
The AhR Ligand, TCDD, Regulates Androgen Receptor Activity Differently in Androgen-Sensitive versus Castration-Resistant Human Prostate Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Protein Isolation and Western Blot Analysis
2.4. Immunocytochemical Staining and Fluorescence Microscopy
2.5. RNA Extraction and Quantitative RT-PCR Analysis
2.6. Statistical Analysis
3. Results
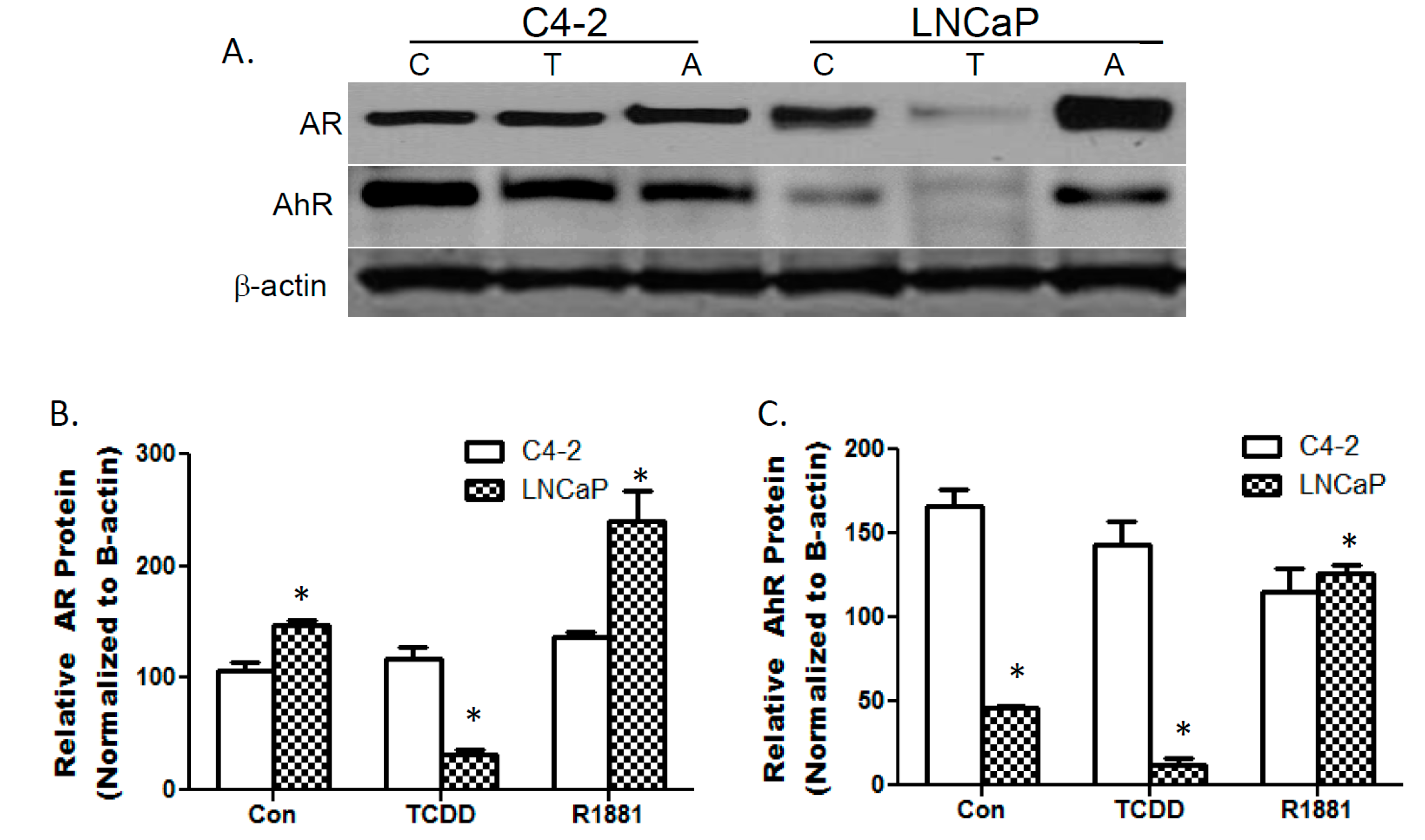
3.1. TCDDs Effect on AhR and AR Protein Expression in LNCaP and C4-2 Prostate Cancer Cells
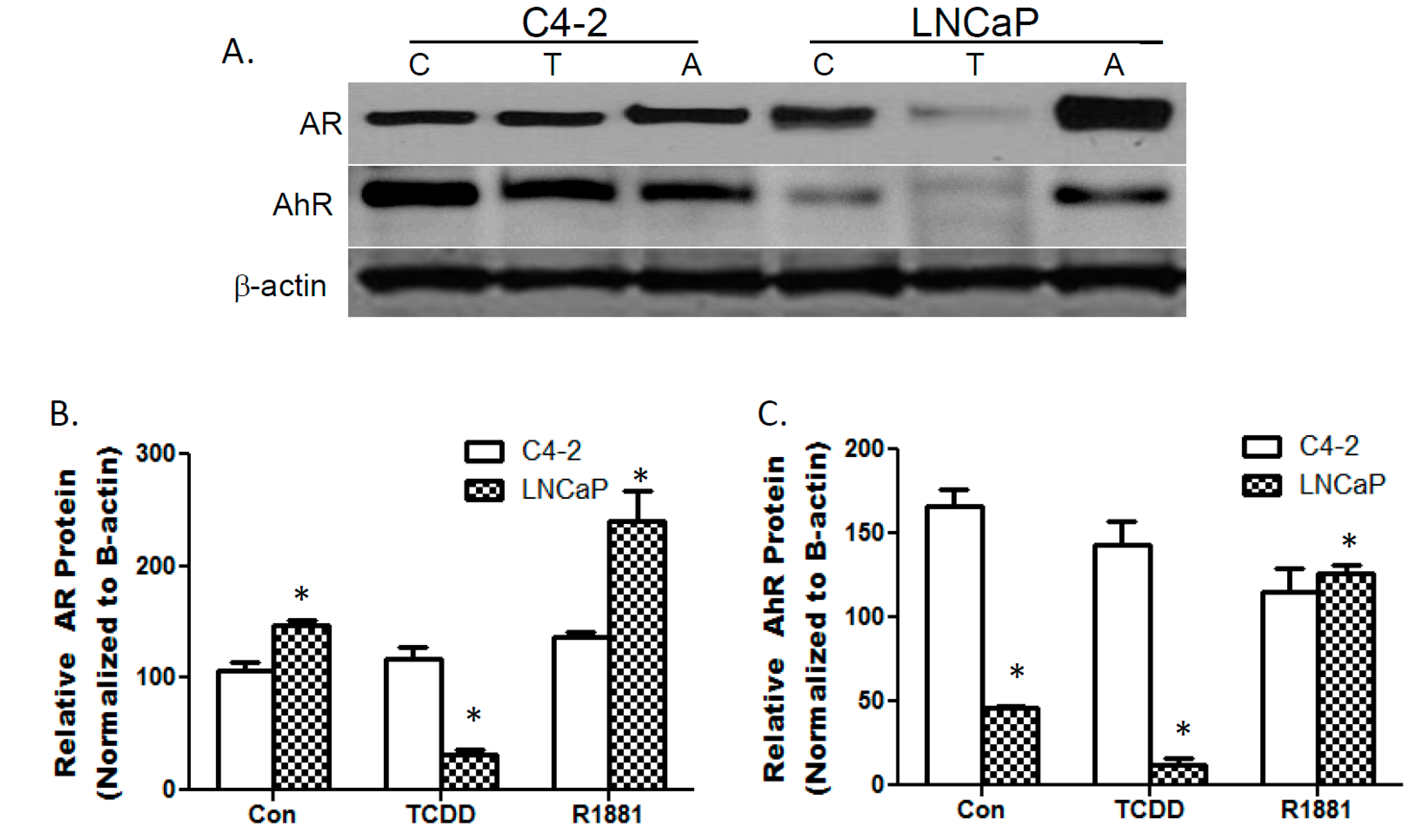
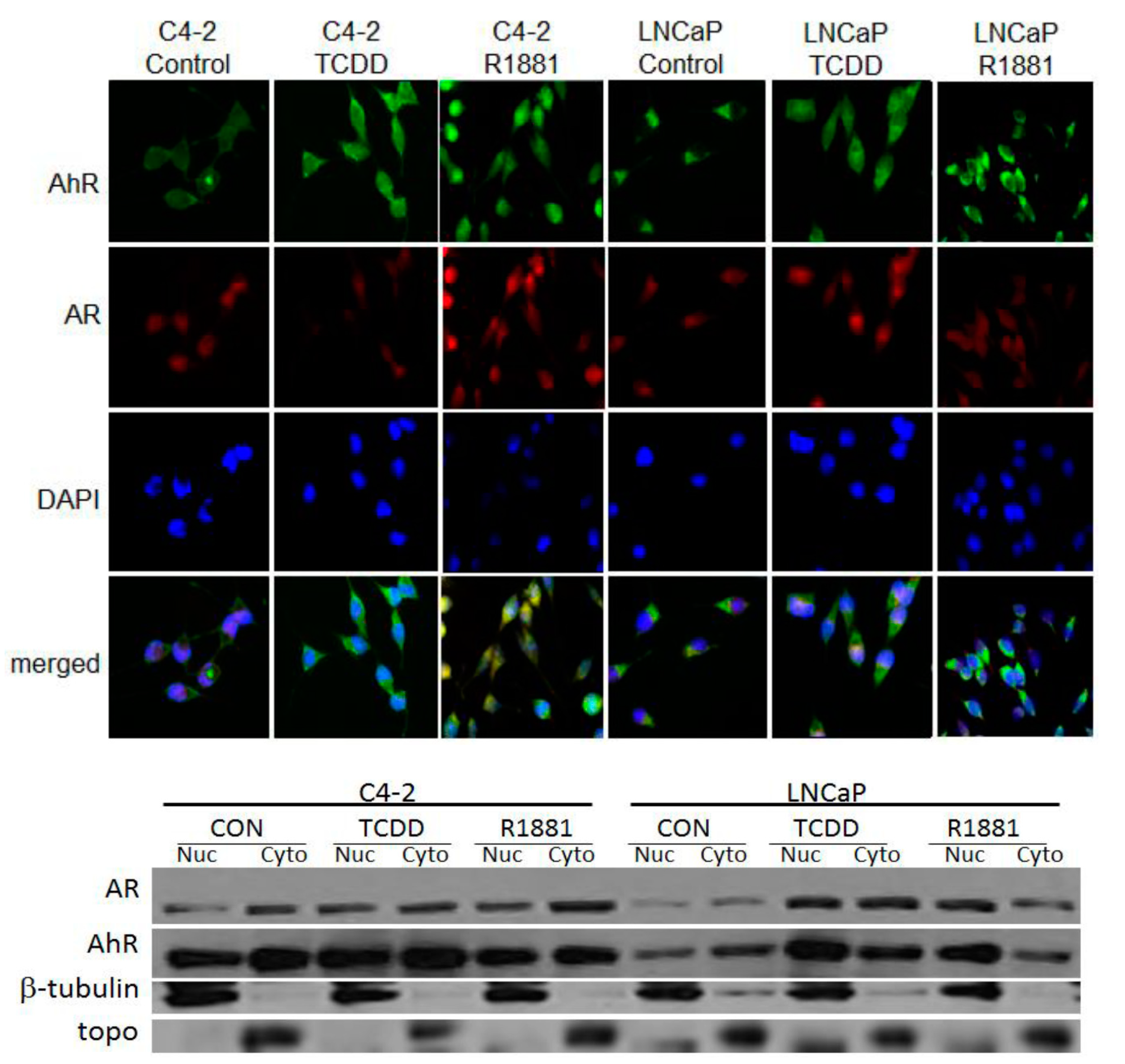
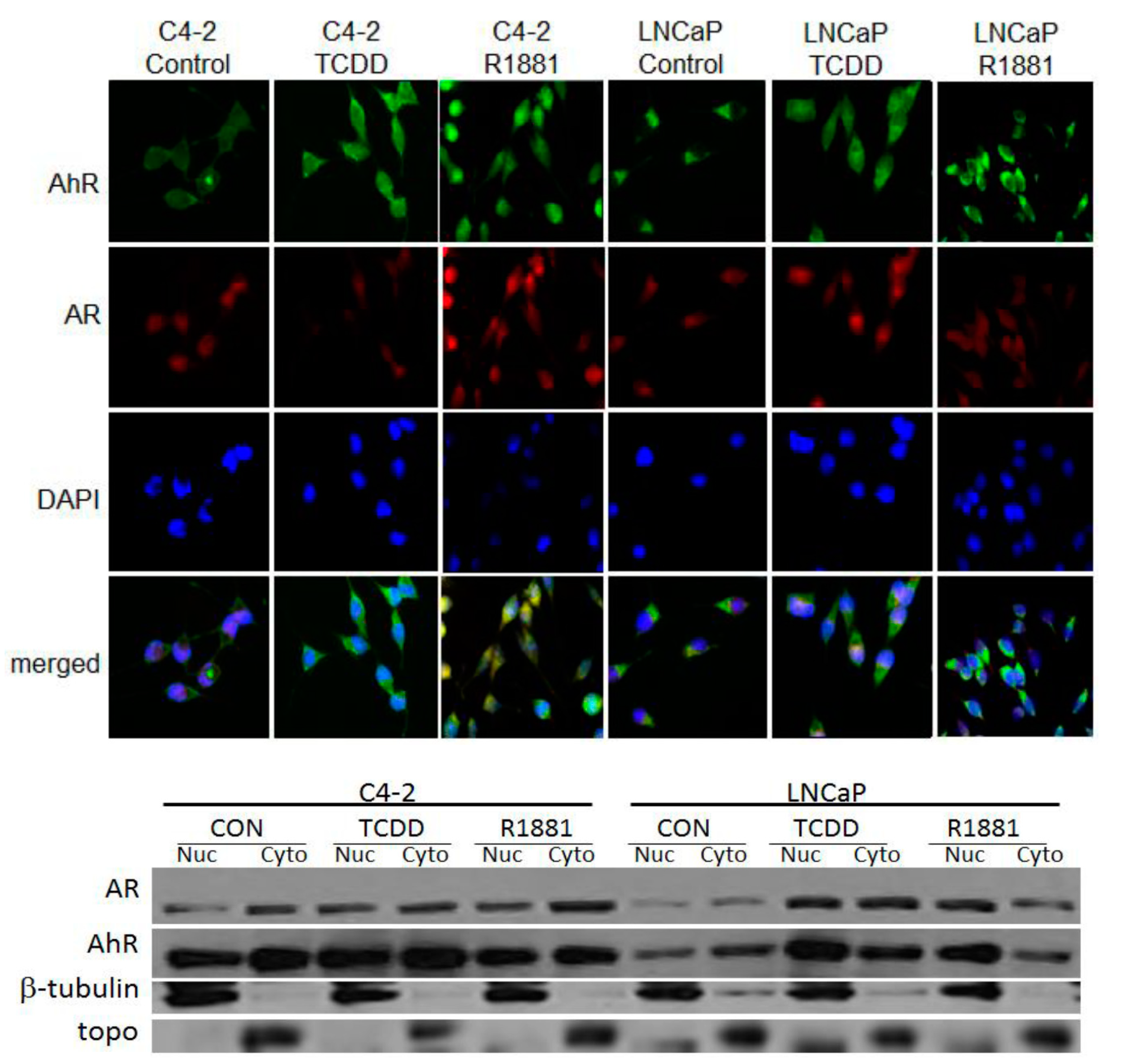
3.2. TCDD Induces Nuclear Localization of AR in LNCaP but Not in C4-2 Prostate Cancer Cells
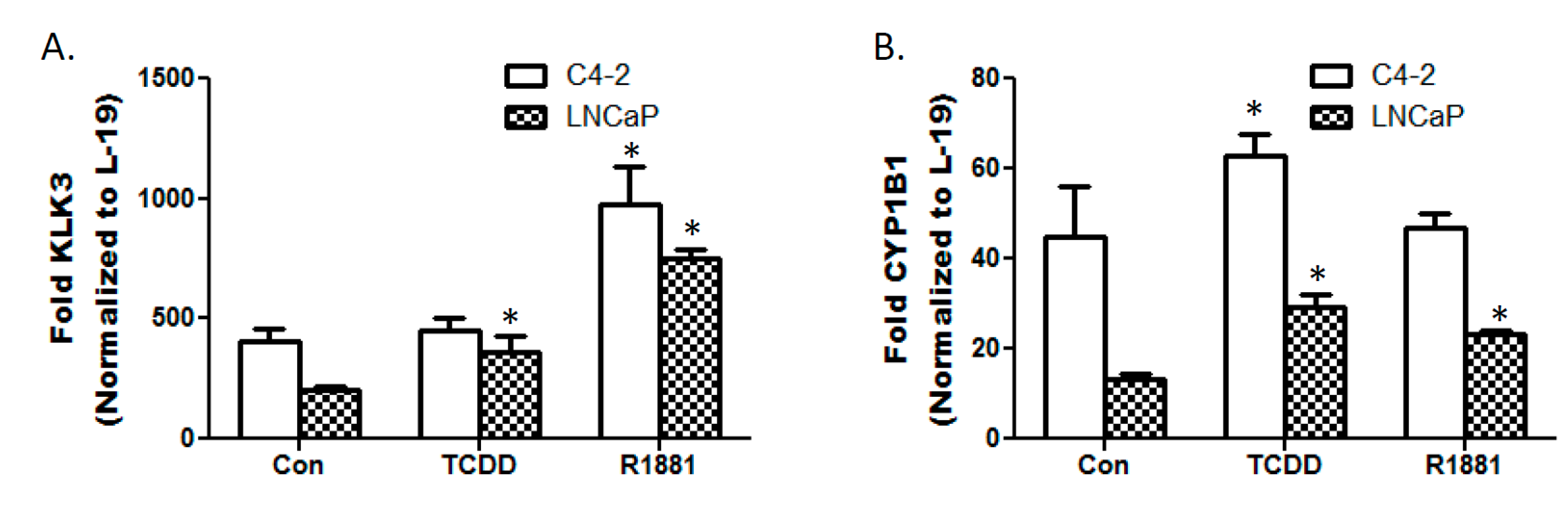
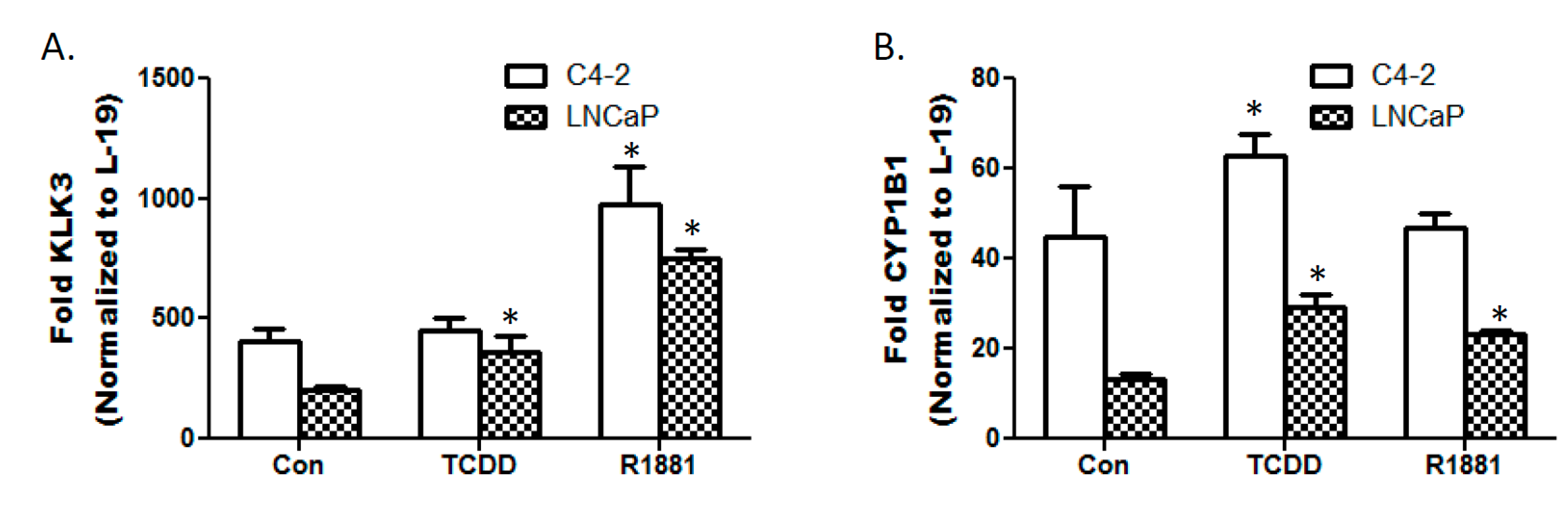
3.3. TCDD Enhances Expression of Androgen Responsive Gene KLK3 in LNCaP Cells
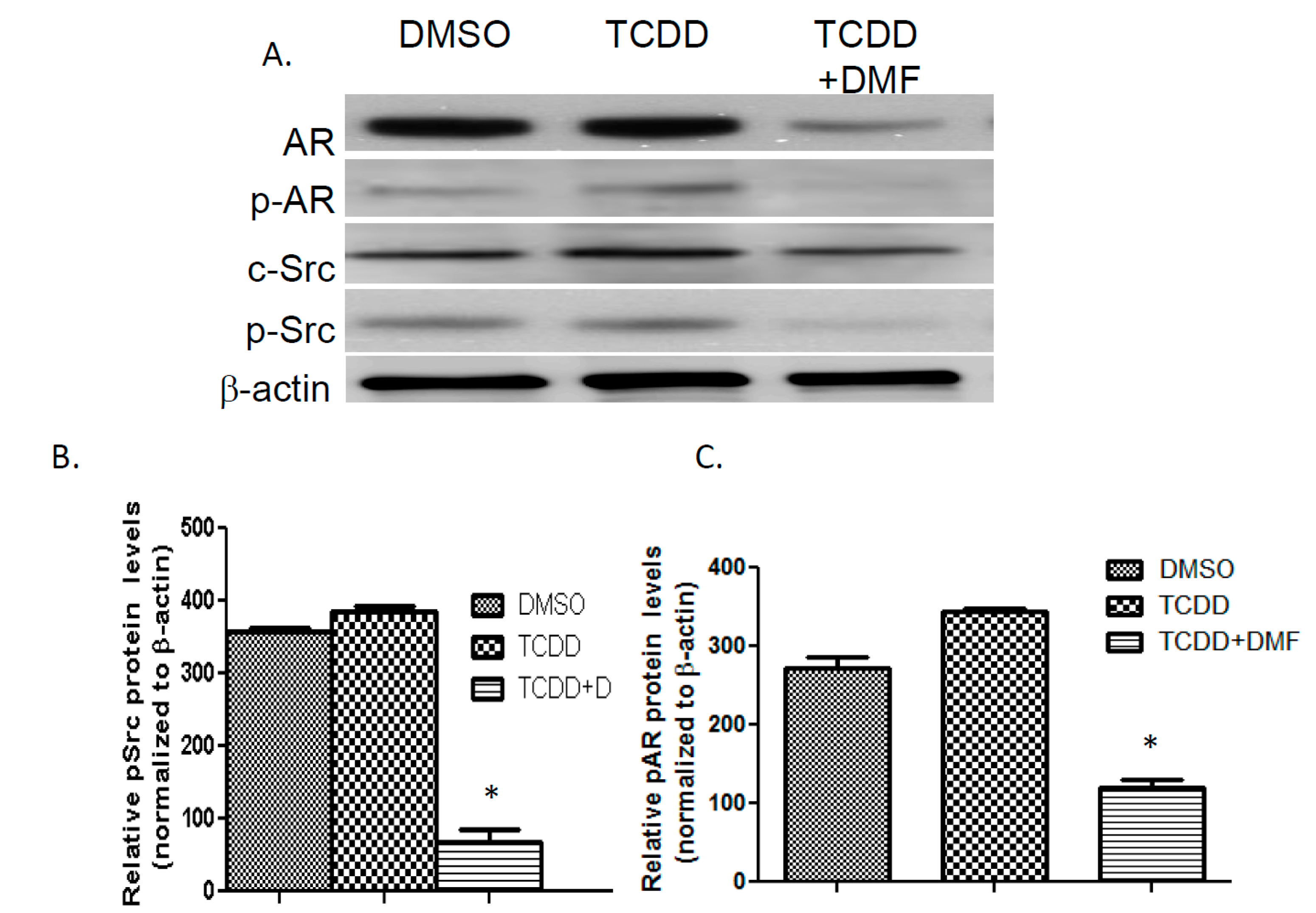
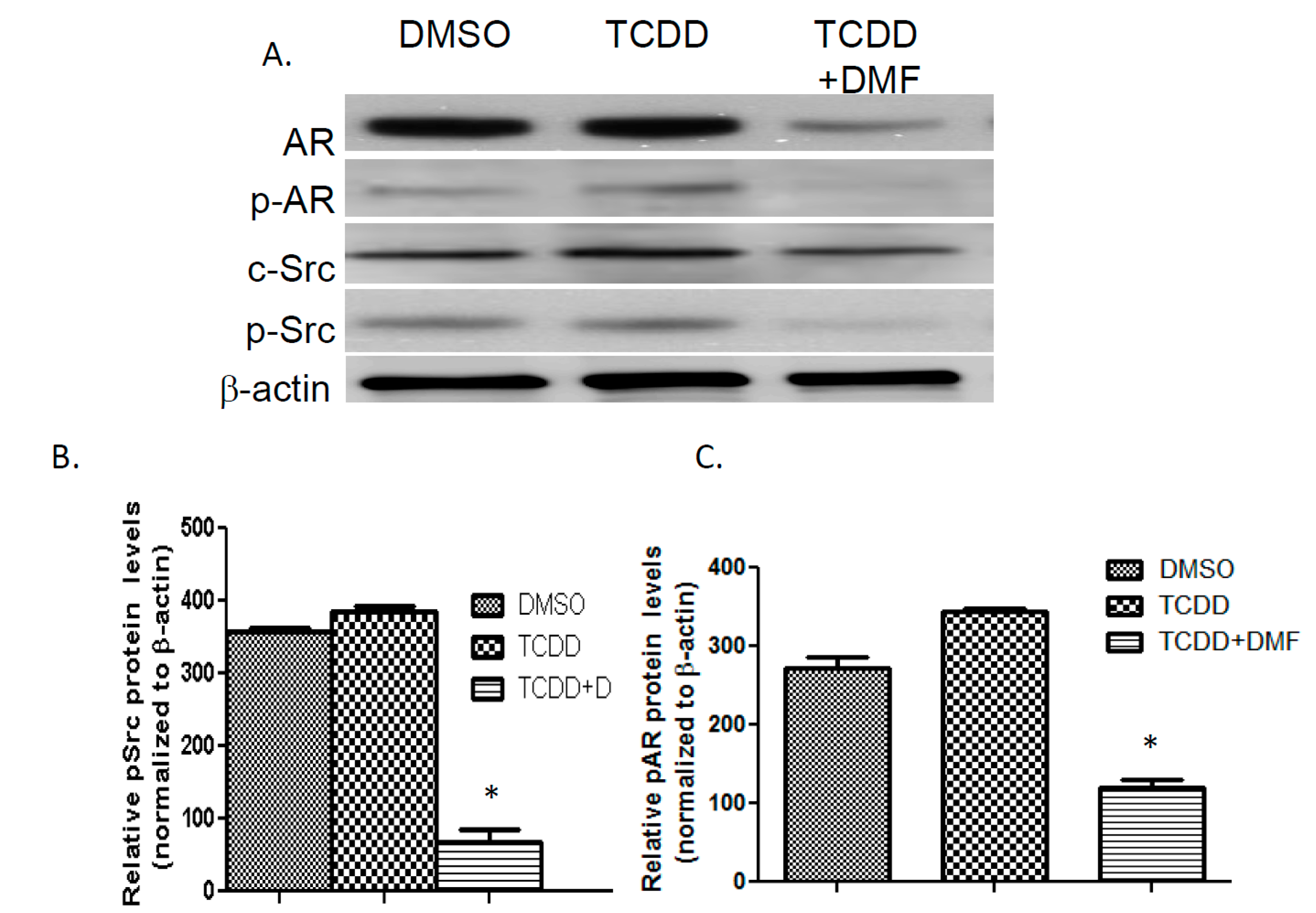
3.4. TCDD Induces AR Phosphorylation in LNCaP Cells
4. Discussion
5. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- (IARC), International Agency for Research on Cancer. Polychlorinated Dibenzo-Para-Dioxins and Polychlorinated Dibenzofurans. In Proceedings of IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, Lyon, France, 4–11 February 1997.
- U.S. Environmental Protection Agency, EPA. Health Assessment Document for Polychlorinated Dibenzo-p-Dioxin; EPA-HQ-SFUND-2009-0907; Environmental Criteria and Assessment Office, Office of Health and Environmental Assessment, Office of Research and Development: Washington, DC, USA, 2009.
- Travis, C.C.; Hattemer-Frey, H.A. Human exposure to dioxin. Sci. Total Environ. 1991, 104, 97–127. [Google Scholar] [CrossRef]
- Safe, S. Development of bioassays and approaches for the risk assessment of 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds. Environ. Health Perspect. 1993, 101 (Suppl. 3), 317–325. [Google Scholar]
- Schecter, A.; Birnbaum, L.; Ryan, J.J.; Constable, J.D. Dioxins: An overview. Environ. Res. 2006, 101, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Knerr, S.; Schrenk, D. Carcinogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in experimental models. Mol. Nutr. Food Res. 2006, 50, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.J.; McCurdy, E.A.; Miller, B.D.; Lucier, G.W.; Tritscher, A.M. Ovarian tumors in rats induced by chronic 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment. Cancer Res. 2000, 60, 5414–5419. [Google Scholar] [PubMed]
- Brunnberg, S.; Andersson, P.; Poellinger, L.; Hanberg, A. The constitutively active Ah receptor (CA-AhR) mouse as a model for dioxin exposure—Effects in reproductive organs. Chemosphere 2011, 85, 1701–1706. [Google Scholar] [CrossRef] [PubMed]
- Jana, N.R.; Sarkar, S.; Ishizuka, M.; Yonemoto, J.; Tohyama, C.; Sone, H. Cross-talk between 2,3,7,8-tetrachlorodibenzo-p-dioxin and testosterone signal transduction pathways in LNCaP prostate cancer cells. Biochem. Biophys. Res. Commun. 1999, 256, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Vorrink, S.U.; Domann, F.E. Regulatory crosstalk and interference between the xenobiotic and hypoxia sensing pathways at the AhR-ARNT-HIF1alpha signaling node. Chem. Biol. Interact. 2014, 218, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; McDougal, A. Mechanism of action and development of selective aryl hydrocarbon receptor modulators for treatment of hormone-dependent cancers (Review). Int. J. Oncol. 2002, 20, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Enan, E.; Matsumura, F. Identification of c-Src as the integral component of the cytosolic Ah receptor complex, transducing the signal of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) through the protein phosphorylation pathway. Biochem. Pharmacol. 1996, 52, 1599–1612. [Google Scholar] [CrossRef]
- Richmond, O.; Ghotbaddini, M.; Allen, C.; Walker, A.; Zahir, S.; Powell, J.B. The aryl hydrocarbon receptor is constitutively active in advanced prostate cancer cells. PLoS One 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Hoivik, D.; Willett, K.; Wilson, C.; Safe, S. Estrogen does not inhibit 2,3,7, 8-tetrachlorodibenzo-p-dioxin-mediated effects in MCF-7 and Hepa 1c1c7 cells. J. Biol. Chem. 1997, 272, 30270–30274. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [PubMed]
- Kharat, I.; Saatcioglu, F. Antiestrogenic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin are mediated by direct transcriptional interference with the liganded estrogen receptor. Cross-talk between aryl hydrocarbon- and estrogen-mediated signaling. J. Biol. Chem. 1996, 271, 10533–10537. [Google Scholar] [PubMed]
- Dong, H.V.; Lee, A.H.; Nga, N.H.; Quang, N.; Chuyen, V.L.; Binns, C.W. Epidemiology and prevention of prostate cancer in Vietnam. Asian Pac. J. Cancer Prev. 2014, 15, 9747–9751. [Google Scholar] [CrossRef] [PubMed]
- Mandair, D.; Rossi, R.E.; Pericleous, M.; Whyand, T.; Caplin, M.E. Prostate cancer and the influence of dietary factors and supplements: A systematic review. Nutr. Metab. (Lond.) 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Schecter, A.; Aragaki, C.C.; Roehrborn, C.G. Dioxin exposure and benign prostatic hyperplasia. J. Occup. Environ. Med. 2006, 48, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.M.; Rasmussen, N.T.; Moore, R.W.; Albrecht, R.M.; Peterson, R.E. 2,3,7,8-tetrachlorodibenzo-p-dioxin inhibits prostatic epithelial bud formation by acting directly on the urogenital sinus. J. Urol. 2004, 172, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Schaufler, K.; Haslmayer, P.; Jager, W.; Pec, M.; Thalhammer, T. The environmental toxin 2,3,7,8-tetrachlorodibenzo-p-dioxin induces cytochrome P450 activity in high passage PC 3 and DU 145 human prostate cancer cell lines. Int. J. Mol. Med. 2002, 9, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.C.; Hsieh, J.T.; Gleave, M.E.; Brown, N.M.; Pathak, S.; Chung, L.W. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: Role of bone stromal cells. Int. J. Cancer 1994, 57, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Nakka, M.; Agoulnik, I.U.; Weigel, N.L. Targeted disruption of the p160 coactivator interface of androgen receptor (AR) selectively inhibits AR activity in both androgen-dependent and castration-resistant AR-expressing prostate cancer cells. Int. J. Biochem. Cell Biol. 2013, 45, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.; Richmond, O.; Aaron, L.; Powell, J.B. Inhibition of constitutive aryl hydrocarbon receptor (AhR) signaling attenuates androgen independent signaling and growth in (C4-2) prostate cancer cells. Biochem. Pharmacol. 2013, 85, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, A.; Castoria, G.; Di Domenico, M.; de Falco, A.; Bilancio, A.; Lombardi, M.; Barone, M.V.; Ametrano, D.; Zannini, M.S.; Abbondanza, C.; et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000, 19, 5406–5417. [Google Scholar] [CrossRef] [PubMed]
- Poland, A.; Knutson, J.C. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: Examination of the mechanism of toxicity. Annu. Rev. Pharmacol. Toxicol. 1982, 22, 517–554. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Whitlock, J.P., Jr. The aromatic hydrocarbon receptor modulates the Hepa 1c1c7 cell cycle and differentiated state independently of dioxin. Mol. Cell Biol. 1996, 16, 2144–2150. [Google Scholar] [PubMed]
- Fernandez-Salguero, P.; Pineau, T.; Hilbert, D.M.; McPhail, T.; Lee, S.S.; Kimura, S.; Nebert, D.W.; Rudikoff, S.; Ward, J.M.; Gonzalez, F.J. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 1995, 268, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Fujii-Kuriyama, Y.; Ema, M.; Mimura, J.; Matsushita, N.; Sogawa, K. Polymorphic forms of the Ah receptor and induction of the CYP1A1 gene. Pharmacogenetics 1995, 5, S149–S153. [Google Scholar] [CrossRef] [PubMed]
- Davarinos, N.A.; Pollenz, R.S. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J. Biol. Chem. 1999, 274, 28708–28715. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Baldwin, K.T. 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway. Role of the transcription activaton and DNA binding of AhR. J. Biol. Chem. 2000, 275, 8432–8438. [Google Scholar] [CrossRef] [PubMed]
- Pollenz, R.S.; Necela, B.; Marks-Sojka, K. Analysis of rainbow trout Ah receptor protein isoforms in cell culture reveals conservation of function in Ah receptor-mediated signal transduction. Biochem. Pharmacol. 2002, 64, 49–60. [Google Scholar] [CrossRef]
- Lin, P.; Chang, J.T.; Ko, J.L.; Liao, S.H.; Lo, W.S. Reduction of androgen receptor expression by benzo[alpha]pyrene and 7,8-dihydro-9,10-epoxy-7,8,9,10-tetrahydrobenzo[alpha]pyrene in human lung cells. Biochem. Pharmacol. 2004, 67, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.; Qin, C.; Smith, R., 3rd.; Safe, S. Aryl hydrocarbon receptor-mediated inhibition of LNCaP prostate cancer cell growth and hormone-induced transactivation. J. Steroid. Biochem. Mol. Biol. 2004, 88, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Barnes-Ellerbe, S.; Knudsen, K.E.; Puga, A. 2,3,7,8-Tetrachlorodibenzo-p-dioxin blocks androgen-dependent cell proliferation of LNCaP cells through modulation of pRB phosphorylation. Mol. Pharmacol. 2004, 66, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Baba, A.; Fujii-Kuriyama, Y.; Kato, S. Intrinsic AhR function underlies cross-talk of dioxins with sex hormone signaling. Biochem. Biophys. Res. Commun. 2008, 370, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Cheng, W.; Li, W.; Zheng, J.; Wu, D.; Matsumura, F.; Vogel, C.F. FRET analysis of protein tyrosine kinase c-Src activation mediated via aryl hydrocarbon receptor. Biochim. Biophys. Acta 2011, 1810, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Yang, L.; Feldman, R.I.; Sun, X.M.; Bhalla, K.N.; Jove, R.; Nicosia, S.V.; Cheng, J.Q. Activation of phosphatidylinositol 3-kinase/Akt pathway by androgen through interaction of p85alpha, androgen receptor, and Src. J. Biol. Chem. 2003, 278, 42992–43000. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Gillard, B.; Gao, L.; Eng, K.H.; Gelman, I.H. Src controls castration recurrence of CWR22 prostate cancer xenografts. Cancer Med. 2013, 2, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Bjork, C.; Giwercman, Y.L. Androgen receptor CAG repeat length modifies the effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on receptor activity in human prostate cells. Reprod. Toxicol. 2013, 35, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.M.; Ko, K.; Moore, R.W.; Simananinen, U.; Oberley, T.; Peterson, R.E. Effects of aryl hydrocarbon receptor null mutation and in utero and lactational 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure on prostate and seminal vesicle development in C57BL/6 mice. Toxicol. Sci. 2002, 68, 479–487. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghotbaddini, M.; Powell, J.B. The AhR Ligand, TCDD, Regulates Androgen Receptor Activity Differently in Androgen-Sensitive versus Castration-Resistant Human Prostate Cancer Cells. Int. J. Environ. Res. Public Health 2015, 12, 7506-7518. https://doi.org/10.3390/ijerph120707506
Ghotbaddini M, Powell JB. The AhR Ligand, TCDD, Regulates Androgen Receptor Activity Differently in Androgen-Sensitive versus Castration-Resistant Human Prostate Cancer Cells. International Journal of Environmental Research and Public Health. 2015; 12(7):7506-7518. https://doi.org/10.3390/ijerph120707506
Chicago/Turabian StyleGhotbaddini, Maryam, and Joann B. Powell. 2015. "The AhR Ligand, TCDD, Regulates Androgen Receptor Activity Differently in Androgen-Sensitive versus Castration-Resistant Human Prostate Cancer Cells" International Journal of Environmental Research and Public Health 12, no. 7: 7506-7518. https://doi.org/10.3390/ijerph120707506
APA StyleGhotbaddini, M., & Powell, J. B. (2015). The AhR Ligand, TCDD, Regulates Androgen Receptor Activity Differently in Androgen-Sensitive versus Castration-Resistant Human Prostate Cancer Cells. International Journal of Environmental Research and Public Health, 12(7), 7506-7518. https://doi.org/10.3390/ijerph120707506