Rhabdomyolysis in a Young Girl with Van Wyk-Grumbach Syndrome due to Severe Hashimoto Thyroiditis



Abstract
:1. Introduction
2. Case Presentation
3. Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Diaz, A.; Lipman Diaz, E.G. Hypothyroidism. Pediatr. Rev. 2014, 35, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Hanley, P.; Lord, K.; Bauer, A.J. Thyroid disorders in children and adolescents: A review. JAMA Pediatr. 2016, 170, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Esen, I.; Demirel, F. Hypothyroidism-associated testicular enlargement: Is it a form of precocious puberty or not? A case report. Turk. J. Pediatr. 2011, 53, 210–212. [Google Scholar] [PubMed]
- Baranowski, E.; Hogler, W. An unusual presentation of acquired hypothyroidism: The Van Wyk Grumbach syndrome. Eur. J. Endocrinol. 2012, 166, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Bassam, T.; Ajlouni, K. A case of ovarian enlargement in severe primary hypothyroidism and review of the literature. Ann. Saudi Med. 2006, 26, 66–68. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.; Xing, L.; Zhang, L.; Fang, S.; Huang, H. Ignored adult primary hypothyroidism presenting chiefly with persistent ovarian cysts: A need for increased awareness. Reprod. Biol. Endocrinol. 2011, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Wormsbecker, A.; Clarson, C. Acquired primary hypothyroidism: Vaginal bleeding in a quiet child. CMAJ 2010, 182, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Geng, N.V.; Wang, Y.M.; Tian, W.Y.; Xue, F.X. Van Wyk and Grumbach syndrome: Two case reports and review of the published work. J. Obstet. Gynaecol. Res. 2014, 40, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Anasti, J.N.; Flack, M.R.; Froehlich, J.; Nelson, L.M.; Nisula, B.C. A potential novel mechanism for precocious puberty in juvenile hypothyroidism. J. Clin. Endocrinol. Metab. 1995, 80, 276–279. [Google Scholar] [PubMed]
- Browne, L.P.; Boswell, H.B.; Crotty, E.J.; O’Hara, S.M.; Birkemeier, K.L.; Guillerman, R.P. Van Wyk and Grumbach syndrome revisited: Imaging and clinical findings in pre- and postpubertal girls. Pediatr. Radiol. 2008, 38, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Benvenga, S.; Toscano, A.; Rodolico, C.; Vita, G.; Trimarchi, F. Endocrine evaluation for muscle pain. J. R. Soc. Med. 2001, 94, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Maity, C. Alteration of serum enzymes in primary hypothyroidism. Clin. Chem. Lab. Med. 2002, 40, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Serranti, D.; Indolfi, G.; Bartolini, E.; Galli, L.; de Martino, M.; Resti, M. Raised serum aminotransferase levels and muscle pseudohypertrophy caused by hypothyroidism. J. Pediatr. Gastroenterol. Nutr. 2013, 56, e48–e49. [Google Scholar] [CrossRef] [PubMed]
- Sindoni, A.; Rodolico, C.; Pappalardo, M.A.; Portaro, S.; Benvenga, S. Hypothyroid myopathy: A peculiar clinical presentation of thyroid failure. Review of the literature. Rev. Endocr. Metab. Disord. 2016, 17, 499–519. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.G.; Moore, K.P. Pathogenesis and treatment of renal dysfunction in rhabdomyolysis. Intensive Care Med. 2001, 27, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Bosch, X.; Poch, E.; Grau, J.M. Rhabdomyolysis and acute kidney injury. N. Engl. J. Med. 2009, 361, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Marzuillo, P.; Grandone, A.; Perrotta, S.; Ruggiero, L.; Capristo, C.; Luongo, C.; Miraglia del Giudice, E.; Perrone, L. Very early onset of autoimmune thyroiditis in a toddler with severe hypothyroidism presentation: A case report. Ital. J. Pediatr. 2016, 42, 61. [Google Scholar] [CrossRef] [PubMed]
- Gunther, D.F.; Chiu, H.K.; Numrych, T.E.; Kletter, G.B. Onset of acquired autoimmune hypothyroidism in infancy: A presentation of delayed gross-motor development and rhabdomyolysis. Eur. J. Pediatr. 2006, 165, 320–322. [Google Scholar] [CrossRef] [PubMed]
- Galli-Tsinopoulou, A.; Stylianou, C.; Kokka, P.; Panagopoulou, P.; Nousia-Arvanitakis, S. Rhabdomyolysis, renal failure, pericardial effusion, and acquired von Willebrand disease resulting from hypothyroidism in a 10-year-old girl. Thyroid 2008, 18, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Cimbek, E.A.; Şen, Y.; Yuca, S.A.; Çam, D.; Gür, C.; Peru, H. Kocher-Debré-Semelaigne syndrome with rhabdomyolysis and increased creatinine. J. Pediatr. Endocrinol. Metab. 2015, 28, 1383–1385. [Google Scholar] [CrossRef] [PubMed]
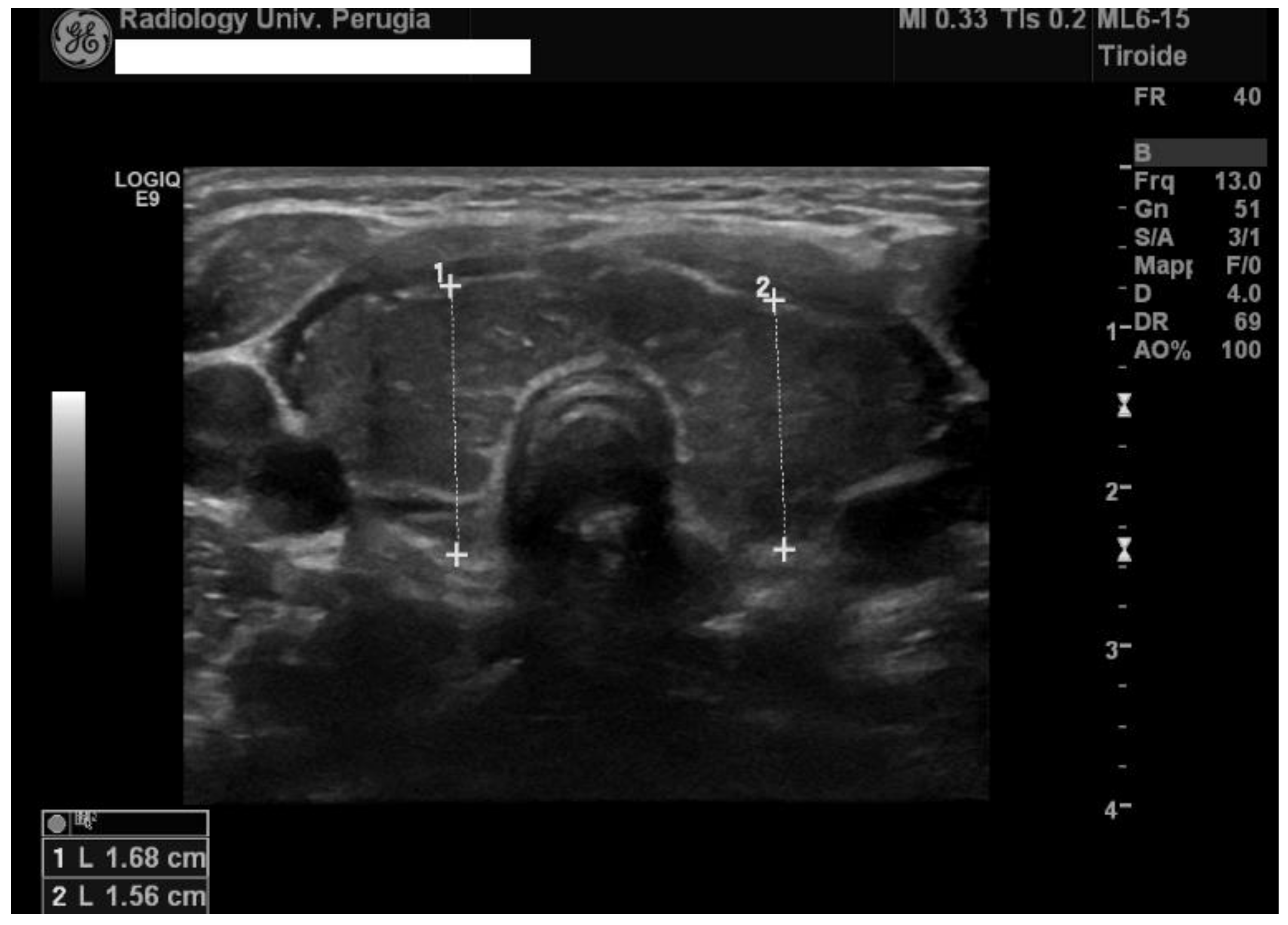

{kind=link}
| Test (Reference Interval) | 0 Day | 1 Week | 1 Month | 3 Months |
|---|---|---|---|---|
| TSH (0.340–5.600 µIU/mL) | 357 | 6.37 | 1.49 | |
| fT4 (0.54–1.24 ng/dL) | 0.26 | 1.00 | 0.89 | |
| Anti-TPO antibodies (0.0–9.0 UI/mL) | 3.4 | |||
| Anti-thyroglobulin antibodies (0–4.9 UI/mL) | 73.7 | |||
| FSH (1.6–9.6 mUI/mL) | 10.2 | |||
| LH (1–11 mIU/mL) | 0.10 | |||
| Estradiol (20–60 pg/mL) | 86 | |||
| SGOT (<45 UI/L) | 105 | 44 | ||
| LDH (225–450 UI/L) | 748 | 496 | 436 | |
| CPK (0–180 UI/L) | 3395 | 392 | 170 | |
| Myoglobin (14.3–65.8 ng/mL) | 76 | 52 | ||
| Total cholesterol (95th percentile: 197 mg/dL) | 294 | 209 | 156 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leonardi, A.; Penta, L.; Cofini, M.; Lanciotti, L.; Principi, N.; Esposito, S. Rhabdomyolysis in a Young Girl with Van Wyk-Grumbach Syndrome due to Severe Hashimoto Thyroiditis. Int. J. Environ. Res. Public Health 2018, 15, 704. https://doi.org/10.3390/ijerph15040704
Leonardi A, Penta L, Cofini M, Lanciotti L, Principi N, Esposito S. Rhabdomyolysis in a Young Girl with Van Wyk-Grumbach Syndrome due to Severe Hashimoto Thyroiditis. International Journal of Environmental Research and Public Health. 2018; 15(4):704. https://doi.org/10.3390/ijerph15040704
Chicago/Turabian StyleLeonardi, Alberto, Laura Penta, Marta Cofini, Lucia Lanciotti, Nicola Principi, and Susanna Esposito. 2018. "Rhabdomyolysis in a Young Girl with Van Wyk-Grumbach Syndrome due to Severe Hashimoto Thyroiditis" International Journal of Environmental Research and Public Health 15, no. 4: 704. https://doi.org/10.3390/ijerph15040704
APA StyleLeonardi, A., Penta, L., Cofini, M., Lanciotti, L., Principi, N., & Esposito, S. (2018). Rhabdomyolysis in a Young Girl with Van Wyk-Grumbach Syndrome due to Severe Hashimoto Thyroiditis. International Journal of Environmental Research and Public Health, 15(4), 704. https://doi.org/10.3390/ijerph15040704