukhopadhyay, S. Glutathione S-transferase M1 and P1 genotype, passive smoking, and peak expiratory flow in asthma. Pediatrics 2006, 118, 710–716. [Google Scholar] [CrossRef]
- Wu, C.C.; Ou, C.Y.; Chang, J.C.; Hsu, T.Y.; Kuo, H.C.; Liu, C.A.; Wang, C.L.; Chuang, C.J.; Chuang, H.; Liang, H.M.; et al. Gender-dependent effect of GSTM1 genotype on childhood asthma associated with prenatal tobacco smoke exposure. Biomed. Res. Int. 2014, 2014, 769452. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, B.S.; Kwon, S.O.; Oh, S.Y.; Shin, H.L.; Jung, Y.H.; Lee, E.; Yang, S.I.; Kim, H.Y.; Seo, J.H.; et al. Modification of additive effect between vitamins and ETS on childhood asthma risk according to GSTP1 polymorphism: A cross-sectional study. BMC Pulm. Med. 2015, 15, 125. [Google Scholar] [CrossRef] [PubMed]
- Wenten, M.; Li, Y.F.; Lin, P.C.; Gauderman, W.J.; Berhane, K.; Avol, E.; Gilliland, F.D. In utero smoke exposure, glutathione S-transferase P1 haplotypes, and respiratory illness-related absence among schoolchildren. Pediatrics 2009, 123, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.; Francis, B.; Wani, N.; Vijverberg, S.; Pino-Yanes, M.; Mukhopadhyay, S.; Tavendale, R.; Palmer, C.; Burchard, E.G.; Merid, S.K.; et al. Variants in genes coding for glutathione S-transferases and asthma outcomes in children. Pharmacogenomics 2018, 19, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Morales, E.; Sunyer, J.; Julvez, J.; Castro-Giner, F.; Estivill, X.; Torrent, M.; De Cid, R. GSTM1 polymorphisms modify the effect of maternal smoking during pregnancy on cognitive functioning in preschoolers. Int. J. Epidemiol. 2009, 38, 690–697. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Filonzi, L.; Magnani, C.; Lavezzi, A.M.; Vaghi, M.; Nosetti, L.; Nonnis Marzano, F. Detoxification genes polymorphisms in SIDS exposed to tobacco smoke. Gene 2018, 648, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.J.; Guo, Y.L.; Lin, T.J.; Chen, P.C.; Wu, Y.N. GSTM1, GSTP1, prenatal smoke exposure, and atopic dermatitis. Ann. Allerg Asthma Immunol. 2010, 105, 124–129. [Google Scholar] [CrossRef]
- Li, X.; Liu, Z.; Deng, Y.; Li, S.; Mu, D.; Tian, X.; Lin, Y.; Yang, J.; Li, J.; Li, N.; et al. Modification of the association between maternal smoke exposure and congenital heart defects by polymorphisms in glutathione S-transferase genes. Sci. Rep. 2015, 5, 14915. [Google Scholar] [CrossRef]
- Dai, X.; Dharmage, S.C.; Bowatte, G.; Waidyatillake, N.T.; Perret, J.L.; Hui, J.; Erbas, B.; Abramson, M.J.; Lowe, A.J.; Burgess, J.A.; et al. Interaction of Glutathione S-Transferase M1, T1, and P1 Genes With Early Life Tobacco Smoke Exposure on Lung Function in Adolescents. Chest 2019, 155, 94–102. [Google Scholar] [CrossRef]
- Wenten, M.; Berhane, K.; Rappaport, E.B.; Avol, E.; Tsai, W.W.; Gauderman, W.J.; McConnell, R.; Dubeau, L.; Gilliland, F.D. TNF-308 modifies the effect of second-hand smoke on respiratory illness-related school absences. Am. J. Respir. Crit. Care Med. 2005, 172, 1563–1568. [Google Scholar] [CrossRef]
- Salam, M.T.; Gauderman, W.J.; McConnell, R.; Lin, P.C.; Gilliland, F.D. Transforming growth factor- 1 C-509T polymorphism, oxidant stress, and early-onset childhood asthma. Am. J. Respir. Crit. Care Med. 2007, 176, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Bernstein, D.I.; LeMasters, G.K.; Huey, N.L.; Ericksen, M.; Villareal, M.; Lockey, J.; Khurana Hershey, G.K. Environmental tobacco smoke and interleukin 4 polymorphism (C-589T) gene: Environment interaction increases risk of wheezing in African-American infants. J. Pediatr. 2008, 152, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Sadeghnejad, A.; Karmaus, W.; Arshad, S.H.; Kurukulaaratchy, R.; Huebner, M.; Ewart, S. IL13 gene polymorphisms modify the effect of exposure to tobacco smoke on persistent wheeze and asthma in childhood, a longitudinal study. Respir. Res. 2008, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.D.; Ramos, R.A.; Tobias, P.S.; Ulevitch, R.J.; Mathison, J.C. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 1990, 249, 1431–1433. [Google Scholar] [CrossRef]
- Bottema, R.W.; Reijmerink, N.E.; Kerkhof, M.; Koppelman, G.H.; Stelma, F.F.; Gerritsen, J.; Thijs, C.; Brunekreef, B.; van Schayck, C.P.; Postma, D.S. Interleukin 13, CD14, pet and tobacco smoke influence atopy in three Dutch cohorts: The allergenic study. Eur. Respir. J. 2008, 32, 593–602. [Google Scholar] [CrossRef]
- Hussein, Y.M.; Shalaby, S.M.; Zidan, H.E.; Sabbah, N.A.; Karam, N.A.; Alzahrani, S.S. CD14 tobacco gene-environment interaction in atopic children. Cell. Immunol. 2013, 285, 31–37. [Google Scholar] [CrossRef]
- Bouzigon, E.; Corda, E.; Aschard, H.; Dizier, M.H.; Boland, A.; Bousquet, J.; Chateigner, N.; Gormand, F.; Just, J.; Le Moual, N.; et al. Effect of 17q21 variants and smoking exposure in early-onset asthma. N. Engl. J. Med. 2008, 359, 1985–1994. [Google Scholar] [CrossRef]
- Flory, J.H.; Sleiman, P.M.; Christie, J.D.; Annaiah, K.; Bradfield, J.; Kim, C.E.; Glessner, J.; Imielinski, M.; Li, H.; Frackelton, E.C.; et al. 17q12-21 variants interact with smoke exposure as a risk factor for pediatric asthma but are equally associated with early-onset versus late-onset asthma in North Americans of European ancestry. J. Allergy Clin. Immunol. 2009, 124, 605–607. [Google Scholar] [CrossRef]
- van der Valk, R.J.; Duijts, L.; Kerkhof, M.; Willemsen, S.P.; Hofman, A.; Moll, H.A.; Smit, H.A.; Brunekreef, B.; Postma, D.S.; Jaddoe, V.W.; et al. Interaction of a 17q12 variant with both fetal and infant smoke exposure in the development of childhood asthma-like symptoms. Allergy 2012, 67, 767–774. [Google Scholar] [CrossRef]
- Kreiner-Moller, E.; Strachan, D.P.; Linneberg, A.; Husemoen, L.L.; Bisgaard, H.; Bonnelykke, K. 17q21 gene variation is not associated with asthma in adulthood. Allergy 2015, 70, 107–114. [Google Scholar] [CrossRef]
- Morita, H.; Nagai, R. Smoking exposure, 17q21 variants, and early-onset asthma. N. Engl. J. Med. 2009, 360, 1255. [Google Scholar] [CrossRef] [PubMed]
- Dizier, M.H.; Bouzigon, E.; Guilloud-Bataille, M.; Siroux, V.; Lemainque, A.; Boland, A.; Lathrop, M.; Demenais, F. Evidence for gene x smoking exposure interactions in a genome-wide linkage screen of asthma and bronchial hyper-responsiveness in EGEA families. Eur. J. Hum. Genet. 2007, 15, 810–815. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dizier, M.H.; Nadif, R.; Margaritte-Jeannin, P.; Barton, S.J.; Sarnowski, C.; Gagne-Ouellet, V.; Brossard, M.; Lavielle, N.; Just, J.; Lathrop, M.; et al. Interaction between the DNAH9 gene and early smoke exposure in bronchial hyperresponsiveness. Eur. Respir. J. 2016, 47, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Dizier, M.H.; Margaritte-Jeannin, P.; Pain, L.; Sarnowski, C.; Brossard, M.; Mohamdi, H.; Lavielle, N.; Babron, M.C.; Just, J.; Lathrop, M.; et al. Interactive effect between ATPase-related genes and early-life tobacco smoke exposure on bronchial hyper-responsiveness detected in asthma-ascertained families. Thorax 2019, 74, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Olivo-Marston, S.E.; Yang, P.; Mechanic, L.E.; Bowman, E.D.; Pine, S.R.; Loffredo, C.A.; Alberg, A.J.; Caporaso, N.; Shields, P.G.; Chanock, S.; et al. Childhood exposure to secondhand smoke and functional mannose binding lectin polymorphisms are associated with increased lung cancer risk. Cancer Epidemiol. Prev. Biomark. 2009, 18, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- Poetsch, M.; Czerwinski, M.; Wingenfeld, L.; Vennemann, M.; Bajanowski, T. A common FMO3 polymorphism may amplify the effect of nicotine exposure in sudden infant death syndrome (SIDS). Int. J. Legal. Med. 2010, 124, 301–306. [Google Scholar] [CrossRef]
- Boneva, R.S.; Botto, L.D.; Moore, C.A.; Yang, Q.; Correa, A.; Erickson, J.D. Mortality associated with congenital heart defects in the United States: Trends and racial disparities, 1979–1997. Circulation 2001, 103, 2376–2381. [Google Scholar] [CrossRef]
- Tang, X.; Hobbs, C.A.; Cleves, M.A.; Erickson, S.W.; MacLeod, S.L.; Malik, S.; the National Birth Defects Prevention Study. Genetic variation affects congenital heart defect susceptibility in offspring exposed to maternal tobacco use. Birth Defects Res. Part A Clin. Mol. Teratol. 2015, 103, 834–842. [Google Scholar] [CrossRef]
- Satokata, I.; Maas, R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat. Genet. 1994, 6, 348–356. [Google Scholar] [CrossRef]
- van den Boogaard, M.J.; de Costa, D.; Krapels, I.P.; Liu, F.; van Duijn, C.; Sinke, R.J.; Lindhout, D.; Steegers-Theunissen, R.P. The MSX1 allele 4 homozygous child exposed to smoking at periconception is most sensitive in developing nonsyndromic orofacial clefts. Hum. Genet. 2008, 124, 525–534. [Google Scholar] [CrossRef]
- NCBI. National Center for Biotechnology Information, U.S. National Library of Medicine, Gene. Available online: https://www.ncbi.nlm.nih.gov/gene/ (accessed on 21 February 2020).
- O’Shaughnessy, P.J.; Monteiro, A.; Bhattacharya, S.; Fowler, P.A. Maternal smoking and fetal sex significantly affect metabolic enzyme expression in the human fetal liver. J. Clin. Endocrinol. Metab. 2011, 96, 2851–2860. [Google Scholar] [CrossRef] [PubMed]
- Drake, A.J.; O’Shaughnessy, P.J.; Bhattacharya, S.; Monteiro, A.; Kerrigan, D.; Goetz, S.; Raab, A.; Rhind, S.M.; Sinclair, K.D.; Meharg, A.A.; et al. In utero exposure to cigarette chemicals induces sex-specific disruption of one-carbon metabolism and DNA methylation in the human fetal liver. BMC Med. 2015, 13, 18. [Google Scholar] [CrossRef] [PubMed]
- Filis, P.; Nagrath, N.; Fraser, M.; Hay, D.C.; Iredale, J.P.; O’Shaughnessy, P.; Fowler, P.A. Maternal Smoking Dysregulates Protein Expression in Second Trimester Human Fetal Livers in a Sex-Specific Manner. J. Clin. Endocrinol. Metab. 2015, 100, E861–E870. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Zamore, P.D. Beginning to understand microRNA function. Cell Res. 2007, 17, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Maccani, M.A.; Avissar-Whiting, M.; Banister, C.E.; McGonnigal, B.; Padbury, J.F.; Marsit, C.J. Maternal cigarette smoking during pregnancy is associated with downregulation of miR-16, miR-21, and miR-146a in the placenta. Epigenetics 2010, 5, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Liu, X.; Millstein, J.; Siegmund, K.D.; Dubeau, L.; Maguire, R.L.; Jim Zhang, J.; Fuemmeler, B.F.; Kollins, S.H.; Hoyo, C.; et al. Self-reported prenatal tobacco smoke exposure, AXL gene-body methylation, and childhood asthma phenotypes. Clin. Epigenet. 2018, 10, 98. [Google Scholar] [CrossRef]
- Mudduluru, G.; Ceppi, P.; Kumarswamy, R.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene 2011, 30, 2888–2899. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, W.; Jing, W. Indoor air pollution aggravates asthma in Chinese children and induces the changes in serum level of miR-155. Int. J. Environ. Health Res. 2019, 29, 22–30. [Google Scholar] [CrossRef]
- Jylhava, J.; Pedersen, N.L.; Hagg, S. Biological Age Predictors. EBioMedicine 2017, 21, 29–36. [Google Scholar] [CrossRef]
- Osorio-Yanez, C.; Clemente, D.B.P.; Maitre, L.; Vives-Usano, M.; Bustamante, M.; Martinez, D.; Casas, M.; Alexander, J.; Thomsen, C.; Chatzi, L.; et al. Early life tobacco exposure and children’s telomere length: The HELIX project. Sci. Total Environ. 2020, 711, 135028. [Google Scholar] [CrossRef]
- Breitling, L.P.; Yang, R.; Korn, B.; Burwinkel, B.; Brenner, H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am. J. Hum. Genet. 2011, 88, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Martino, D.; Prescott, S. Epigenetics and prenatal influences on asthma and allergic airways disease. Chest 2011, 139, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Ganu, R.S.; Harris, R.A.; Collins, K.; Aagaard, K.M. Early origins of adult disease: Approaches for investigating the programmable epigenome in humans, nonhuman primates, and rodents. ILAR J. 2012, 53, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, P.D.; Buss, C.; Entringer, S.; Swanson, J.M. Developmental origins of health and disease: Brief history of the approach and current focus on epigenetic mechanisms. Semin. Reprod. Med. 2009, 27, 358–368. [Google Scholar] [CrossRef]
- Nielsen, C.H.; Larsen, A.; Nielsen, A.L. DNA methylation alterations in response to prenatal exposure of maternal cigarette smoking: A persistent epigenetic impact on health from maternal lifestyle? Arch. Toxicol. 2016, 90, 231–245. [Google Scholar] [CrossRef]
- Breton, C.V.; Byun, H.M.; Wenten, M.; Pan, F.; Yang, A.; Gilliland, F.D. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am. J. Respir. Crit. Care Med. 2009, 180, 462–467. [Google Scholar] [CrossRef]
- Suter, M.; Abramovici, A.; Showalter, L.; Hu, M.; Shope, C.D.; Varner, M.; Aagaard-Tillery, K. In utero tobacco exposure epigenetically modifies placental CYP1A1 expression. Metabolism 2010, 59, 1481–1490. [Google Scholar] [CrossRef]
- Suter, M.; Ma, J.; Harris, A.; Patterson, L.; Brown, K.A.; Shope, C.; Showalter, L.; Abramovici, A.; Aagaard-Tillery, K.M. Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics 2011, 6, 1284–1294. [Google Scholar] [CrossRef]
- Murphy, S.K.; Adigun, A.; Huang, Z.Q.; Overcash, F.; Wang, F.; Jirtle, R.L.; Schildkraut, J.M.; Murtha, A.P.; Iversen, E.S.; Hoyo, C. Gender-specific methylation differences in relation to prenatal exposure to cigarette smoke. Gene 2012, 494, 36–43. [Google Scholar] [CrossRef]
- Wang, I.J.; Chen, S.L.; Lu, T.P.; Chuang, E.Y.; Chen, P.C. Prenatal smoke exposure, DNA methylation, and childhood atopic dermatitis. Clin. Exp. Allergy 2013, 43, 535–543. [Google Scholar] [CrossRef]
- Novakovic, B.; Ryan, J.; Pereira, N.; Boughton, B.; Craig, J.M.; Saffery, R. Postnatal stability, tissue, and time specific effects of AHRR methylation change in response to maternal smoking in pregnancy. Epigenetics 2014, 9, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Joubert, B.R.; Haberg, S.E.; Nilsen, R.M.; Wang, X.; Vollset, S.E.; Murphy, S.K.; Huang, Z.; Hoyo, C.; Midttun, O.; Cupul-Uicab, L.A.; et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ. Health Perspect. 2012, 120, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Markunas, C.A.; Xu, Z.; Harlid, S.; Wade, P.A.; Lie, R.T.; Taylor, J.A.; Wilcox, A.J. Identification of DNA methylation changes in newborns related to maternal smoking during pregnancy. Environ. Health Perspect. 2014, 122, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Zakarya, R.; Adcock, I.; Oliver, B.G. Epigenetic impacts of maternal tobacco and e-vapour exposure on the offspring lung. Clin. Epigenet. 2019, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Den Dekker, H.T.; Burrows, K.; Felix, J.F.; Salas, L.A.; Nedeljkovic, I.; Yao, J.; Rifas-Shiman, S.L.; Ruiz-Arenas, C.; Amin, N.; Bustamante, M.; et al. Newborn DNA-methylation, childhood lung function, and the risks of asthma and COPD across the life course. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Runyon, R.S.; Cachola, L.M.; Rajeshuni, N.; Hunter, T.; Garcia, M.; Ahn, R.; Lurmann, F.; Krasnow, R.; Jack, L.M.; Miller, R.L.; et al. Asthma discordance in twins is linked to epigenetic modifications of T cells. PLoS ONE 2012, 7, e48796. [Google Scholar] [CrossRef]
- Reinius, L.E.; Gref, A.; Saaf, A.; Acevedo, N.; Joerink, M.; Kupczyk, M.; D’Amato, M.; Bergstrom, A.; Melen, E.; Scheynius, A.; et al. DNA methylation in the Neuropeptide S Receptor 1 (NPSR1) promoter in relation to asthma and environmental factors. PLoS ONE 2013, 8, e53877. [Google Scholar] [CrossRef]
- Haghighi, A.; Melka, M.G.; Bernard, M.; Abrahamowicz, M.; Leonard, G.T.; Richer, L.; Perron, M.; Veillette, S.; Xu, C.J.; Greenwood, C.M.; et al. Opioid receptor mu 1 gene, fat intake and obesity in adolescence. Mol. Psychiatry 2014, 19, 63–68. [Google Scholar] [CrossRef]
- Haghighi, A.; Schwartz, D.H.; Abrahamowicz, M.; Leonard, G.T.; Perron, M.; Richer, L.; Veillette, S.; Gaudet, D.; Paus, T.; Pausova, Z. Prenatal exposure to maternal cigarette smoking, amygdala volume, and fat intake in adolescence. JAMA Psychiatry 2013, 70, 98–105. [Google Scholar] [CrossRef]
- Lee, K.W.; Abrahamowicz, M.; Leonard, G.T.; Richer, L.; Perron, M.; Veillette, S.; Reischl, E.; Bouchard, L.; Gaudet, D.; Paus, T.; et al. Prenatal exposure to cigarette smoke interacts with OPRM1 to modulate dietary preference for fat. J. Psychiatry Neurosci. 2015, 40, 38–45. [Google Scholar] [CrossRef][Green Version]
- Oken, E.; Levitan, E.B.; Gillman, M.W. Maternal smoking during pregnancy and child overweight: Systematic review and meta-analysis. Int. J. Obes. 2008, 32, 201–210. [Google Scholar] [CrossRef] [PubMed]
- COPHES. Consortium to Perform Human Biomonitoring on a European Scale. 2012. Available online: http://eu-hbm.info/cophes (accessed on 16 April 2020).
- Joas, R.; Casteleyn, L.; Biot, P.; Kolossa-Gehring, M.; Castano, A.; Angerer, J.; Schoeters, G.; Sepai, O.; Knudsen, L.E.; Joas, A.; et al. Harmonised human biomonitoring in Europe: Activities towards an EU HBM framework. Int. J. Hyg. Environ. Health 2012, 215, 172–175. [Google Scholar] [CrossRef] [PubMed]
- DiMoPEx. Diagnosis, Monitoring and Prevention of Exposure Related Non-Communicable Diseases, DiMoPEx (CA 15129). 2020. Available online: http://dimopex.eu/about/ (accessed on 16 April 2020).
- Budnik, L.T.; Adam, B.; Albin, M.; Banelli, B.; Baur, X.; Belpoggi, F.; Bolognesi, C.; Broberg, K.; Gustavsson, P.; Goen, T.; et al. Diagnosis, monitoring and prevention of exposure-related non-communicable diseases in the living and working environment: DiMoPEx-project is designed to determine the impacts of environmental exposure on human health. J. Occup. Med. Toxicol. 2018, 13, 6. [Google Scholar] [CrossRef] [PubMed]


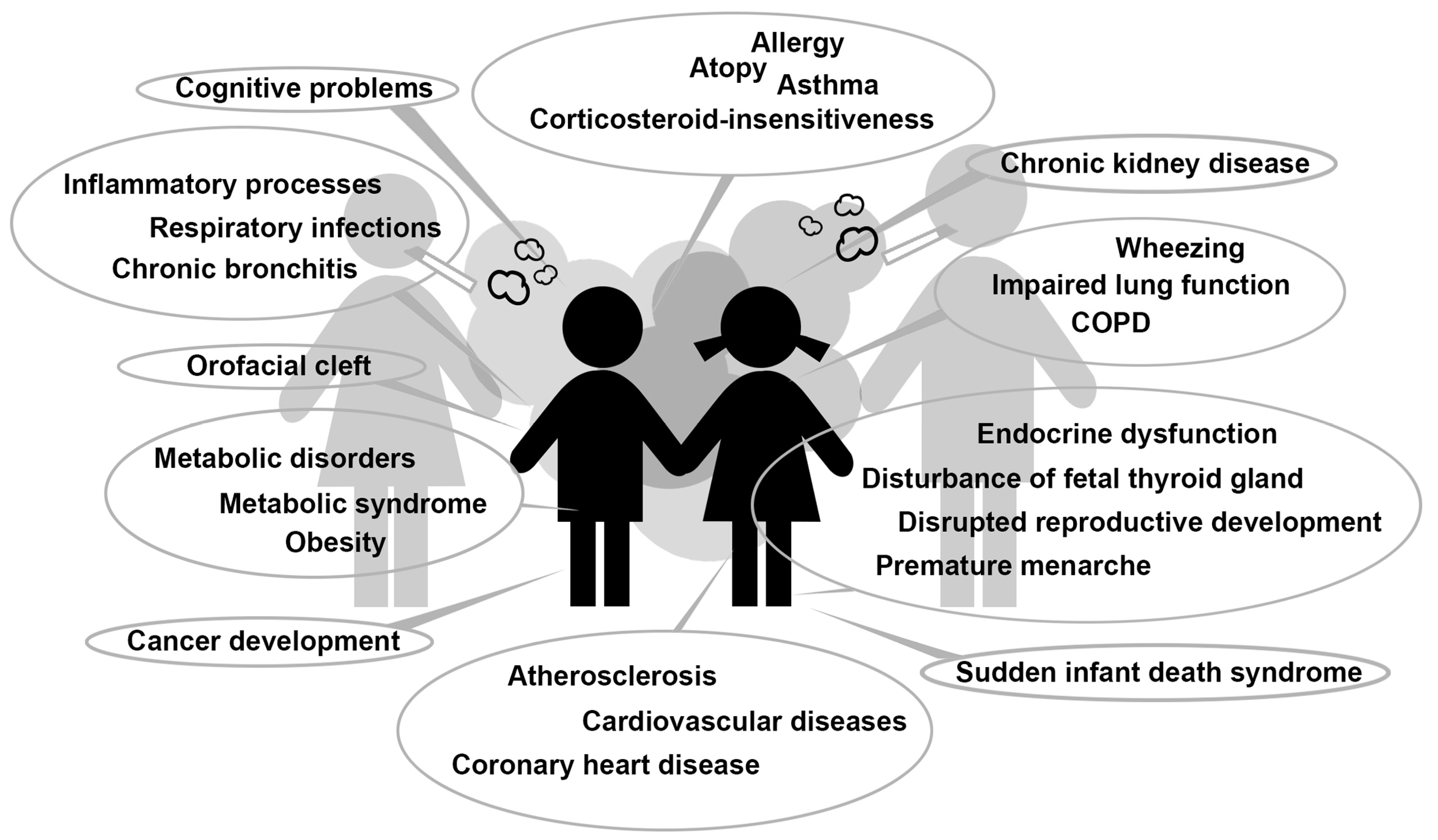{kind=link}
| Topic | Effects on Children | Test Material | Source | Association |
|---|---|---|---|---|
| Biomarkers | ||||
| Matrix metalloproteinase-9 (MMP-9) | MMP-9 increased | Nasal secretion | SHS | Allergy, asthma, chronic bronchitis [26], no effect [29] |
| Cytokine Interleukin (IL) | IL-1β increased | Saliva | SHS | Inflammatory processes [35,36] |
| IL-1β decreased | Blood | SHS | Inflammatory processes [34] | |
| IL-4 decreased | Blood | SHS | Inflammatory processes [34] | |
| IL-5 decreased | Blood | SHS | Inflammatory processes [34] | |
| IL-6 increased | Saliva | SHS | Inflammatory processes [36] | |
| IL-8 increased | New-born dried blood | PTS | Inflammatory processes [33] | |
| IL-8 | Nasal secretion | SHS | no effect by SHS [29] | |
| IL-8 | Saliva | SHS | no effect by SHS [36] | |
| IL-13 increased | Airway secretion | SHS | Inflammatory processes [37] | |
| IL-17 | Nasal secretion | SHS | no effect by SHS [29] | |
| Cytokine Interferon gamma (IFN-γ) | IFN-γ decreased | Blood | SHS | Inflammatory processes [34] |
| Tumour necrosis factor alpha (TNF-α) | TNF-α increased | Saliva | SHS | Inflammatory processes [36] |
| Urinary leukotriene E4 (uLTE4) | uLTE4 increased | Urine | SHS | Asthma [42,43] |
| Estimated glomerular filtration rate (eGFR) | eGFR decreased | Serum | SHS | Kidney function, proteinuria [55,56] |
| Intercellular adhesion molecule 1 (s-ICAM1) | s-ICAM1 increased | Serum | SHS | Endothelial stress [61] |
| Intima-media thickness (IMT) | IMT increased | Ultrasonography | SHS, PTS | Atherosclerosis [64,65] |
| C-reactive protein (CRP) | CRP increased | Serum | SHS | Inflammation response [69,71,72] |
| Immune status | ||||
| Regulatory T-cells (Tregs) | Treg cell number decreased | Cord blood, blood | PTS, SHS | Atopy, asthma [74,75,78] |
| T-helper 17 (Th17) cells | Th17 cell number increased | Blood | SHS | Asthma severity [78] |
| T-cells subsets | Circulating CD3+ and CD4+ memory T-cell number decreased | Blood | SHS | Systemic immunological response [80] |
| Circulating CD3+ and CD4+ naïve T-cell number increased | Blood | SHS | Systemic immunological response [80] | |
| CD4+CD45RA+ T-cell number increased | Blood | SHS | Systemic immunological response [80] | |
| CD8+ T-cell number decreased | Adenoids | SHS | Systemic immunological response [81] | |
| Immunoglobulins A and M (IgA, IgM) | IgA and IgM increased | Adenoids | SHS | Systemic immunological response [82] |
| Immunoglobulin E (IgE) | Immune response to allergens increased | Serum | SHS | Allergy [83,84,85] |
| Lipid profile | ||||
| High-density lipoprotein-cholesterol (HDL-C) | HDL-C decreased | Blood | SHS, PTS | Arteriosclerosis, obesity, metabolic syndrome [86,87,88,89,90,91,93], no effect by SHS [95] |
| Low-density lipoprotein-cholesterol (LDL-C) | LDL-C increased | Blood | SHS, PTS | Arteriosclerosis, obesity, metabolic syndrome [87], no effect by SHS [95] |
| Triglycerides | Triglycerides increased | Blood | SHS, PTS | Arteriosclerosis, obesity, metabolic syndrome [87,88,93], no effect by SHS [95] |
| Apolipoprotein A-1 (ApoA-1) | ApoA-1 decreased | Blood | SHS | Arteriosclerosis, obesity, metabolic syndrome [70] |
| Apolipoprotein B (ApoB) | ApoB increased | Blood | SHS | Arteriosclerosis, obesity, metabolic syndrome [92] |
| Oxidative stress (OS) increased | In general: cell, tissue and organ injury, cell death; asthma, COPD, cardiovascular events, metabolic syndrome [100,101,102,107] | |||
| Nicotinamide adenine dinucleotide phosphate oxidase-2 (Nox2) | Nox2 increased | Serum | SHS | Artery dilation [104] |
| Adiponectin | Adiponectin decreased | Cord blood | PTS | Lipid peroxidation increased [108] |
| Pre-B-cell colony enhancing factor (Visfatin) | Visfatin increased | Cord blood | PTS | Lipid peroxidation increased [108] |
| Urinary 15-F2t-isoprostane | Urinary 15-F2t-isoprostane increased | Urine | SHS | Lower lung function parameters [109] |
| Histone deacetylase-2 (HDAC2) | HDAC2 decreased | Broncho-alveolar lavage fluid | SHS | Corticosteroid-insensitiveness leads to impairment of severe asthma treatment [110] |
| Hormonal changes | In general: Metabolic and endocrine dysfunction (foetal, in childhood, and later life) [114,116] | |||
| Ghrelin | Ghrelin increased until early adulthood by PTS exposure | Plasma | PTS | Metabolic disorders [115] |
| Leptin | Leptin increased | Plasma | SHS | Impairing of vascular function, BMI [70] |
| Adiponectin | Adiponectin decreased | Cord blood | PTS | OS increased, lipid peroxidation increased [108] |
| Luteinizing hormone (LH) | In girls, LH decreased by PTS exposure but increased by current SHS exposure | Blood | PTS, SHS | Reproductive development [117] |
| Inhibin B (InB) | In girls, InB decreased by PTS exposure with no effect by current SHS exposure | Blood | PTS, SHS | Reproductive development [117] |
| Thyrotropin (TSH) | TSH decreased | Serum | SHS | Hypothyroidism, BMI [119] |
| Foetal triiodothyronine (T3), thyroxine (T4) and TSH | T3, T4 and TSH decreased (possibly by downregulation of foetal thyroid transcripts GATA6 and NKX2-1) | Foetal plasma | PTS | Disorder of foetal thyroid development and endocrine function [120] |
| Foetal corticotropin-releasing hormone (CRH), adrenocorticotrophin (ACTH), cortisol, gonadotropins, androgens, oestrogens | Changes in foetal steroidogenesis | Foetal plasma | PTS | Multiple pathophysiological effects (foetal and later in life) by endocrine dysfunction [116] |
| Gene (Chromosome) | SNP | Risk Allele | Association |
|---|---|---|---|
| GSTP1 Exon 5 (11q13) | rs1695 (Val-105 or Ile105Val) | AG (Ile105Val) | Early childhood wheezing [123]; protection against respiratory illness was lost by PTS exposure [128]; no effect in asthma [129] |
| GG (Val105Val) | Asthma [125]; no effect in asthma [129]; current and ever wheezing [124] | ||
| AA (Ile105Ile) | + low vitamin A intake: asthma [127]; no effect in asthma [129]; current wheezing [124]; atopic dermatitis [132]; lung function impairment in later life [134] | ||
| GSTP1 Intron 5 (11q13) | rs749174 | TT | Early childhood wheezing [123] |
| GSTP1 Intron 6 (11q13) | rs1871042 | TT | Early childhood wheezing [123] |
| TNF Promoter (6p21) | rs1800629 (-308) | AA/AG | Respiratory illness [135] |
| TNF Promoter (6p21) | rs1799724 (T-857C) | CC | Early childhood wheezing [123] |
| TNF Intron 1 (6p21) | rs1800610 | CC | Early childhood wheezing [123] |
| TNF Intron 3 (6p21) | rs3093664 | AG/GG | Early childhood wheezing [123] |
| TGFB1 Promoter (19q13) | rs4803457 (C-509T) | TT | Asthma [136] |
| IL-4 (5q31) | rs2243250 (C-589T) | TT/CT | Wheezing [137] |
| IL-13 Exon 4 (5q31) | rs20541 (G/A) | GG | Early onset persistent wheeze and persistent asthma [138] |
| IL-13 haplotype pair (Promoter, Intron 1, Exon 4) (5q31) | rs1800925 (C/T), rs2066960 (C/A), rs20541 (G/A) | CCG/CCG | Early onset persistent wheeze and persistent asthma [138] |
| CD14 (5q31) | 3’untranslated region (UTR) | AA | Lower IgE levels [140] |
| CD14 Promoter (5q31) | rs2569190 (C-159T) | TT | Elevated IgE levels, atopy [141] |
| CD14 (5q31) | C-550T | TT | Elevated IgE levels, atopy [141] |
| IKZF3 Intron 3 (17q21) | rs9303277 | C | Increased risk of early-onset asthma enhanced by SHS [142]; confirmed in Caucasians without age of onset [143] |
| ZPBP2 Exon 2 (17q21) | rs11557467 (I151S) | G | Increased risk of early-onset asthma enhanced by SHS [142]; confirmed in Caucasians without age of onset [143] |
| GSDMB Exon 8 (17q21) | rs2305480 (P298S) | G | Increased risk of early-onset asthma enhanced by SHS [142]; confirmed in Caucasians without age of onset [143]; asthma-like symptoms [144] |
| GSDMB Exon 8 (17q21) | rs2305479 (G291R) | C | Increased risk of early-onset asthma enhanced by SHS [142] |
| GSDMB Intron (17q21) | rs4795400 | C | Increased risk of early-onset asthma enhanced by SHS [142] |
| GSDMB Intron (17q21) | rs9303281 | A | Increased risk of early-onset asthma enhanced by SHS [142] |
| GSDMB Intron 1 (17q21) | rs7219923 | T | Increased risk of early-onset asthma enhanced by SHS [142] |
| GSDMB Intron 2 (17q21) | rs2290400 | C | Increased risk of asthma in Caucasians enhanced by SHS [143] |
| GSDMB Intron 2 (17q21) | rs7216389 | T | Increased risk of asthma in Caucasians enhanced by SHS [143,145] |
| GSDMA Exon 2 (17q21) | rs3894194 | A | Increased risk of asthma in Caucasians enhanced by SHS [143] |
| GSDMA Intron 6 (17q21) | rs3859192 | ? | Increased risk of asthma in Caucasians enhanced by SHS [143] |
| ORMDL3 Intron (17q21) | rs8076131 | A | Increased risk of early-onset asthma enhanced by SHS [142] |
| LRRC3C Intron (17q21) | rs8079416 | ? | Increased risk of asthma in Caucasians enhanced by SHS [143] |
| Intergenic region (17q21) | rs8069176 | G | Increased risk of early-onset asthma enhanced by SHS [142] |
| Intergenic region (17q21) | rs4795405 | C | Increased risk of early-onset asthma enhanced by SHS [142]; confirmed in Caucasians without age of onset [143] |
| Intergenic region (17q21) | rs4794820 | G | Increased risk of early-onset asthma enhanced by SHS [142] |
| Intergenic region (17q21) | rs8067378 | ? | Increased risk of asthma in Caucasians enhanced by SHS [143] |
| DNAH9 Intron (17p11) | rs7225157 | ? | Bronchial hyperresponsiveness [148] |
| ATP8A1 Intron (4p13) | rs17448506 | ? | Bronchial hyperresponsiveness [149] |
| ABCA1 Intron (9q31) | rs2253304 | ? | Bronchial hyperresponsiveness [149] |
| MBL2 (10q21) | rs5030737 | AA | Increased risk of lung cancer in later life [150] |
| MBL2 Intron (10q21) | rs1838066 | CC | Increased risk of lung cancer in later life [150] |
| MBL2 Intron (10q21) | rs7095891 | TT | Increased risk of lung cancer in later life [150] |
| MBL2 (10q21) | rs2165810 | TT | Increased risk of lung cancer in later life [150] |
| FMO3 (1q24) | rs2266782 (G472A) | AA | Risk factor for sudden infant death syndrome [151] |
| OSGEP Intron (14q11) | rs1320150 | AG | Increased risk of congenital heart defects [153] |
| OSGEP Intron (14q11) | rs938881 | ? | Increased risk of congenital heart defects [153] |
| OSGEP (14q11) | rs2275007 | ? | Increased risk of congenital heart defects [153] |
| OSGEP Intron (14q11) | rs883037 | ? | Increased risk of congenital heart defects [153] |
| MSX1 Intron allele 4 (4p16) | Homozygosity of 9 repeats of the A4 CA marker | Increased risk of nonsyndromic orofacial clefts [155] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braun, M.; Klingelhöfer, D.; Oremek, G.M.; Quarcoo, D.; Groneberg, D.A. Influence of Second-Hand Smoke and Prenatal Tobacco Smoke Exposure on Biomarkers, Genetics and Physiological Processes in Children—An Overview in Research Insights of the Last Few Years. Int. J. Environ. Res. Public Health 2020, 17, 3212. https://doi.org/10.3390/ijerph17093212
Braun M, Klingelhöfer D, Oremek GM, Quarcoo D, Groneberg DA. Influence of Second-Hand Smoke and Prenatal Tobacco Smoke Exposure on Biomarkers, Genetics and Physiological Processes in Children—An Overview in Research Insights of the Last Few Years. International Journal of Environmental Research and Public Health. 2020; 17(9):3212. https://doi.org/10.3390/ijerph17093212
Chicago/Turabian StyleBraun, Markus, Doris Klingelhöfer, Gerhard M. Oremek, David Quarcoo, and David A. Groneberg. 2020. "Influence of Second-Hand Smoke and Prenatal Tobacco Smoke Exposure on Biomarkers, Genetics and Physiological Processes in Children—An Overview in Research Insights of the Last Few Years" International Journal of Environmental Research and Public Health 17, no. 9: 3212. https://doi.org/10.3390/ijerph17093212
APA StyleBraun, M., Klingelhöfer, D., Oremek, G. M., Quarcoo, D., & Groneberg, D. A. (2020). Influence of Second-Hand Smoke and Prenatal Tobacco Smoke Exposure on Biomarkers, Genetics and Physiological Processes in Children—An Overview in Research Insights of the Last Few Years. International Journal of Environmental Research and Public Health, 17(9), 3212. https://doi.org/10.3390/ijerph17093212