Analysis of Short-Term Smoking Effects in PBMC of Healthy Subjects—Preliminary Study

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Blood Collection, PBMCs, and Serum Isolation
2.3. Experimental Design
2.4. RNA Extraction and cDNA Synthesis
2.5. Analysis of Gene Expression
2.6. Estimation of SOD Level
2.7. Estimation of Thiol Groups
2.8. Estimation of TBARs
2.9. Statistical Analysis
3. Results
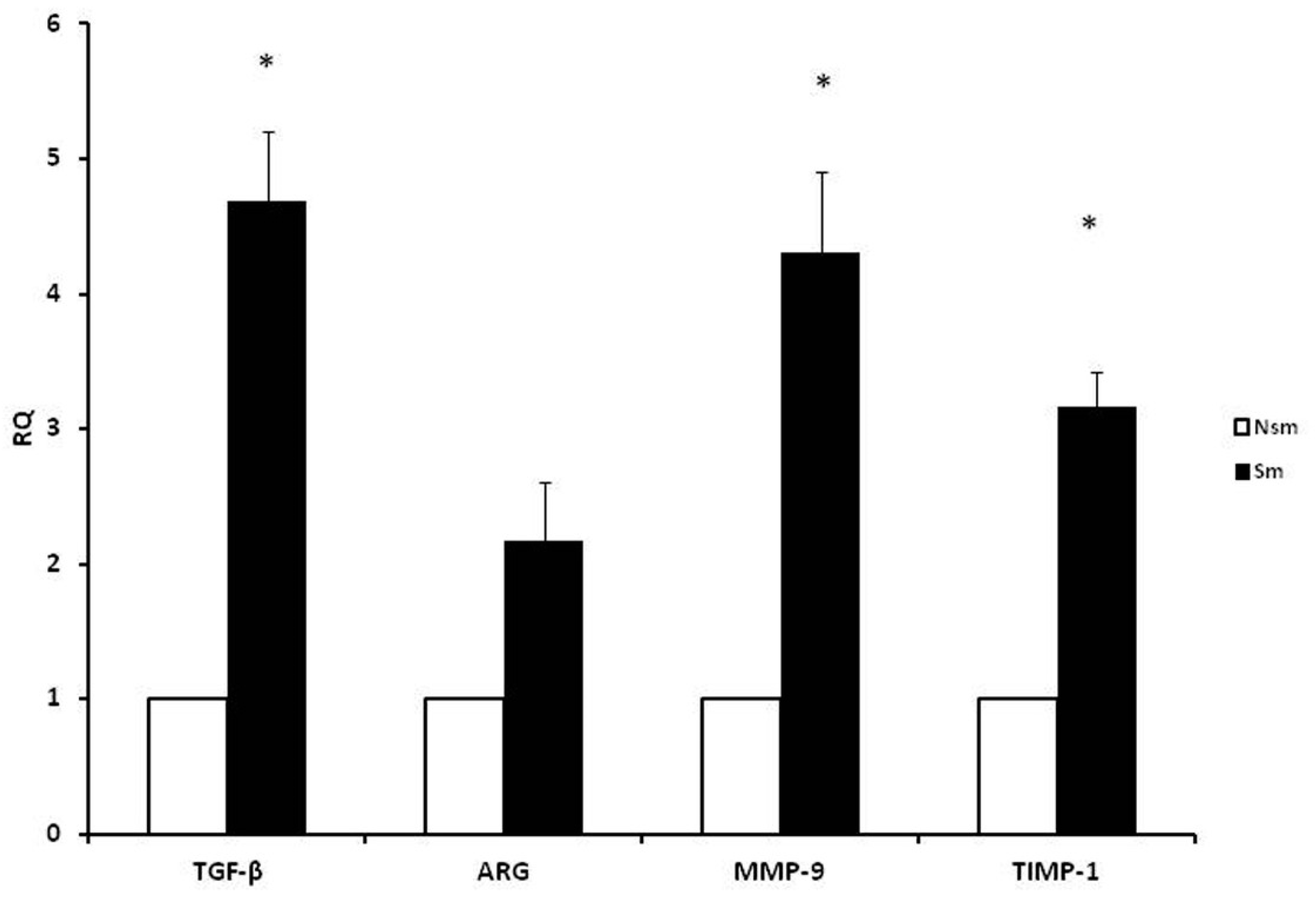
3.1. PBMCs of Smoking Subjects Respond with Higher mRNA Expression
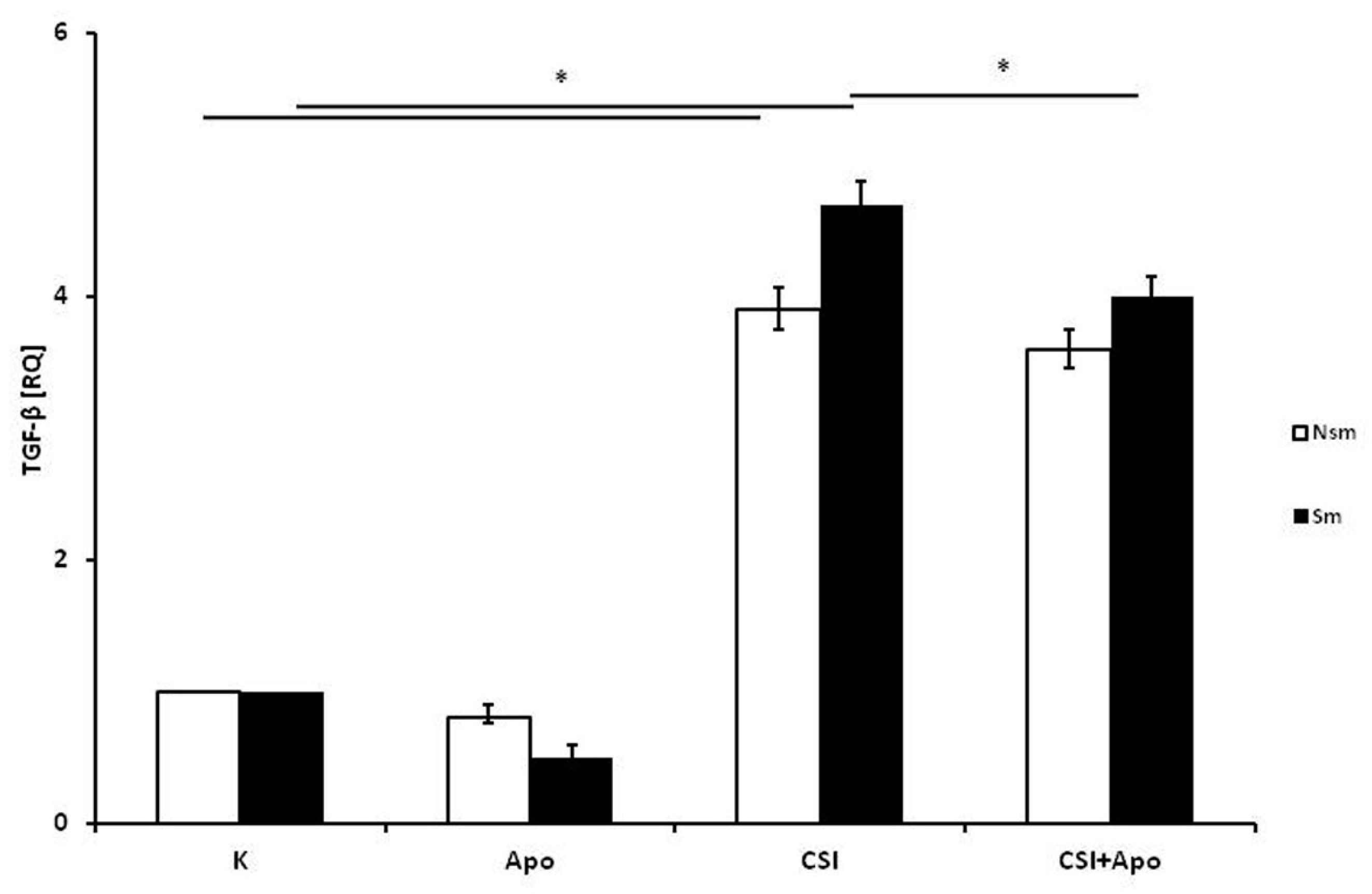
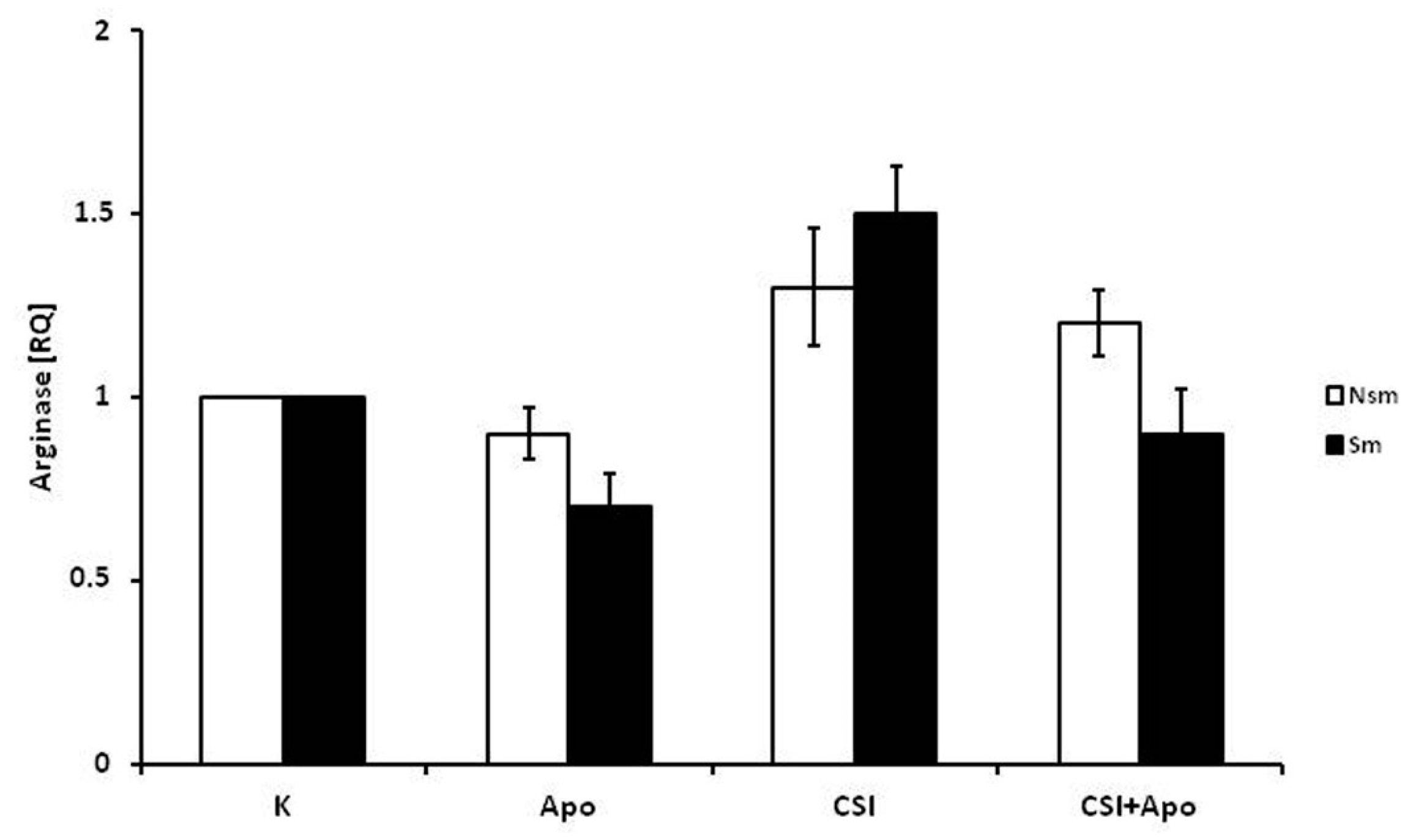
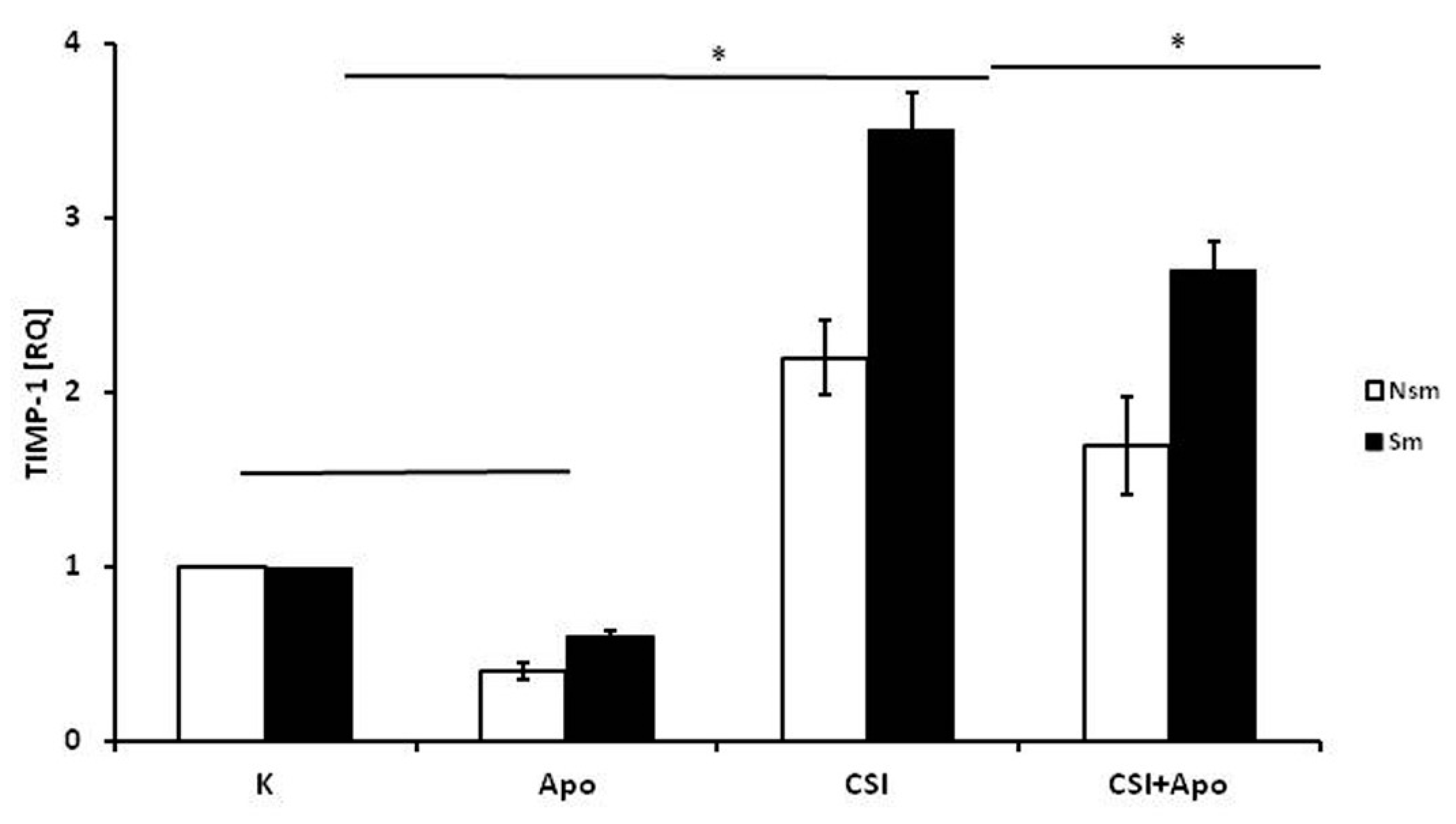
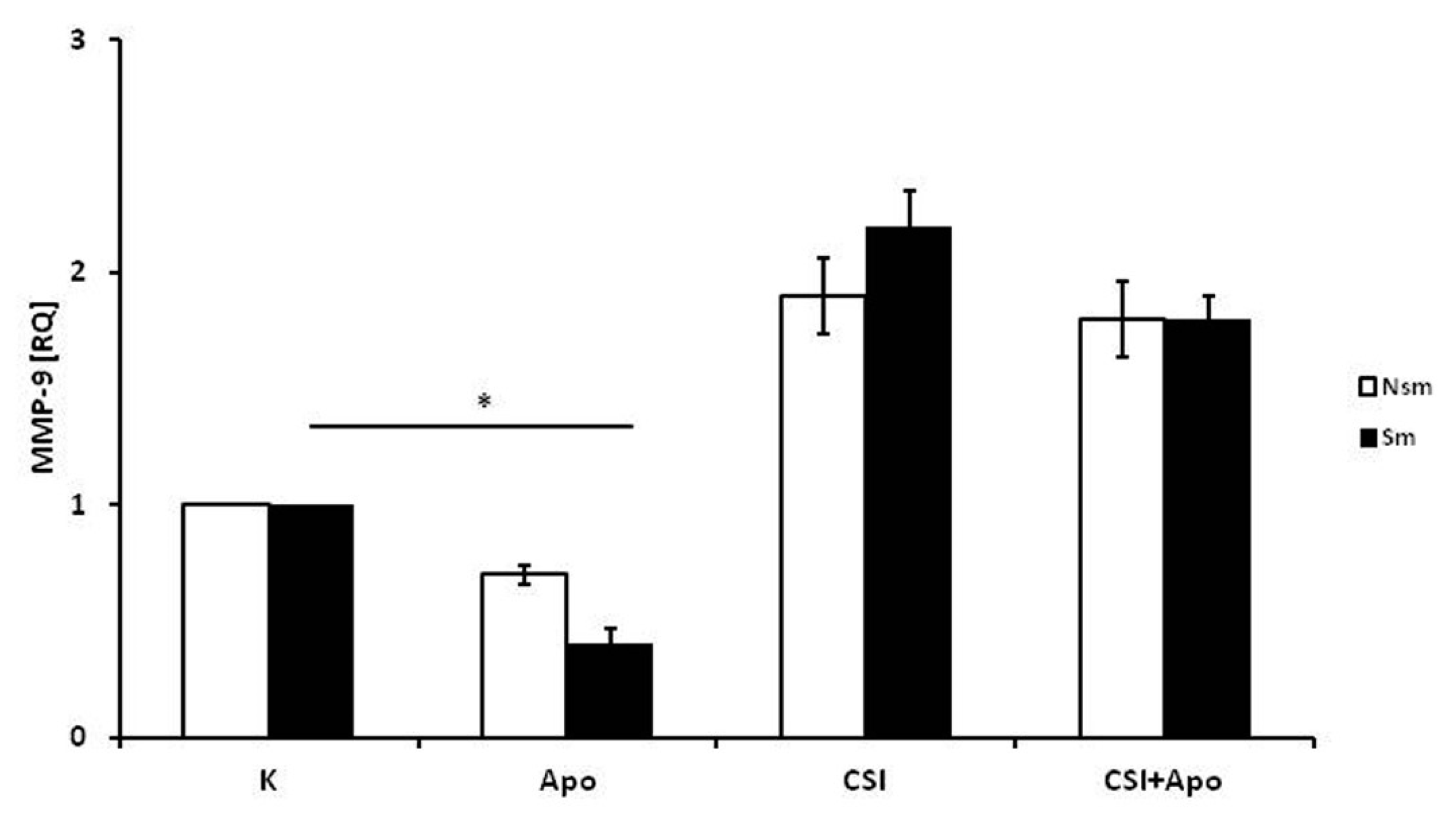
3.2. Results of qPCR in Smoking and Nonsmoking Groups
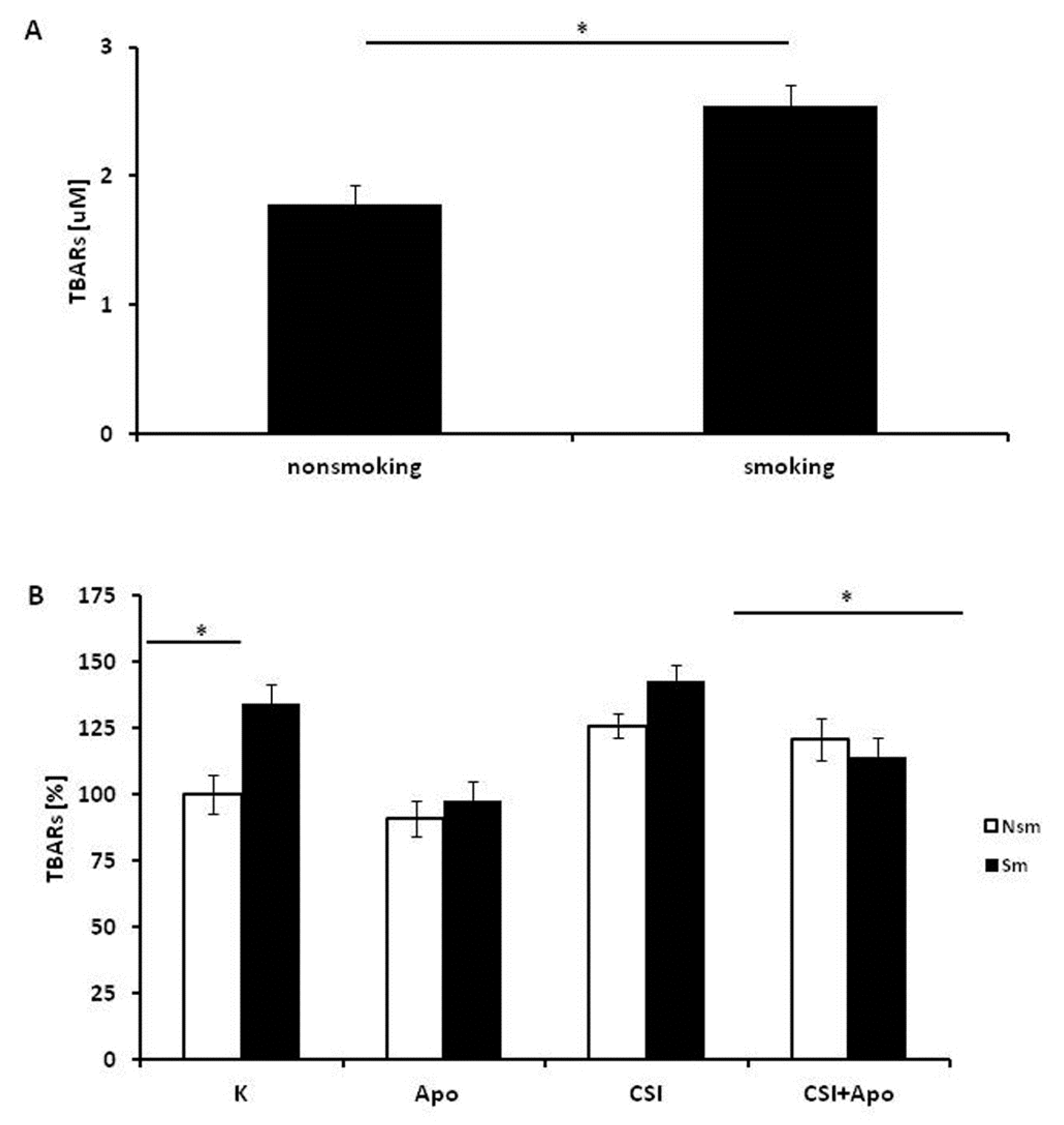
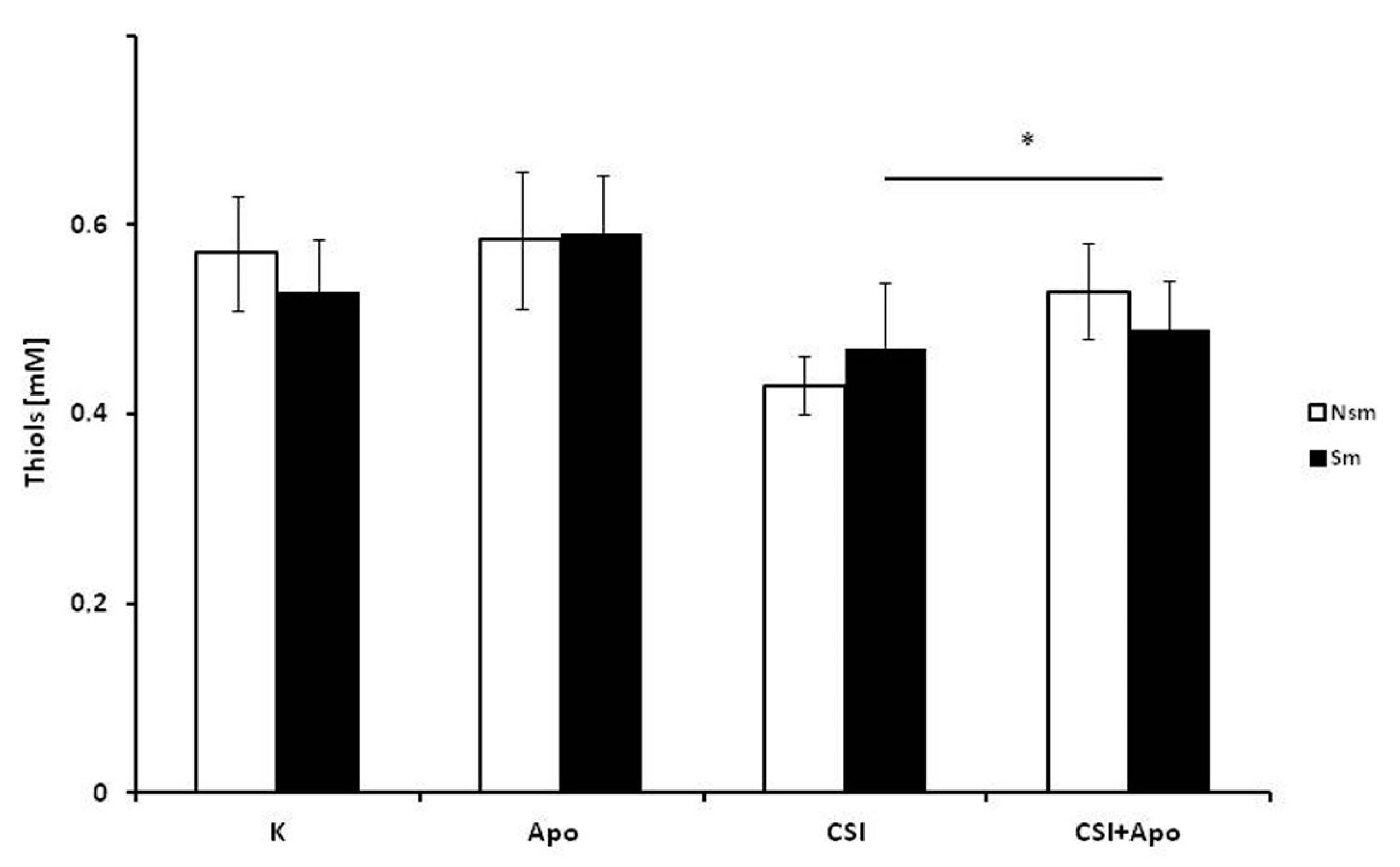
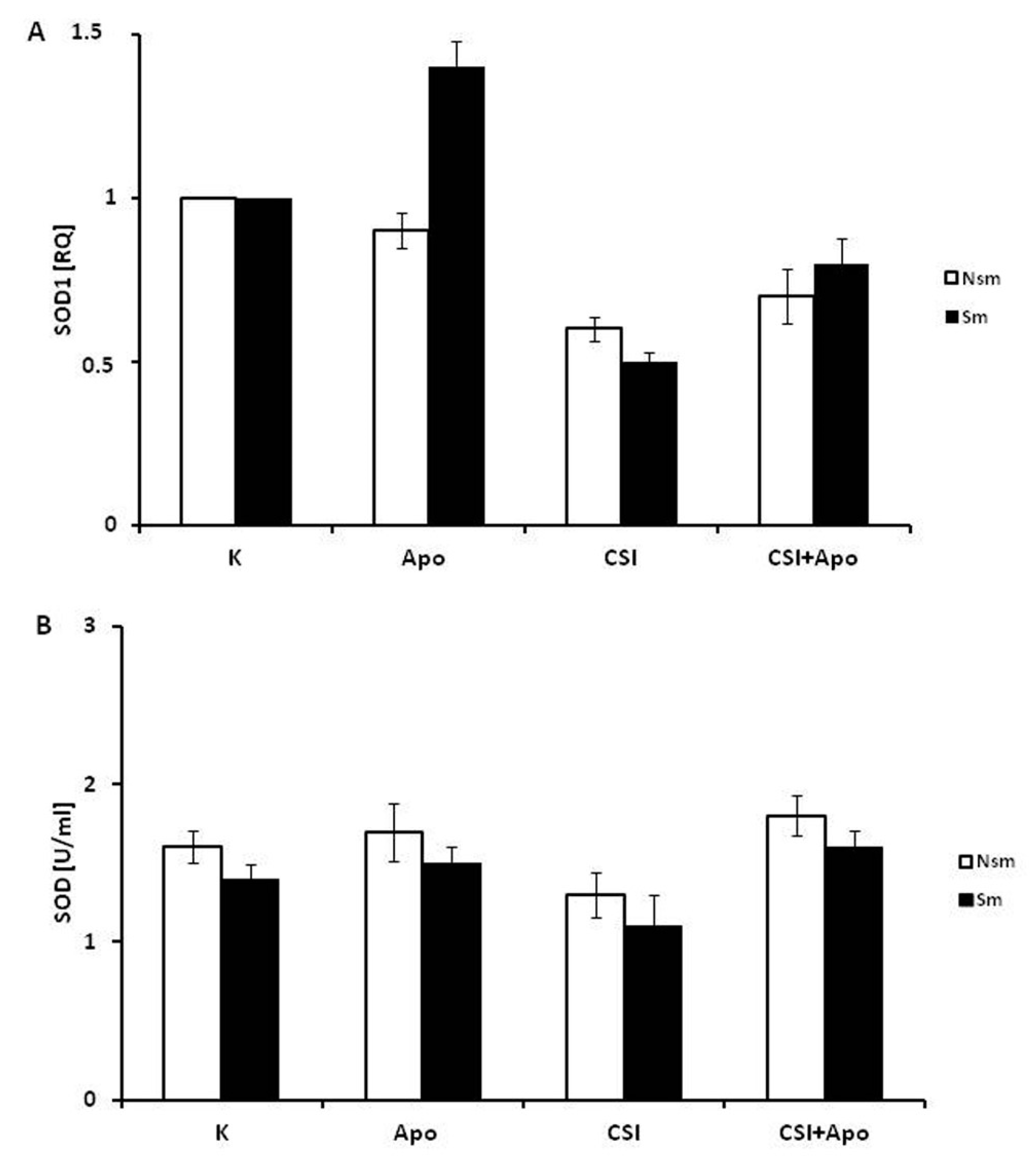
3.3. Oxidative and Antioxidative Properties of PBMC in Smokers and Never Smokers
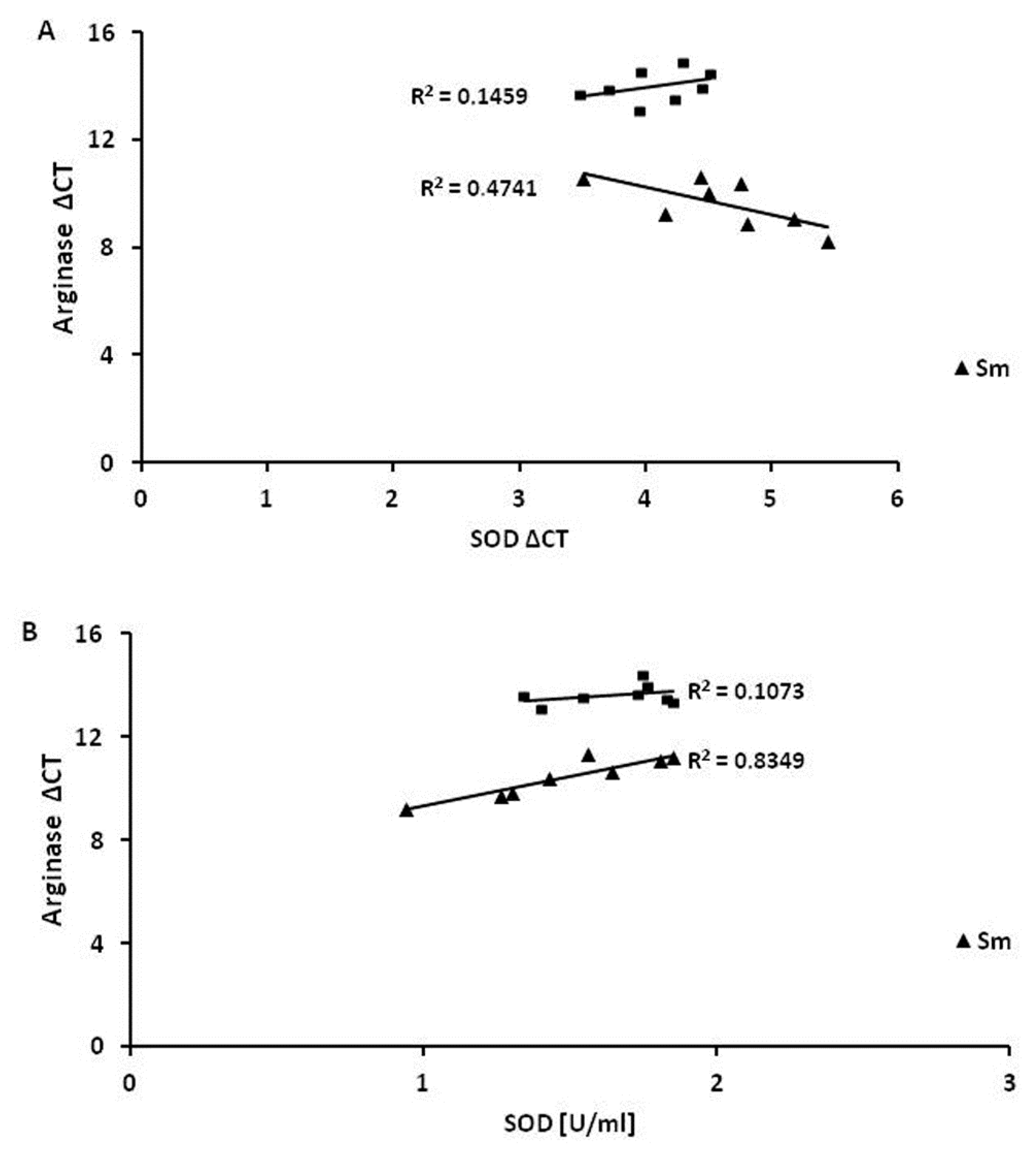
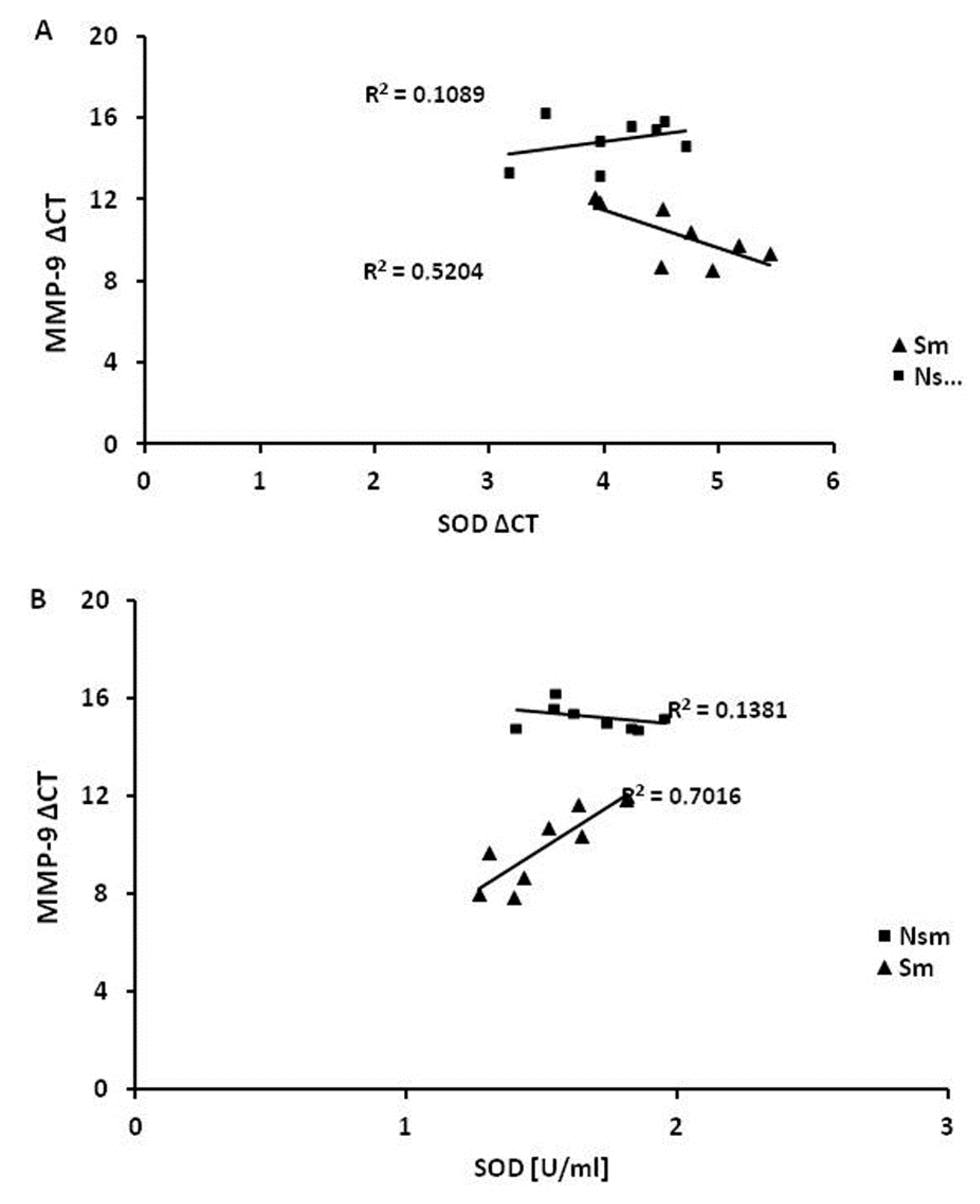
3.4. Superoxide Dismutase 1 Correlates with Arginase and MMP-9 mRNA Expression
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Aslan, R.; Kutlu, R.; Civi, S.; Tasyurek, E. The correlation of the total antioxidant status (TAS), total oxidant status (TOS) and paraoxonase activity (PON1) with smoking. Clin. Biochem. 2014, 47, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Masjedi, M.R.; Rahman, I. Outcome of Smoking Cessation on Airway Remodeling and Pulmonary Inflammation in COPD Patients. Tanaffos 2011, 10, 7–11. [Google Scholar] [PubMed]
- Brunnemann, K.D.; Hoffmann, D. Analytical studies on tobacco-specific N-nitrosamines in tobacco and tobacco smoke. Crit. Rev. Toxicol. 1991, 21, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Peinado, V.I.; Galdiz, J.B.; Ferrer, E.; Marin-Corral, J.; Sanchez, F.; Gea, J.; Barberà, J.A. Cigarette smoke-induced oxidative stress: A role in chronic obstructive pulmonary disease skeletal muscle dysfunction. Am. J. Respir. Crit. Care Med. 2010, 182, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Conti, V.; Corbi, G.; Manzo, V.; Pelaia, G.; Filippelli, A.; Vatrella, A. Sirtuin 1 and aging theory for chronic obstructive pulmonary disease. Anal. Cell Pathol. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Le Rouzic, O.; Pichavant, M.; Frealle, E.; Guillon, A.; Si-Tahar, M.; Gosset, P. Th17 cytokines: Novel potential therapeutic targets for COPD pathogenesis and exacerbations. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, M.; Zhang, L.; Cheng, Y.; Tu, X.; Lu, Z. Klotho Regulates Cigarette Smoke-Induced Autophagy: Implication in Pathogenesis of COPD. Lung 2017, 195, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Vij, N.; Chandramani-Shivalingappa, P.; Van Westphal, C.; Hole, R.; Bodas, M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am. J. Physiol. Cell Physiol. 2018, 314, C73–C87. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, F.; Hylkema, M.N.; Melgert, B.N.; Timens, W.; Postma, D.S.; ten Hacken, N.H. Smoking and nonsmoking asthma: Differences in clinical outcome and pathogenesis. Expert Rev. Respir. Med. 2011, 5, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, C.; Boulet, L.P.; Page, N.; Laviolette, M.; Zimmermann, N.; Rothenberg, M.E.; Hamid, Q. Influence of cigarette smoke on the arginine pathway in asthmatic airways: Increased expression of arginase I. J. Allergy Clin. Immunol. 2007, 119, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Poljsak, B. Strategies for reducing or preventing the generation of oxidative stress. Oxid. Med. Cell. Longev. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Balcerczyk, A.; Bartosz, G. Thiols are main determinants of total antioxidant capacity of cellular homogenates. Free Radic Res. 2003, 37, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Okoh, V.O.; Garba, N.A.; Penney, R.B.; Das, J.; Deoraj, A.; Singh, K.P.; Sarkar, S.; Felty, Q.; Yoo, C.; Jackson, R.M.; et al. Redox signalling to nuclear regulatory proteins by reactive oxygen species contributes to oestrogen-induced growth of breast cancer cells. Br. J. Cancer 2015, 112, 1687–1702. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, S.; Chen, P.L.; Greenshields, A.L.; Nightingale, M.; Hoskin, D.W.; Bedard, K. Resveratrol, piperine and apigenin differ in their NADPH-oxidase inhibitory and reactive oxygen species-scavenging properties. Phytomedicine 2016, 23, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Orosz, Z.; Csiszar, A.; Labinskyy, N.; Smith, K.; Kaminski, P.M.; Ferdinandy, P.; Wolin, M.S.; Rivera, A.; Ungvari, Z. Cigarette smoke-induced proinflammatory alterations in the endothelial phenotype: Role of NAD(P)H oxidase activation. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H130–H139. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, P.K. The roles of leukotrienes and the effects of leukotriene receptor antagonists in the inflammatory response and remodeling of allergic asthma. Clin. Exp. Allergy Rev. 2008. [Google Scholar] [CrossRef]
- Grzela, K.; Litwiniuk, M.; Zagorska, W.; Grzela, T. Airway Remodeling in Chronic Obstructive Pulmonary Disease and Asthma: The Role of Matrix Metalloproteinase-9. Arch. Immunol. Ther. Exp. 2016, 64, 47–55. [Google Scholar] [CrossRef] [PubMed]
- James, A. Airway remodeling in asthma. Curr. Opin. Pulm. Med. 2005, 11, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, M.Y.; Markesbery, W.R. Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer’s disease. Neurosci. Lett. 2001, 302, 141–145. [Google Scholar] [CrossRef]
- World Health Organization. Chronic respiratory diseases: Burden of COPD. Available online: http://www.who.int/respiratory/copd/burden/en/ (accessed on 3 September 2018).
- Wang, R.D.; Tai, H.; Xie, C.; Wang, X.; Wright, J.L.; Churg, A. Cigarette smoke produces airway wall remodeling in rat tracheal explants. Am. J. Respir. Crit. Care Med. 2003, 168, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, P.K. Remodeling in asthma and chronic obstructive lung disease. Am. J. Respir. Crit. Care Med. 2001, 164, S28–S38. [Google Scholar] [CrossRef] [PubMed]
- Saetta, M.; Finkelstein, R.; Cosio, M.G. Morphological and cellular basis for airflow limitation in smokers. Eur. Respir. J. 1994, 7, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, M.Q.; Reid, D.; Ward, C.; Muller, H.K.; Knight, D.A.; Sohal, S.S.; Walters, E.H. Transforming growth factor (TGF) beta1 and Smad signalling pathways: A likely key to EMT-associated COPD pathogenesis. Respirology 2017, 22, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Helmig, S.; Stephan, P.; Dohrel, J.; Schneider, J. TNF-alpha mRNA expression correlates with TGF-beta mRNA expression in vivo. Inflammation 2011, 34, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ge, Q.; Tjin, G.; Alkhouri, H.; Deng, L.; Brandsma, C.A.; Adcock, I.; Timens, W.; Postma, D.; Burgess, J.K.; et al. Effects of cigarette smoke extract on human airway smooth muscle cells in COPD. Eur. Respir. J. 2014, 44, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.J.; Pedersen, B.; Frederiksen, R.; Dahl, R. Prospective study on the effect of smoking and nicotine substitution on leucocyte blood counts and relation between blood leucocytes and lung function. Thorax 1998, 53, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, H.; Tanaka, M.; Takami, K.; Ohtoshi, T.; Ito, K.; Satoh, M.; Okada, Y.; Yamasawa, F.; Nakahara, K.; Umeda, A. Increased expression of transforming growth factor-beta1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Am. J. Respir. Crit. Care Med. 2001, 163, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Checa, M.; Hagood, J.S.; Velazquez-Cruz, R.; Ruiz, V.; Garcia-De-Alba, C.; Rangel-Escareno, C.; Urrea, F.; Becerril, C.; Montaño, M.; García-Trejo, S.; et al. Cigarette Smoke Enhances the Expression of Profibrotic Molecules in Alveolar Epithelial Cells. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.M.; Li, Y.L.; Chen, H.; Guo, S.L.; Shui, L.L.; Chen, Y.J. Cigarette smoke extract alters the cell cycle via the phospholipid transfer protein/transforming growth factor-beta1/CyclinD1/CDK4 pathway. Eur. J. Pharmacol. 2016, 786, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Robertson, J.; Maharaj, S.; Waldhauser, L.; Niu, J.; Wang, J.; Farkas, L.; Kolb, M.; Gauldie, J. Oxidative stress contributes to the induction and persistence of TGF-beta1 induced pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2011, 43, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, X.; Umino, T.; Sköld, C.M.; Zhu, Y.; Kohyama, T.; Spurzem, J.R.; Romberger, D.J.; Rennard, S.I. Cigarette smoke inhibits human bronchial epithelial cell repair processes. Am. J. Respir. Cell Mol. Biol. 2001, 25, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, D.M.; Gottschalk, R.W.; van Herwijnen, M.H.; Moonen, E.J.; Kleinjans, J.C.; van Delft, J.H. Differential gene expression in human peripheral blood mononuclear cells induced by cigarette smoke and its constituents. Toxicol. Sci. 2005, 86, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Rockett, J.C.; Burczynski, M.E.; Fornace, A.J.; Herrmann, P.C.; Krawetz, S.A.; Dix, D.J. Surrogate tissue analysis: Monitoring toxicant exposure and health status of inaccessible tissues through the analysis of accessible tissues and cells. Toxicol. Appl. Pharmacol. 2004, 194, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Abrigo, J.; Rivera, J.C.; Simon, F.; Cabrera, D.; Cabello-Verrugio, C. Transforming growth factor type beta (TGF-beta) requires reactive oxygen species to induce skeletal muscle atrophy. Cell Signal. 2016, 28, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Abrigo, J.; Morales, M.G.; Simon, F.; Cabrera, D.; Di Capua, G.; Cabello-Verrugio, C. Apocynin inhibits the upregulation of TGF-beta1 expression and ROS production induced by TGF-beta in skeletal muscle cells. Phytomedicine 2015, 22, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.-B. Aging, exercise, and extracellular matrix in the heart. J. Exerc. Rehabil. 2013, 9, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.K.; Cawston, T.E.; Hazleman, B.L. Transforming growth factor beta stimulates the production of the tissue inhibitor of metalloproteinases (TIMP) by human synovial and skin fibroblasts. Biochem. Biophys. Acta 1991, 1094, 207–210. [Google Scholar] [CrossRef]
- Yang, Z.Z.; Zou, A.P. Homocysteine enhances TIMP-1 expression and cell proliferation associated with NADH oxidase in rat mesangial cells. Kidney Int. 2003, 63, 1012–1020. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stanojkovic, I.; Kotur-Stevuljevic, J.; Spasic, S.; Milenkovic, B.; Vujic, T.; Stefanovic, A.; Ivanisevic, J. Relationship between bone resorption, oxidative stress and inflammation in severe COPD exacerbation. Clin. Biochem. 2013, 46, 1678–1682. [Google Scholar] [CrossRef] [PubMed]
- Tollefson, A.K.; Oberley-Deegan, R.E.; Butterfield, K.T.; Nicks, M.E.; Weaver, M.R.; Remigio, L.K.; Decsesznak, J.; Chu, H.W.; Bratton, D.L.; Riches, D.W.; et al. Endogenous enzymes (NOX and ECSOD) regulate smoke-induced oxidative stress. Free Radic. Biol. Med. 2010, 49, 1937–1946. [Google Scholar] [CrossRef] [PubMed]
- Alberg, A. The influence of cigarette smoking on circulating concentrations of antioxidant micronutrients. Toxicology 2002, 180, 121–137. [Google Scholar] [CrossRef]
- Haj-Mouhamed, D.; Ezzaher, A.; Neffati, F.; Douki, W.; Gaha, L.; Najjar, M.F. Effect of cigarette smoking on plasma uric acid concentrations. Environ. Health Prev. Med. 2011, 16, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Vardavas, C.I.; Linardakis, M.K.; Hatzis, C.M.; Malliaraki, N.; Saris, W.H.; Kafatos, A.G. Smoking status in relation to serum folate and dietary vitamin intake. Tob. Induc. Dis. 2008, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Pasupathi, P.; Saravanan, G.; Chinnaswamy, P.; Bakthavathsalam, G. Effect of chronic smoking on lipid peroxidation and antioxidant status in gastric carcinoma patients. Indian J. Gastroenterol. 2009, 28, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Stolarek, R.A.; Kasielski, M.; Rysz, J.; Bialasiewicz, P.; Nowak, D. Differential effect of cigarette smoking on hydrogen peroxide and thiobarbituric acid reactive substances exhaled in patients with community acquired pneumonia. Monaldi. Arch. Chest. Dis. 2006, 65, 19–25. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bloomer, R.J. Decreased blood antioxidant capacity and increased lipid peroxidation in young cigarette smokers compared to nonsmokers: Impact of dietary intake. Nutr. J. 2007, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.A.; Amaral, T.A.; Carvalho, L.C.; Ognibene, D.T.; da Silva, A.F.; Moss, M.B.; Valença, S.S.; de Moura, R.S.; Resende, A.C. Antioxidant treatment with tempol and apocynin prevents endothelial dysfunction and development of renovascular hypertension. Am. J. Hypertens. 2009, 22, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Heumuller, S.; Wind, S.; Barbosa-Sicard, E.; Schmidt, H.H.; Busse, R.; Schröder, K.; Brandes, R.P. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 2008, 51, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.H.; Chuang, C.H.; Liu, S.L.; Lee, T.S.; Kou, Y.R.; Zhang, H. Apocynin attenuates ventilator-induced lung injury in an isolated and perfused rat lung model. Intensive Care Med. 2011, 37, 1360–1367. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, M.; Stefanska, J.; Wodz-Naskiewicz, K.; Cieslak, M.; Pawliczak, R. Cytosolic phospholipase A2 group IVA is overexpressed in patients with persistent asthma and regulated by the promoter microsatellites. J. Allergy Clin. Immunol. 2010, 125, 1393–1395. [Google Scholar] [CrossRef] [PubMed]
- Stefanska, J.; Sarniak, A.; Wlodarczyk, A.; Sokolowska, M.; Doniec, Z.; Bialasiewicz, P.; Nowak, D.; Pawliczak, R. Hydrogen peroxide and nitrite reduction in exhaled breath condensate of COPD patients. Pulm Pharmacol. Ther. 2012, 25, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Lu, Y.; Ping, N.N.; Li, X.; Lin, Y.X.; Li, C.F. Apocynin ameliorates pressure overload-induced cardiac remodeling by inhibiting oxidative stress and apoptosis. Physiol. Res. 2017, 66, 741–752. [Google Scholar] [PubMed]
- Li, Y.Q.; Li, X.B.; Guo, S.J.; Chu, S.L.; Gao, P.J.; Zhu, D.L.; Niu, W.Q.; Jia, N. Apocynin attenuates oxidative stress and cardiac fibrosis in angiotensin II-induced cardiac diastolic dysfunction in mice. Acta Pharmacol. Sin. 2013, 34, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhao, T.; Chen, Y.; Ahokas, R.A.; Sun, Y. Oxidative stress mediates cardiac fibrosis by enhancing transforming growth factor-beta1 in hypertensive rats. Mol. Cell Biochem. 2008, 317, 43–50. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Smokers (n = 8) | Nonsmokers (n = 8) |
|---|---|---|
| Women | 4 | 5 |
| Men | 4 | 3 |
| Age (yrs) [min-max] | 27 ± 6 | 25 ± 6 |
| BMI | 23 ± 2 | 22 ± 3 |
| Cigarettes per day | 13 ± 8 | NA |
| Years smoking | 6 ± 5 | NA |
| Smoking Pack-Years | 4 ± 3 | NA |
| FEV1 [%] | 95 | 101 |
| FVC [%] | 93 | 99 |
| Cells Treatment | Cell Viability [%] | |
|---|---|---|
| Smokers | Nonsmokers | |
| Control | 95 | 96 |
| Apocynin | 94 | 94 |
| CSI | 86 | 87 |
| TGF-β | ARG | MMP-9 | TIMP-1 | |||||
|---|---|---|---|---|---|---|---|---|
| Nsm | Sm | Nsm | Sm | Nsm | Sm | Nsm | Sm | |
| TBARs PBMC | −0.13 | −0.2 | 0.3 | 0.2 | −0.5 | 0.14 | −0.03 | 0.09 |
| TBARs serum | −0.01 | −0.47 | 0.1 | 0.1 | −0.5 | −0.21 | 0.2 | 0.07 |
| thiols | 0.18 | 0.38 | 0.5 | 0.3 | −0.02 | −0.01 | 0.2 | −0.1 |
| SOD1 | 0.6 | 0.13 | 0.3 | 0.91 * | −0.3 * | 0.83 * | −0.1 | −0.2 |
| SOD1 mRNA | 0.4 | 0.02 | 0.38 * | −0.6 * | 0.33 | −0.72 | 0.3 | 0.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wieczfinska, J.; Kowalczyk, T.; Sitarek, P.; Skała, E.; Pawliczak, R. Analysis of Short-Term Smoking Effects in PBMC of Healthy Subjects—Preliminary Study. Int. J. Environ. Res. Public Health 2018, 15, 1021. https://doi.org/10.3390/ijerph15051021
Wieczfinska J, Kowalczyk T, Sitarek P, Skała E, Pawliczak R. Analysis of Short-Term Smoking Effects in PBMC of Healthy Subjects—Preliminary Study. International Journal of Environmental Research and Public Health. 2018; 15(5):1021. https://doi.org/10.3390/ijerph15051021
Chicago/Turabian StyleWieczfinska, Joanna, Tomasz Kowalczyk, Przemyslaw Sitarek, Ewa Skała, and Rafal Pawliczak. 2018. "Analysis of Short-Term Smoking Effects in PBMC of Healthy Subjects—Preliminary Study" International Journal of Environmental Research and Public Health 15, no. 5: 1021. https://doi.org/10.3390/ijerph15051021
APA StyleWieczfinska, J., Kowalczyk, T., Sitarek, P., Skała, E., & Pawliczak, R. (2018). Analysis of Short-Term Smoking Effects in PBMC of Healthy Subjects—Preliminary Study. International Journal of Environmental Research and Public Health, 15(5), 1021. https://doi.org/10.3390/ijerph15051021