Cannabidiol Protects Dopaminergic Neuronal Cells from Cadmium

 ,
, 
 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Line
2.2. Treatment
2.3. MTT Assay
2.4. ROS Evaluation
2.5. Western Blotting Analysis
2.6. Immunofluorescent Staining
2.7. Statistical Analysis
3. Results
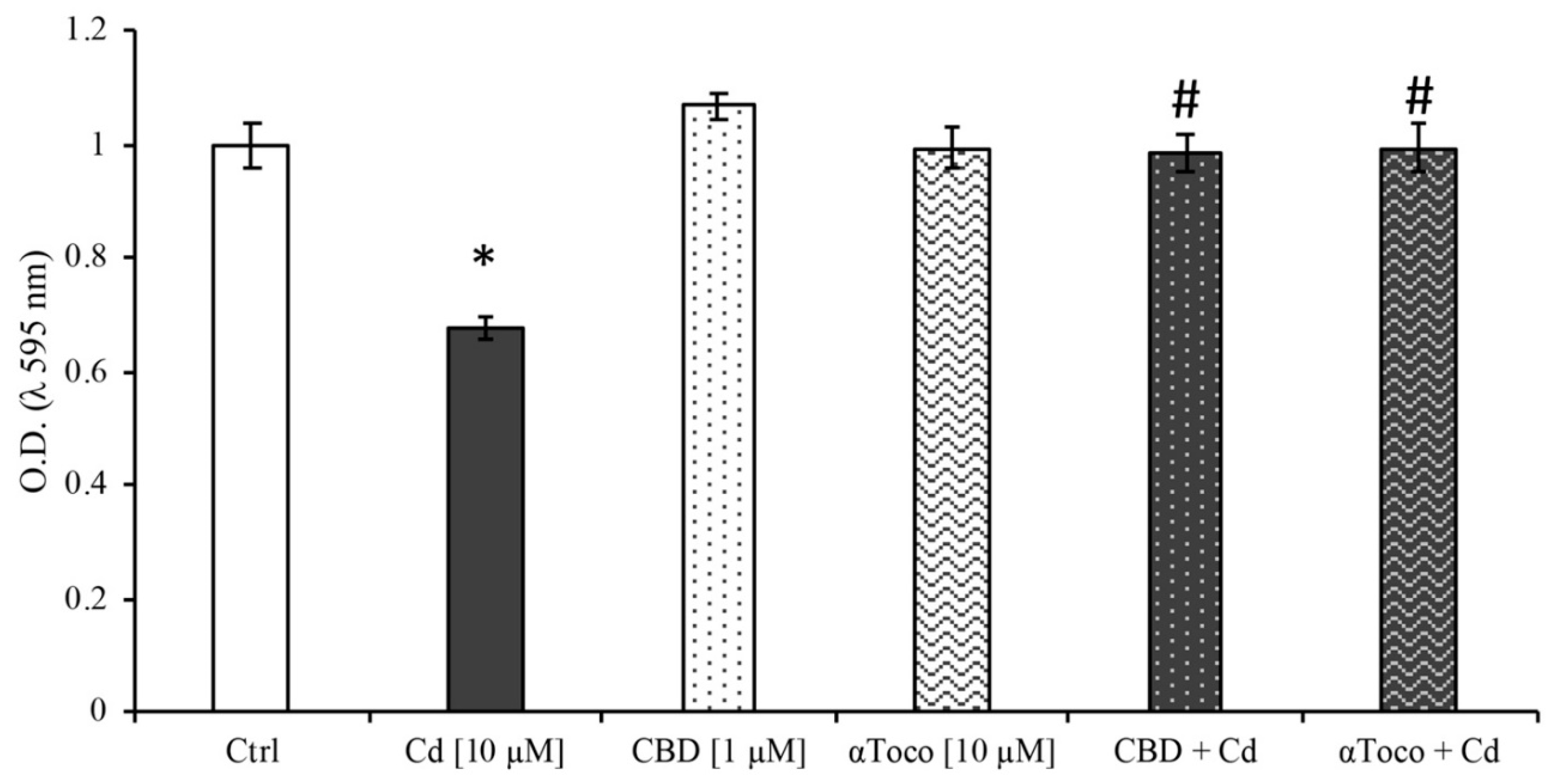
3.1. Cell Viability
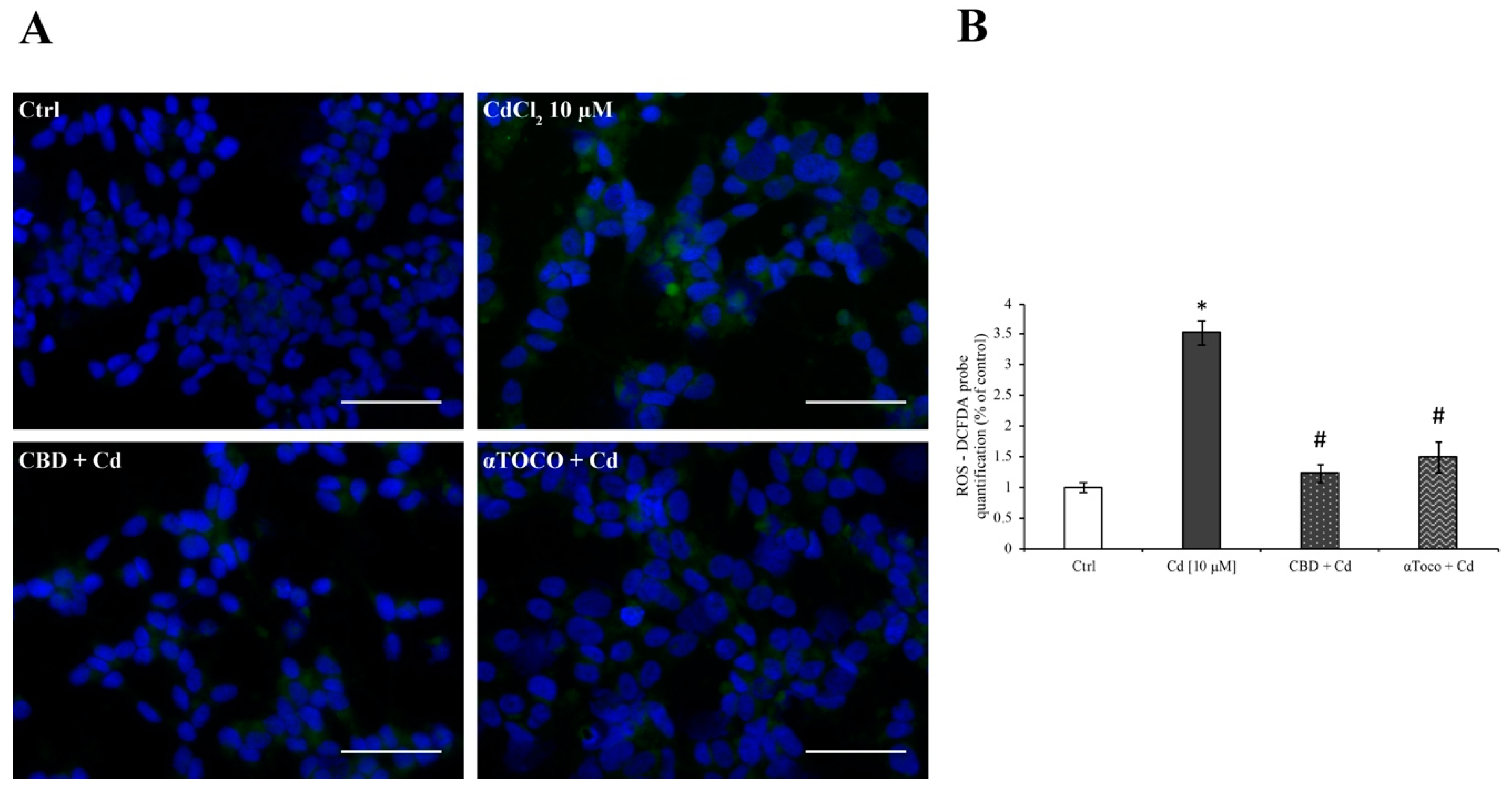
3.2. SH-SY5Y ROS Production
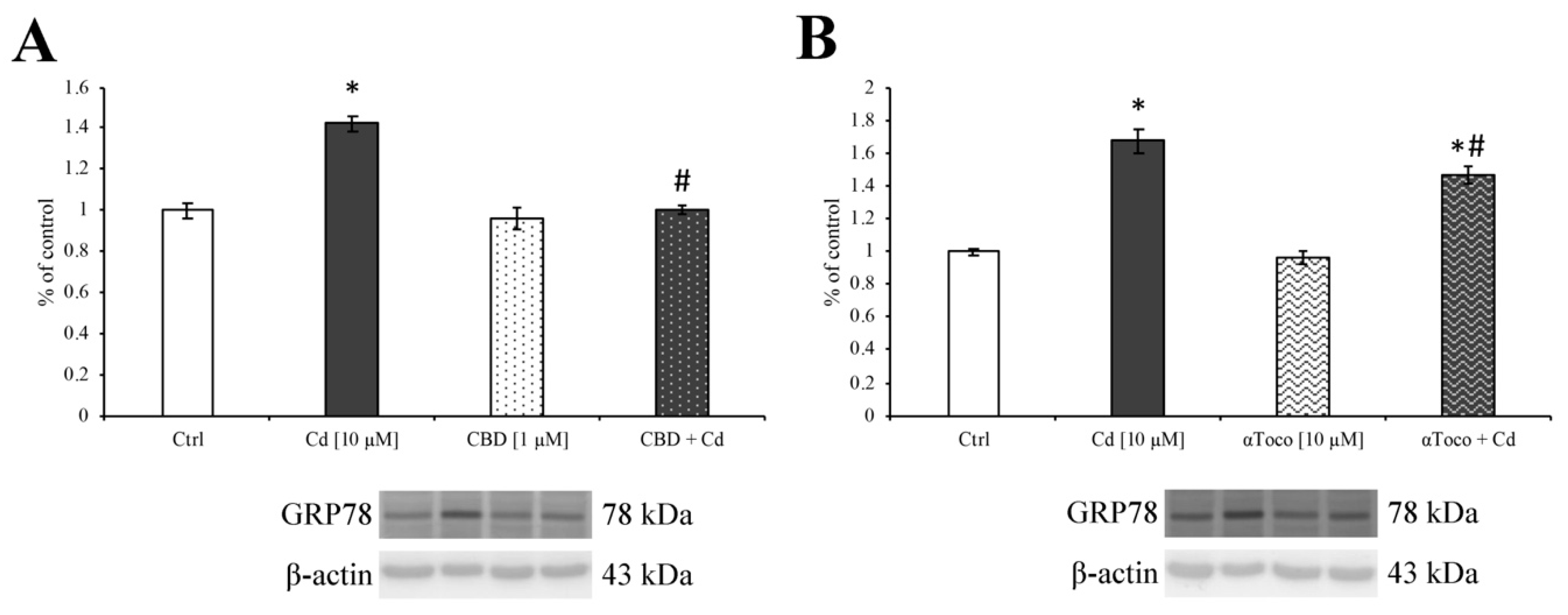
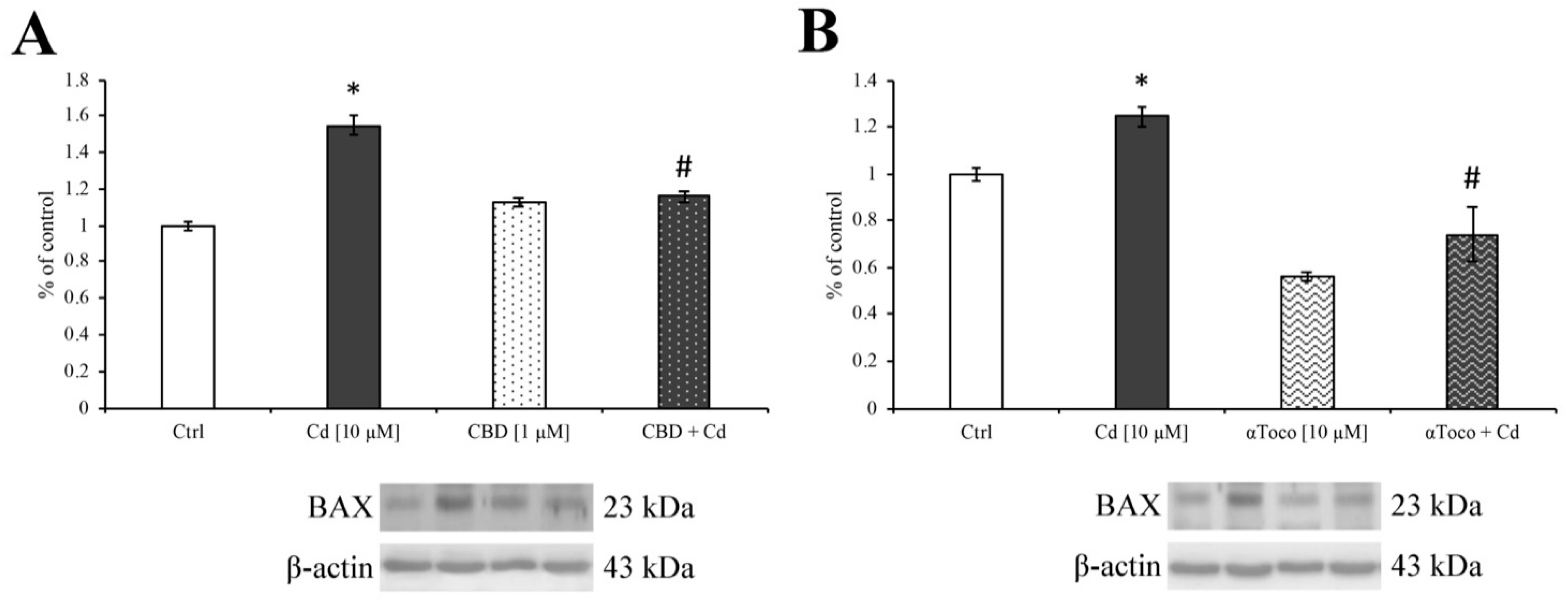
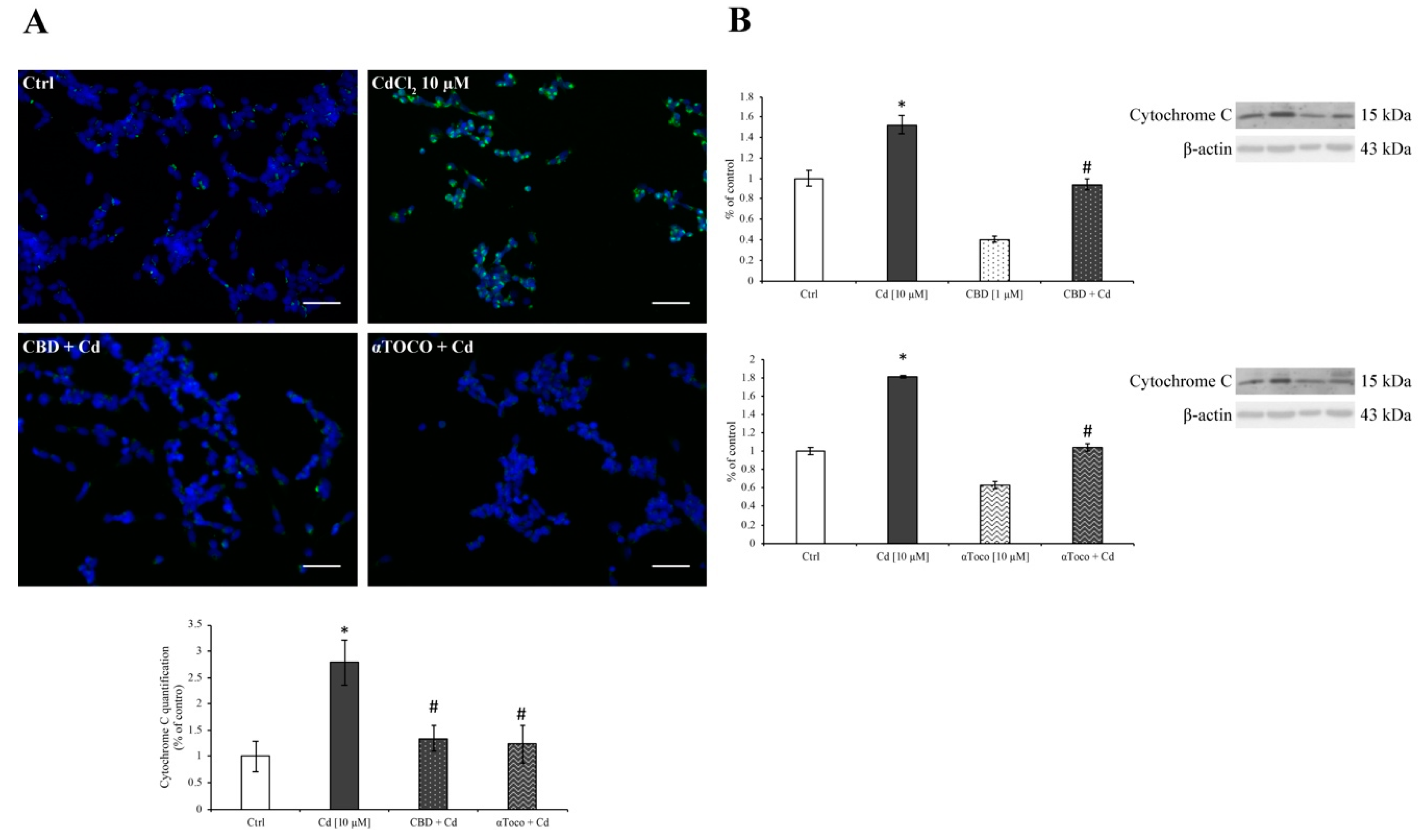
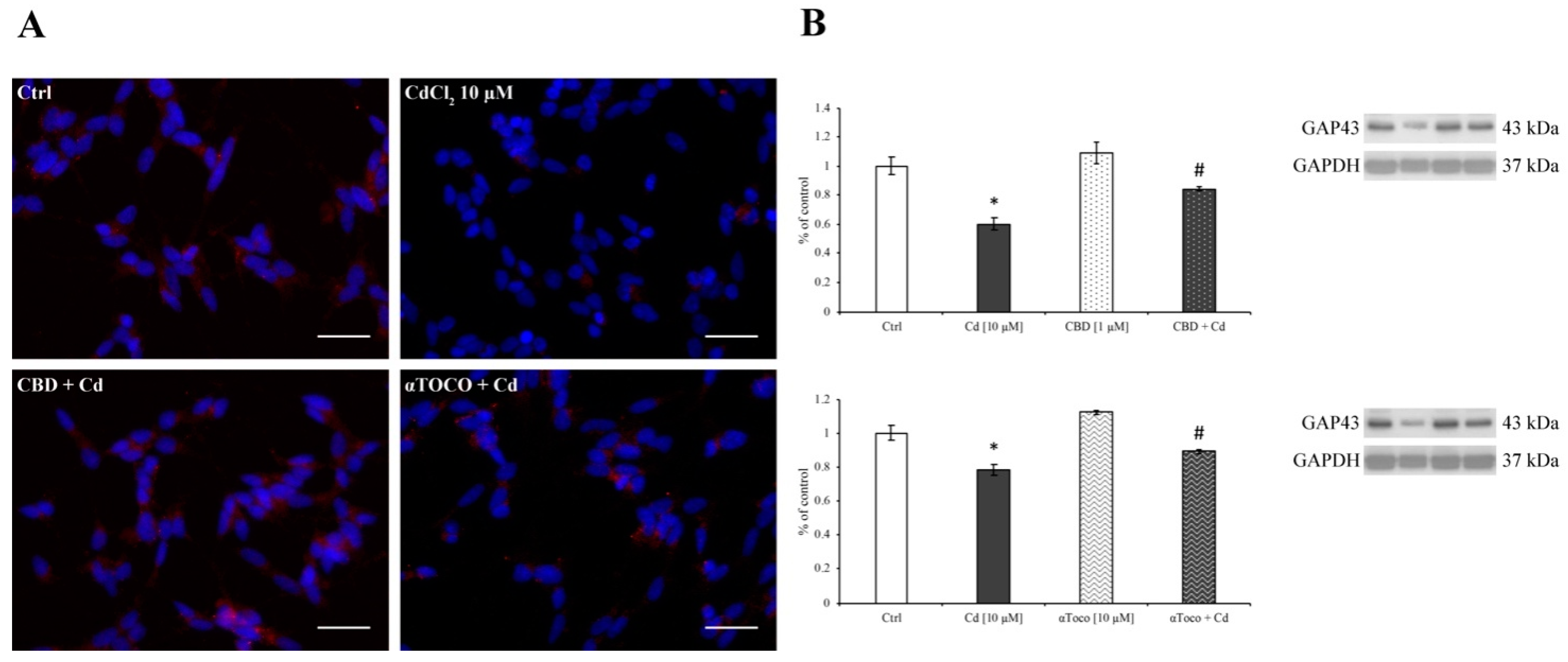
3.3. Cd-Induced ER Stress and Apoptotic Cascade Signaling
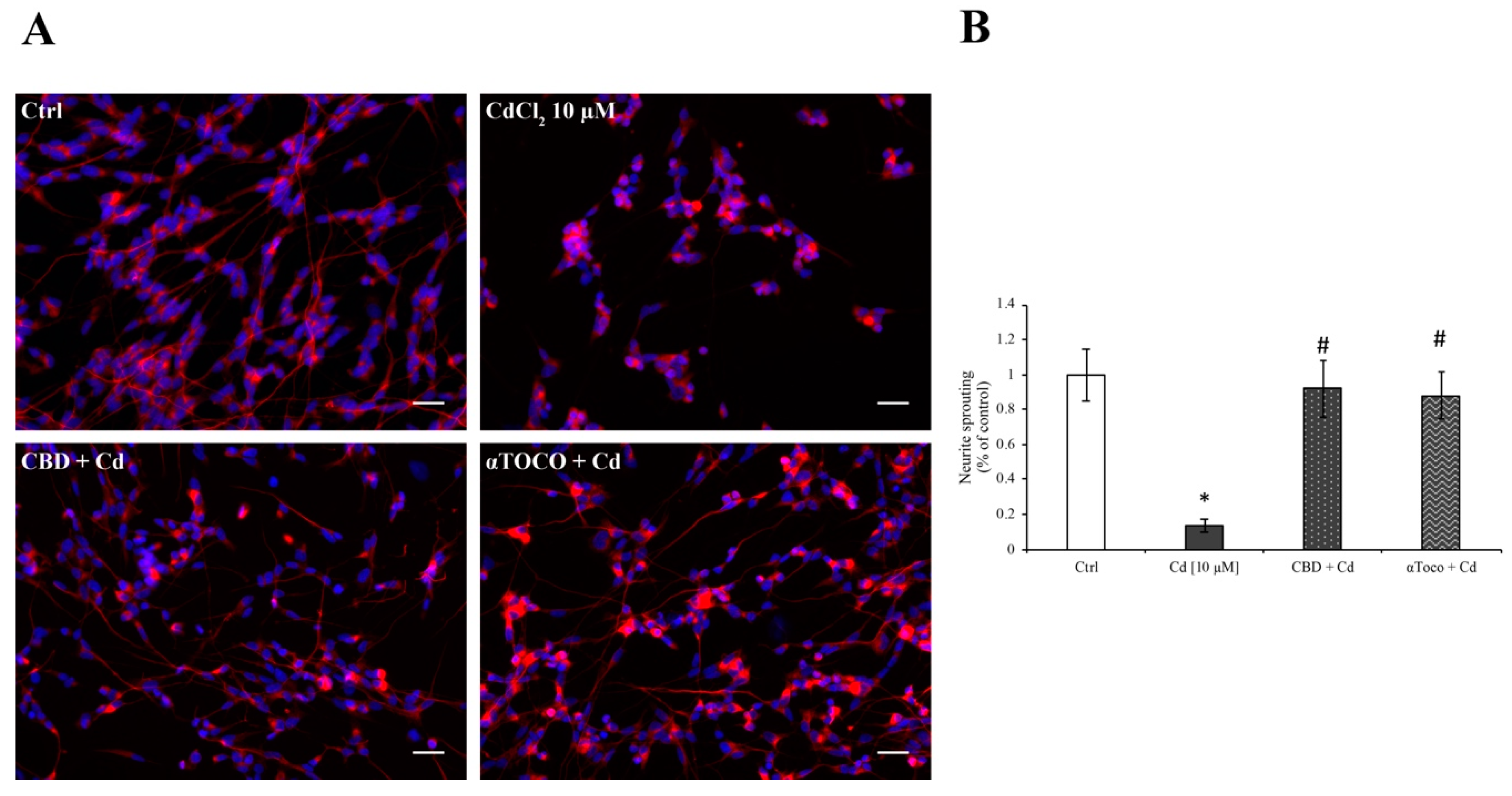
3.4. Neuronal Sprouting
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mead, M.N. Cadmium confusion: Do consumers need protection? Environ. Health Perspect. 2010, 118, a528–a534. [Google Scholar] [CrossRef] [PubMed]
- Egan, S.K.; Bolger, P.M.; Carrington, C.D. Update of US FDA’s total diet study food list and diets. J. Expo. Sci. Environ. Epidemiol. 2007, 17, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Järup, L.; Akesson, A. Current status of cadmium as an environmental health problem. Toxicol. Appl. Pharm. 2009, 238, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Baker, J.R.; Urbenjapol, S.; Haswell-Elkins, M.; Reilly, P.E.; Williams, D.J.; Moore, M.R. A global perspective on cadmium pollution and toxicity in non-occupationally exposed population. Toxicol. Lett. 2003, 137, 65–83. [Google Scholar] [CrossRef]
- Waalkes, M.P. Cadmium carcinogenesis in review. J. Inorg. Biochem. 2000, 79, 241–244. [Google Scholar] [CrossRef]
- Pesch, B.; Haerting, J.; Ranft, U.; Klimpel, A.; Oelschlägel, B.; Schill, W. Occupational risk factors for renal cell carcinoma: Agent-specific results from a case-control study in Germany. MURC Study Group. Multicenter urothelial and renal cancer study. Int. J. Epidemiol. 2000, 29, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.; Muchnok, T.K.; Klishis, M.L.; Roberts, J.R.; Antonini, J.M.; Whong, W.Z.; Ong, T. Cadmium induced cell transformation and tumorigenesis are associated with transcriptional activation of c-fos, c-jun, and c-myc protooncogenes: Role of cellular calcium and reactive oxygen species. Toxicol. Sci. 2001, 61, 295–303. [Google Scholar] [CrossRef] [PubMed]
- López, E.; Figueroa, S.; Oset-Gasque, M.J.; González, M.P. Apoptosis and necrosis: Two distinct events induced by cadmium in cortical neurons in culture. Br. J. Pharm. 2003, 138, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Bao, Q.S.; Lu, C.Y.; Song, H.; Wang, M.; Ling, W.; Chen, W.Q.; Deng, X.Q.; Hao, Y.T.; Rao, S. Behavioural development of school-aged children who live around a multi-metal sulphide mine in Guangdong province, China: A cross-sectional study. BMC Public Health 2009, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Armenta, M.; Ríos, C. Cadmium neurotoxicity. Environ. Toxicol. Pharmacol. 2007, 23, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Morucci, G.; Pacini, A. Cadmium-induced neurotoxicity: Still much ado. Neural Regen. Res. 2018, 13, 1879–1882. [Google Scholar] [PubMed]
- Pihl, R.O.; Parkes, M. Hair element content in learning disabled children. Science 1977, 198, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.D.; Moon, C.K.; Eun, S.-Y.; Ryu, P.D.; Jo, S.A. Identification of ASK1, MKK4, JNK, c-Jun, and caspase-3 as a signaling cascade involved in cadmium-induced neuronal cell apoptosis. Biochem. Biophys. Res. Commun. 2005, 328, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Monroe, R.K.; Halvorsen, S.W. Cadmium blocks receptormediated Jak/STAT signaling in neurons by oxidative stress. Free Radic. Biol. Med. 2006, 41, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Du, Y. Cadmium and its neurotoxic effects. Oxid. Med. Cell. Longev. 2013, 2013, 898034. [Google Scholar] [CrossRef] [PubMed]
- Arvidson, B.; Tjälve, H. Distribution of 109Cd in the nervous system of rats after intravenous injection. Acta Neuropathol. 1986, 69, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Takefuta, S.; Ijiro, H.; Okada, S.; Oku, N. 109Cd transport in rat brain. Brain Res. Bull. 1999, 49, 453–457. [Google Scholar] [CrossRef]
- Shukla, A.; Shukla, G.S.; Srimal, R.C. Cadmium-induced alterations in blood-brain barrier permeability and its possible correlation with decreased microvessel antioxidant potential in rat. Hum. Exp. Toxicol. 1996, 15, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.L.; Quinn, J.F. Alzheimer’s disease and the Blood-Brain Barrier: Past, Present and Future. Aging Health 2008, 4, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal blood–brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Morucci, G.; Maresca, M.; Tenci, B.; Cascella, R.; Paternostro, F.; Ghelardini, C.; Gulisano, M.; Di Cesare Mannelli, L.; Pacini, A. Selenium and zinc: Two key players against cadmium-induced neuronal toxicity. Toxicol In Vitro 2018, 48, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Minami, A.; Takeda, A.; Nishibaba, D.; Takefuta, S.; Oku, N. Cadmium toxicity in synaptic neurotransmission in the brain. Brain Res. 2001, 894, 336–339. [Google Scholar] [CrossRef]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Brotchie, J.; Fitzer-Attas, C. Mechanisms compensating for dopamine loss in early Parkinson disease. Neurology 2009, 72, S32–S38. [Google Scholar] [CrossRef] [PubMed]
- Wirdefeldt, K.; Adami, H.O.; Cole, P.; Trichopoulos, D.; Mandel, J. Epidemiology and etiology of Parkinson’s disease: A review of the evidence. Eur. J. Epidemiol. 2011, 26, S1–S58. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharm. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fang, J.; Leonard, S.S.; Rao, K.M.K. Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic. Biol. Med. 2004, 36, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial oxidative stress: Implications for cell death. Annu. Rev. Pharm. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.R.; DeGheselle, O.; Smeets, K.; van Kerkhove, E.; Cuypers, A. Cadmium-induced pathologies: Where is the oxidative balance lost (or not)? Int. J. Mol. Sci. 2013, 14, 6116–6143. [Google Scholar] [CrossRef] [PubMed]
- Manca, D.; Ricard, A.C.; Trottier, B.; Chevalier, G. Studies on lipid peroxidation in rat tissues following administration of low and moderate doses of cadmium chloride. Toxicology 1991, 67, 303–323. [Google Scholar] [CrossRef]
- Méndez-Armenta, M.; Villeda-Hernández, J.; Barroso-Moguel, R.; Nava-Ruíz, C.; Jiménez-Capdeville, M.E.; Ríos, C. Brain regional lipid peroxidation and metallothionein levels of developing rats exposed to cadmium and dexamethasone. Toxicol. Lett. 2003, 144, 151–157. [Google Scholar] [CrossRef]
- El-Maraghy, S.A.; Gad, M.Z.; Fahim, A.T.; Hamdy, M.A. Effect of cadmium and aluminum intake on the antioxidant status and lipid peroxidation in rat tissues. J. Biochem. Mol. Toxicol. 2001, 15, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Jiang, C.Y.; Xu, H.; Sun, Y.; Hu, F.F.; Bian, J.C.; Liu, X.Z.; Gu, J.H.; Liu, Z.P. Cadmium-induced apoptosis in primary rat cerebral cortical neurons culture is mediated by a calcium signaling pathway. PLoS ONE 2013, 8, e64330. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Zhang, S.L.; Liu, Z.Y.; Tian, Y.; Sun, Q. Cadmium toxicity induces ER stress and apoptosis via impairing energy homoeostasis in cardiomyocytes. Biosci. Rep. 2015, 35, e00214. [Google Scholar] [CrossRef] [PubMed]
- Nemmiche, S.; Chabane-Sari, D.; Guiraud, P. Role of alpha-tocopherol in cadmium-induced oxidative stress in Wistar rat’s blood, liver and brain. Chem. Biol. Interact. 2007, 170, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Abdel Moneim, A.E.; Bauomy, A.A.; Diab, M.M.; Shata, M.T.; Al-Olayan, E.M.; El-Khadragy, M.F. The protective effect of Physalis peruviana L. against cadmium-induced neurotoxicity in rats. Biol. Trace Elem. Res. 2014, 160, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Dong, C. Protective Effect of Proanthocyanidins in Cadmium Induced Neurotoxicity in Mice. Drug. Res. (Stuttg.) 2015, 65, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Adefegha, S.A.; Oboh, G.; Omojokun, O.S.; Adefegha, O.M. Alterations of Na+/K+-ATPase, cholinergic and antioxidant enzymes activity by protocatechuic acid in cadmium-induced neurotoxicity and oxidative stress in Wistar rats. Biomed. Pharm. 2016, 83, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Cassol-Jr, O.J.; Comim, C.M.; Silva, B.R.; Hermani, F.V.; Constantino, L.S.; Felisberto, F.; Petronilho, F.; Hallak, J.E.; De Martinis, B.S.; Zuardi, A.W.; et al. Treatment with cannabidiol reverses oxidative stress parameters, cognitive impairment and mortality in rats submitted to sepsis by cecal ligation and puncture. Brain Res. 2010, 1348, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Karniol, I.G.; Shirakawa, I.; Kasinski, N.; Pfeferman, A.; Carlini, E.A. Cannabidiol interferes with the effects of delta 9-tetrahydrocannabinol in man. Eur. J. Pharm. 1974, 28, 172–177. [Google Scholar] [CrossRef]
- Grlic, L. A comparative study on some chemical and biological characteristics of various samples of cannabis resin. Bull. Narc. 1976, 14, 37–46. [Google Scholar]
- Izzo, A.A.; Borrelli, F.; Capasso, R.; Di Marzo, V.; Mechoulam, R. Non-psychotropic plant cannabinoids: New therapeutic opportunities from an ancient herb. Trends Pharm. Sci. 2009, 30, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Bossong, M.G.; Niesink, R.J.M. Adolescent brain maturation, the endogenous cannabinoid system and the neurobiology of cannabisinduced schizophrenia. Prog. Neurobiol. 2010, 92, 370–385. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, J.; Sagredo, O.; Pazos, M.R.; García, C.; Pertwee, R.; Mechoulam, R.; Martínez-Orgado, J. Cannabidiol for neurodegenerative disorders: Important new clinical applications for this phytocannabinoid? Br. J. Clin. Pharm. 2013, 75, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Borges, R.S.; Batista, J., Jr.; Viana, R.B.; Baetas, A.C.; Orestes, E.; Andrade, M.A.; Honório, K.M.; da Silva, A.B. Understanding the molecular aspects of tetrahydrocannabinol and cannabidiol as antioxidants. Molecules 2013, 18, 12663–12674. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.C.; Brant, F.; Miranda, A.S.; Machado, F.S.; Teixeira, A.L. Cannabidiol increases survival and promotes rescue of cognitive function in a murine model of cerebral malaria. Neuroscience 2015, 289, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Silveira, J.W.; Issy, A.C.; Castania, A.; Salmon, C.E.; Nogueira-Barbosa, M.H.; Guimaraes, F.S.; Defino, H.L.; Del Bel, E. Protective effects of cannabidiol on lesion-induced intervertebral disc degeneration. PLoS ONE 2014, 9, e113161. [Google Scholar] [CrossRef] [PubMed]
- Schiavon, A.P.; Soares, L.M.; Bonato, J.M.; Milani, H.; Guimaraes, F.S.; Weffort de Oliveira, R.M. Protective effects of cannabidiol against hippocampal cell death and cognitive impairment induced by bilateral common carotid artery occlusion in mice. Neurotox. Res. 2014, 26, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.; Benitez, S.U.; Cartarozzi, L.P.; Del Bel, E.; Guimaraes, F.S.; Oliveira, A.L. Neuroprotection and reduction of glial reaction by cannabidiol treatment after sciatic nerve transection in neonatal rats. Eur. J. Neurosci. 2013, 38, 3424–3434. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkoski, M.; Guimaraes, F.S.; Del-Bel, E. Cannabidiol-treated rats exhibited higher motor score after cryogenic spinal cord injury. Neurotox. Res. 2012, 21, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.C.; Fogaca, M.V.; Sonego, A.B.; Guimaraes, F.S. Cannabidiol, neuroprotection and neuropsychiatric disorders. Pharm. Res. 2016, 112, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Harvey, B.S.; Ohlsson, K.S.; Maag, J.L.; Musgrave, I.F.; Smid, S.D. Contrasting protective effects of cannabinoids against oxidative stress and amyloid-beta evoked neurotoxicity in vitro. Neurotoxicology 2012, 33, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, V.K.; de Freitas, B.S.; da Silva Dornelles, A.; Nery, L.R.; Falavigna, L.; Ferreira, R.D.; Bogo, M.R.; Hallak, J.E.; Zuardi, A.W.; Crippa, J.A.; et al. Cannabidiol normalizes caspase 3, synaptophysin, andmitochondrial fission protein DNM1L expression levels in rats with brain iron overload: Implications for neuroprotection. Mol. Neurobiol. 2013, 49, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Korecka, J.A.; van Kesteren, R.E.; Blaas, E.; Spitzer, S.O.; Kamstra, J.H.; Smit, A.B.; Swaab, D.F.; Verhaagen, J.; Bossers, K. Phenotypic characterization of retinoic acid differentiated SH-SY5Y cells by transcriptional profiling. PLoS ONE 2013, 28, e63862. [Google Scholar] [CrossRef] [PubMed]
- Lopes, F.M.; Schröder, R.; da Frota, M.L., Jr.; Zanotto-Filho, A.; Müller, C.B.; Pires, A.S.; Meurer, R.T.; Colpo, G.D.; Gelain, D.P.; Kapczinski, F.; et al. Comparison between proliferative and neuron-like SH-SY5Y cells as an in vitro model for Parkinson disease studies. Brain Res. 2010, 1337, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Rigobello, M.P.; Scutari, G.; Friso, A.; Barzon, E.; Artusi, S.; Bindoli, A. Mitochondrial permeability transition and release of cytochrome c induced by retinoic acids. Biochem. Pharmacol. 1999, 58, 665–670. [Google Scholar] [CrossRef]
- Xun, Z.; Lee, D.Y.; Lim, J.; Canaria, C.A.; Barnebey, A.; Yanonne, S.M.; McMurray, C.T. Retinoic acid-induced differentiation increases the rate of oxygen consumption and enhances the spare respiratory capacity of mitochondria in SH-SY5Y cells. Mech. Ageing Dev. 2012, 133, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Del Pino, J.; Zeballos, G.; Anadon, M.J.; Capo, M.A.; Díaz, M.J.; García, J.; Frejo, M.T. Higher sensitivity to cadmium induced cell death of basal forebrain cholinergic neurons: A cholinesterase dependent mechanism. Toxicology 2014, 325, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.; Drysdale, A.J.; Lafourcade, C.; Pertwee, R.G.; Platt, B. Cannabidiol targets mitochondria to regulate intracellular Ca2+ levels. J. Neurosci. 2009, 29, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Borges, R.S.; da Silva, A.B.F. Chapter e12, Cannabidiol as an antioxidant. In Handbook of Cannabis and Related Pathologies; Preedy, V.R., Ed.; Elsevier Academic Press: London, UK, 2017; pp. e122–e130. [Google Scholar]
- Gugliandolo, A.; Chiricosta, L.; Silvestro, S.; Bramanti, P.; Mazzon, E. α-Tocopherol Modulates Non-Amyloidogenic Pathway and Autophagy in an In Vitro Model of Alzheimer’s Disease: A Transcriptional Study. Brain Sci. 2019, 9, 196. [Google Scholar] [CrossRef] [PubMed]
- Capitini, C.; Conti, S.; Perni, M.; Guidi, F.; Cascella, R.; De Poli, A.; Penco, A.; Relini, A.; Cecchi, C.; Chiti, F. TDP-43 Inclusion bodies formed in bacteria are structu-rally amorphous, non-amyloid and inherently toxic to neuroblastoma cells. PLoS ONE 2014, 9, e86720. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.W.; Wang, Y.; Wang, T.; Zhang, K.B.; Yuan, Y.; Bian, J.C.; Liu, X.Z.; Gu, J.H.; Zhu, J.Q.; Liu, Z.P. Cadmium-induced autophagy is mediated by oxidative signaling in PC-12 cells and is associated with cytoprotection. Mol. Med. Rep. 2015, 12, 4448–4454. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Bradl, H. Heavy Metals in the Environment: Origin, Interaction and Remediation, 1st ed.; Academic Press: London, UK, 2002. [Google Scholar]
- He, Z.L.; Yang, X.E.; Stoffella, P.J. Trace elements in agroecosystems and impacts on the environment. J. Trace Elem. Med. Biol. 2005, 19, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Goyer, R.A.; Clarkson, T.W. Toxic Effects of Metals. In Casarett and Doull’s Toxicology: The Basic Science of Poisons, 6th ed.; Klaasen, C.D., Ed.; McGraw-Hill: New York, NY, USA, 2001; pp. 861–867. [Google Scholar]
- Herawati, N.; Suzuki, S.; Hayashi, K.; Rivai, I.F.; Koyoma, H. Cadmium, copper and zinc levels in rice and soil of Japan, Indonesia and China by soil type. Bull. Environ. Contam. Toxicol. 2000, 64, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Shallari, S.; Schwartz, C.; Hasko, A.; Morel, J.L. Heavy metals in soils and plants of serpentine and industrial sites of Albania. Sci. Total Environ. 1998, 209, 133–142. [Google Scholar] [CrossRef]
- WHO—World Health Organization, World Health Report 2006. Available online: https://www.who.int/whr/2006/en/ (accessed on 19 July 2019).
- ATSDR—Tox Guide for Cadmium. CAS#7440-43-9. October 2011. Available online: http://www.atsdr.cdc.gov/toxprofiles/index.asp (accessed on 27 June 2019).
- Wilson, D.N. Association Cadmium. Cadmium-Market Trends and Influences. In Proceedings of the 6th International Cadmium Conference, London, UK, 4–6 February 1988; pp. 9–16. [Google Scholar]
- Almenara, C.C.; Broseghini-Filho, G.B.; Vescovi, M.V.; Angeli, J.K.; Faria Tde, O.; Stefanon, I.; Vassallo, D.V.; Padilha, A.S. Chronic cadmium treatment promotes oxidative stress and endothelial damage in isolated rat aorta. PLoS ONE 2013, 8, e68418. [Google Scholar] [CrossRef] [PubMed]
- Caudle, W.M.; Guillot, T.S.; Lazo, C.R.; Miller, G.W. Industrial toxicants and Parkinson’s disease. Neurotoxicology 2012, 33, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, C.D.; Amdur, M.O.; Doull, J. Principles of toxicology. In Casarett and Doull’s Toxicology: The Basis Science of Poisons, 3rd ed.; Klaassen, C.D., Amdur, M.O., Doull, J., Eds.; Macmillan Publishing Company: New York, NY, USA, 1986. [Google Scholar]
- Kaye, S. Handbook of Emergency Toxicology: A Guide for the Identification, Diagnosis and Treatment of Poisoning, 4th ed.; Charles C Thomas Publisher: Springfield, IL, USA, 1980; pp. 232–235. [Google Scholar]
- Hung, Y.M.; Chung, H.M. Acute self-poisoning by ingestion of cadmium and barium. Nephrol. Dial. Transpl. 2004, 19, 1308–1309. [Google Scholar] [CrossRef] [PubMed]
- Poisindex, Thomson Micromedex. Available online: https://www.micromedexsolutions.com/home/dispatch (accessed on 19 July 2019).
- Alli, L.A. Blood level of cadmium and lead in occupationally exposed persons in Gwagwalada, Abuja, Nigeria. Interdiscip. Toxicol. 2015, 8, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Gupta, R.; Reddy, H.; Sinha, S.; Muley, A.; Kolluru, G.K.; Chatterjee, S. Cadmium attenuates bradykinin-driven nitric oxide production by interplaying with the localization pattern of endothelial nitric oxide synthase. Biochem. Cell Biol. 2009, 87, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Chen, S.; Luo, Y.; Chen, Z.; Liu, L.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Chen, L.; et al. Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PLoS ONE 2011, 6, e19052. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, N.; Zhang, H.; Liu, C.; Dong, X.; Wang, X.; Zhu, Y.; Xu, C.; Liu, L.; Yang, S.; et al. Celastrol prevents cadmium-induced neuronal cell death by blocking reactive oxygen species-mediated mammalian target of rapamycin pathway. Br. J. Pharm. 2017, 174, 82–100. [Google Scholar] [CrossRef] [PubMed]
- Pak, E.J.; Son, G.D.; Yoo, B.S. Cadmium inhibits neurite outgrowth in differentiating human SH-SY5Y neuroblastoma cells. Int. J. Toxicol. 2014, 33, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Demirakca, T.; Sartorius, A.; Ende, G.; Meyer, N.; Welzel, H.; Skopp, G.; Mann, K.; Hermann, D. Diminished gray matter in the hippocampus of cannabis users: Possible protective effects of cannabidiol. Drug Alcohol Depend. 2011, 114, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Niesink, R.J.M.; van Laar, M.W. Does cannabidiol protect against adverse psychological effects of THC? Front. Psychiatry 2013, 4, 130. [Google Scholar] [CrossRef] [PubMed]
- Pagotto, U.; Marsicano, G.; Cota, D.; Lutz, B.; Pasquali, R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr. Rev. 2006, 27, 73–100. [Google Scholar] [CrossRef] [PubMed]
- Elphick, M.R.; Egertová, M. The neurobiology and evolution of cannabinoid signalling. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2001, 356, 381–408. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. Br. J. Pharm. 2008, 153, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Onaivi, E.S. Neuropsychobiological evidence for the functional presence and expression of cannabinoid CB2 receptors in the brain. Neuropsychobiology 2006, 54, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Stempel, A.V.; Stumpf, A.; Zhang, H.Y.; Özdoğan, T.; Pannasch, U.; Theis, A.K.; Otte, D.M.; Wojtalla, A.; Rácz, I.; Ponomarenko, A.; et al. Cannabinoid Type 2 Receptors Mediate a Cell Type-Specific Plasticity in the Hippocampus. Neuron 2016, 90, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Maccarrone, M.; De Petrocellis, L.; Jarrahian, A.; Finazzi-Agro, A.; Hillard, C.; Di Mazo, V. The uptake by cells of 2-arachidonoylglycerol, an endogenous agonist of cannabinoid receptors. Eur. J. Biochem. 2001, 268, 1982–1989. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.B.; Burnett, A.; Hall, B.; Parker, K.K. Agonistic properties of cannabidiol at 5-HT1a receptors. Neurochem. Res. 2005, 30, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.C.; Guimarães, F.S. Involvement of 5HT1A receptors in the anxiolytic-like effects of cannabidiol injected into the dorsolateral periaqueductal gray of rats. Psychopharmacology (Berlin) 2008, 199, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P. Cannabidiol is a partial agonist at dopamine D2High receptors, predicting its antipsychotic clinical dose. Transl. Psychiatry 2016, 6, e920. [Google Scholar] [CrossRef] [PubMed]
- Bruni, N.; Della Pepa, C.; Oliaro-Bosso, S.; Pessione, E.; Gastaldi, D.; Dosio, F. Cannabinoid Delivery Systems for Pain and Inflammation Treatment. Molecules 2018, 23, 2478. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.J.; Williams, C.M.; Whalley, B.J.; Stephens, G.J. Phytocannabinoids as novel therapeutic agents in CNS disorders. Pharmacol. Ther. 2012, 133, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; Bissonnette, C.J.; McGeer, P.L. Reduction of human monocytic cell neurotoxicity and cytokine secretion by ligands of the cannabinoid-type CB2 receptor. Br. J. Pharmacol. 2003, 139, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Svensson, A.C.; Johansson, M.; Persson, E.; Carchenilla, M.S.; Jacobsson, S.O. Expression of functional CB1 cannabinoid receptors in retinoic acid-differentiated P19 embryonal carcinoma cells. J. Neurosci. Res. 2006, 83, 1128–1140. [Google Scholar] [CrossRef] [PubMed]
- Presgraves, S.P.; Ahmed, T.; Borwege, S.; Joyce, J.N. Terminally differentiated SH-SY5Y cells provide a model system for studying neuroprotective effects of dopamine agonists. Neurotox. Res. 2004, 5, 579–598. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.T.; Lau, W.K.; Yu, M.S.; Lai, C.S.; Yeung, S.C.; So, K.F.; Chang, R.C. Effects of all-trans-retinoic acid on human SH-SY5Y neuroblastoma as in vitro model in neurotoxicity research. Neurotoxicology 2009, 30, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [PubMed]
- Iuvone, T.; Esposito, G.; Esposito, R.; Santamaria, R.; Di Rosa, M.; Izzo, A.A. Neuroprotective effect of cannabidiol, a non-psychoactive component from Cannabis sativa, on beta-amyloid-induced toxicity in PC12 cells. J. Neurochem. 2004, 89, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.Q.; Gao, J.; Zhu, Z.R.; Xiong, Y.; Liu, J. Mitochondrial dysfunction enhances susceptibility to oxidative stress by down-regulation of thioredoxin in human neuroblastoma cells. Neurochem. Res. 2008, 33, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.; Lee, K.H.; Park, J.H.; Geum, D.; Kim, K. Mitochondrial membrane depolarization and the selective death of dopaminergic neurons by rotenone: Protective effect of coenzyme Q (10). J. Neurochem. 2005, 93, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, J.S.; Klaunig, J.E. Role of the mitochondrial membrane permeability transition (MPT) in rotenone-induced apoptosis in liver cells. Toxicol. Sci. 2000, 53, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Doran, E.; Halestrap, A.P. Cytochrome c release from isolated rat liver mitochondria can occur independently of outer membrane rupture: Possible role of contact sites. Biochem. J. 2000, 348, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Qie, X.; Wen, D.; Guo, H.; Xu, G.; Liu, S.; Shen, Q.; Liu, Y.; Zhang, W.; Cong, B.; Ma, C. Endoplasmic reticulum stress mediates methamphetamine-induced blood-brain barrier damage. Front. Pharm. 2017, 8, 639. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhang, K.; Kaufman, R.J. The unfolded protein response–a stress signaling pathway of the endoplasmic reticulum. J. Chem. Neuroanat. 2004, 28, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 2008, 105, 18525–18530. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Mecha, M.; Torrao, A.S.; Mestre, L.; Carrillo-Salinas, F.J.; Mechoulam, R.; Guaza, C. Cannabidiol protects oligodendrocyte progenitor cells from inflammation-induced apoptosis by attenuating endoplasmic reticulum stress. Cell Death Dis. 2012, 3, e331. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Hu, F.; Wu, J.; Zhang, S. Cannabidiol attenuates OGD/R-induced damage by enhancing mitochondrial bioenergetics and modulating glucose metabolism via pentose-phosphate pathway in hippocampal neurons. Redox Biol. 2017, 11, 577–585. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Branca, J.J.V.; Morucci, G.; Becatti, M.; Carrino, D.; Ghelardini, C.; Gulisano, M.; Di Cesare Mannelli, L.; Pacini, A. Cannabidiol Protects Dopaminergic Neuronal Cells from Cadmium. Int. J. Environ. Res. Public Health 2019, 16, 4420. https://doi.org/10.3390/ijerph16224420
Branca JJV, Morucci G, Becatti M, Carrino D, Ghelardini C, Gulisano M, Di Cesare Mannelli L, Pacini A. Cannabidiol Protects Dopaminergic Neuronal Cells from Cadmium. International Journal of Environmental Research and Public Health. 2019; 16(22):4420. https://doi.org/10.3390/ijerph16224420
Chicago/Turabian StyleBranca, Jacopo Junio Valerio, Gabriele Morucci, Matteo Becatti, Donatello Carrino, Carla Ghelardini, Massimo Gulisano, Lorenzo Di Cesare Mannelli, and Alessandra Pacini. 2019. "Cannabidiol Protects Dopaminergic Neuronal Cells from Cadmium" International Journal of Environmental Research and Public Health 16, no. 22: 4420. https://doi.org/10.3390/ijerph16224420
APA StyleBranca, J. J. V., Morucci, G., Becatti, M., Carrino, D., Ghelardini, C., Gulisano, M., Di Cesare Mannelli, L., & Pacini, A. (2019). Cannabidiol Protects Dopaminergic Neuronal Cells from Cadmium. International Journal of Environmental Research and Public Health, 16(22), 4420. https://doi.org/10.3390/ijerph16224420