
NNK, a Tobacco-Specific Carcinogen, Inhibits the Expression of Lysyl Oxidase, a Tumor Suppressor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Cell Culture and NNK Exposure
2.3. Assay for LOX Activities
2.4. Western Blot Analysis
2.5. Reverse Transcription (RT) and Quantitative Real-Time PCR Analysis
2.6. The Nuclear Run-On Assay
2.7. Cell Transfection and Assays for Reporter Gene Products
2.8. Assay for Methylation of the LOX Core Promoter Region
2.9. Chromatin Immunoprecipitation (ChIP) Assay
3. Results and Discussion
3.1. NNK Effects on LOX Expression at Catalytic and Protein Levels
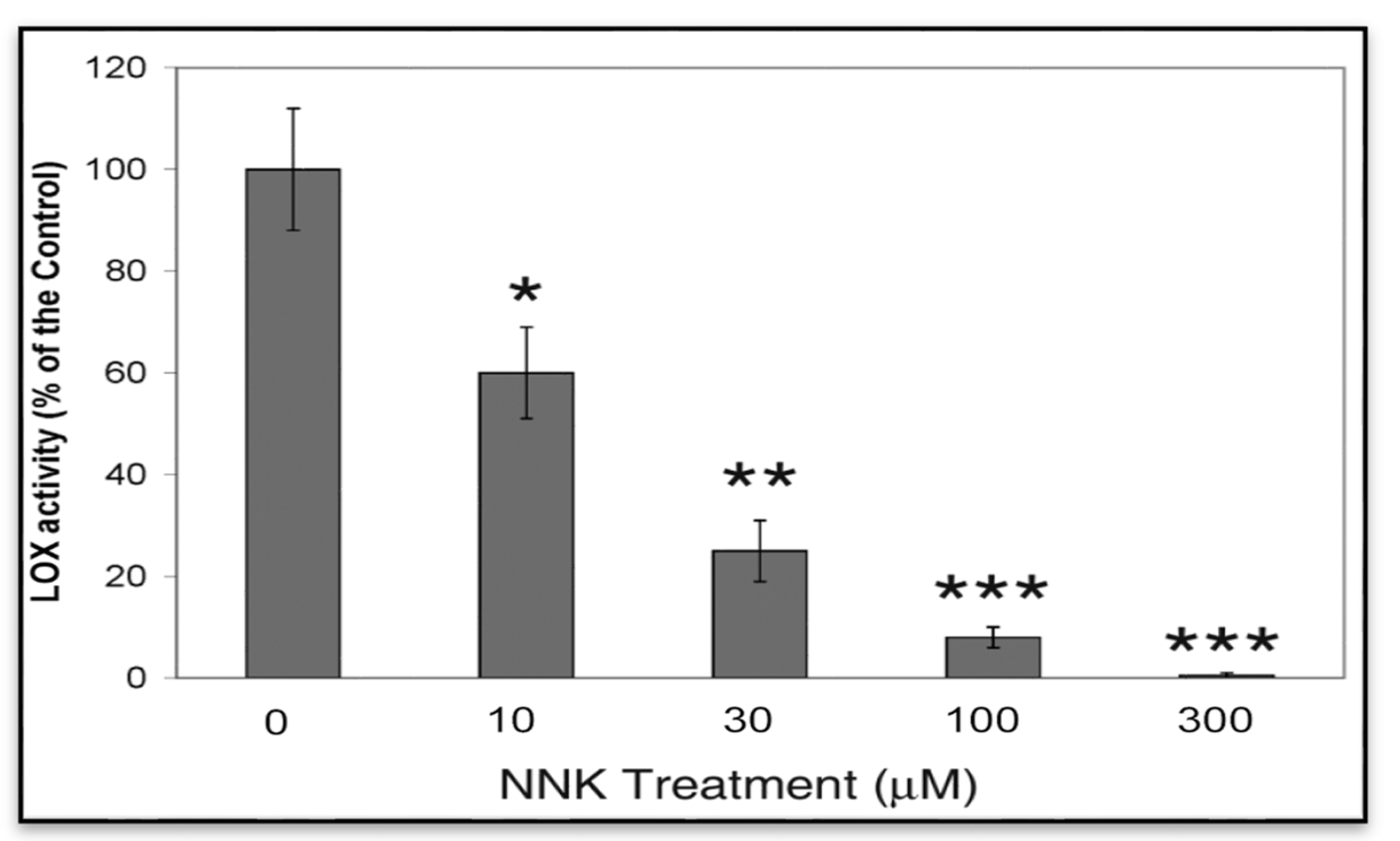
3.1.1. NNK Inhibition of LOX Catalytic Activity in NNK Treated Cells

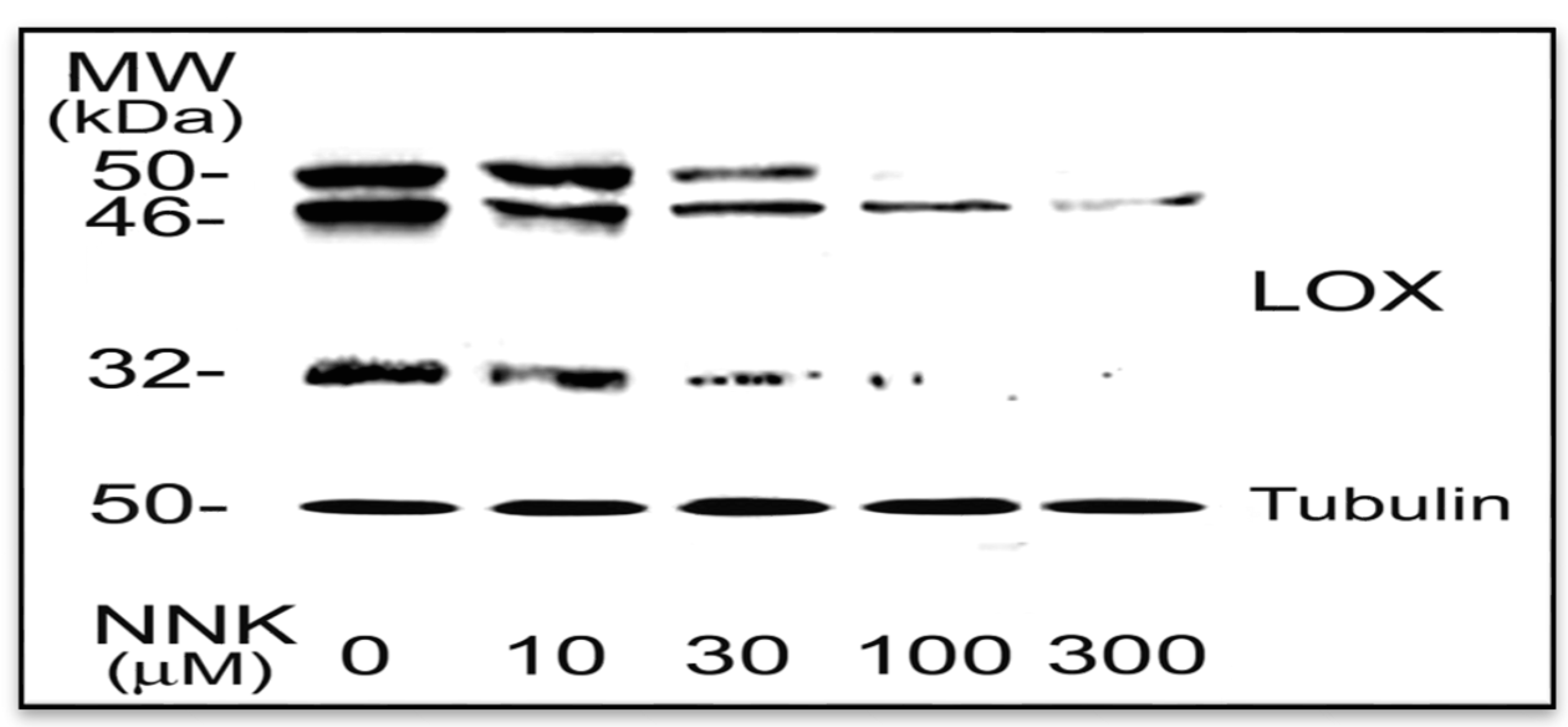
3.1.2. NNK Inhibition LOX Synthesis and Processing in Treated Cells
3.2. NNK Effects on LOX Transcriptional Levels
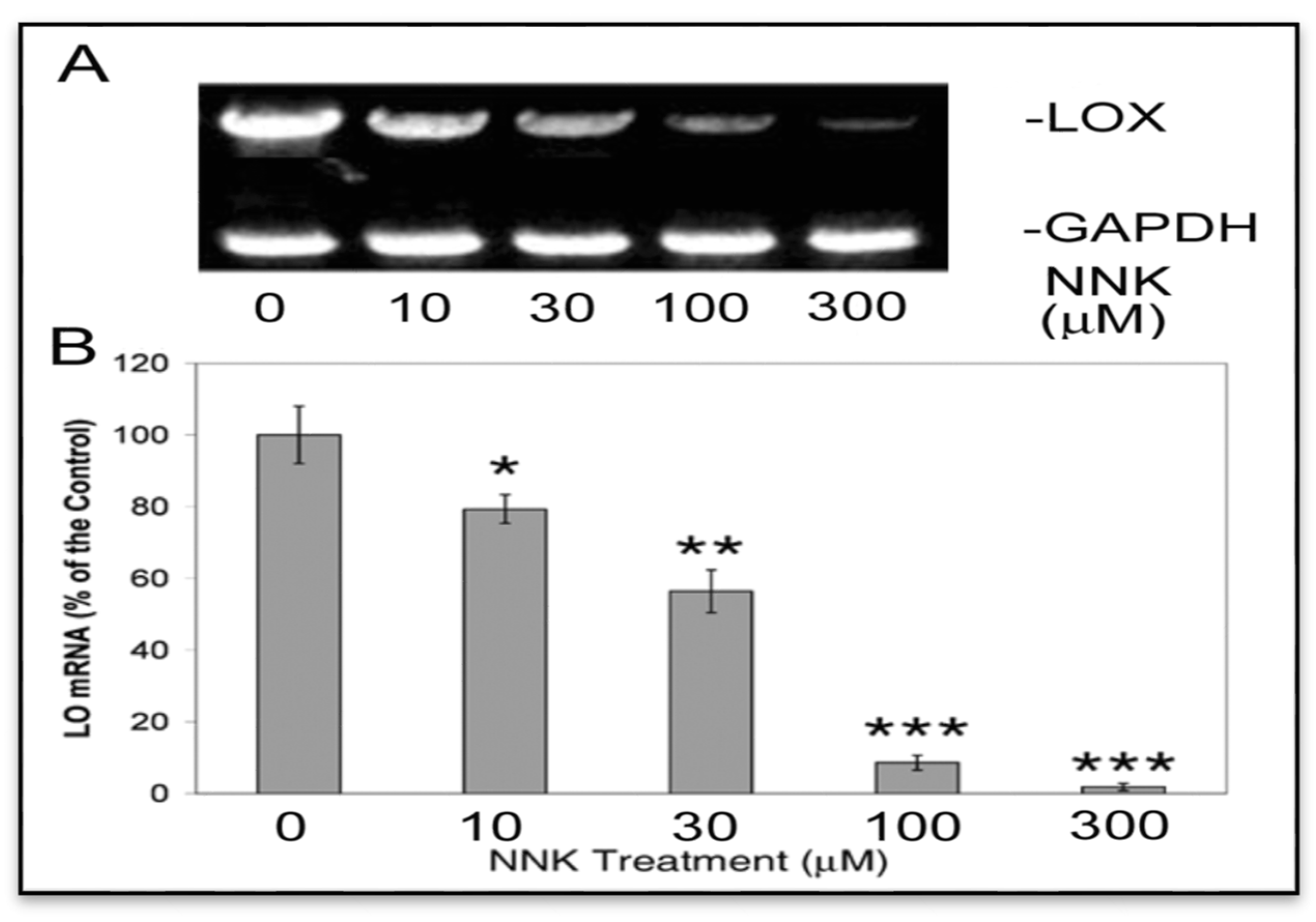
3.2.1. NNK Inhibition of the Steady-State mRNA Levels of LOX in Treated Cells
3.2.2. NNK Inhibition the Initial Transcription Rate of LOX in Treated Cells
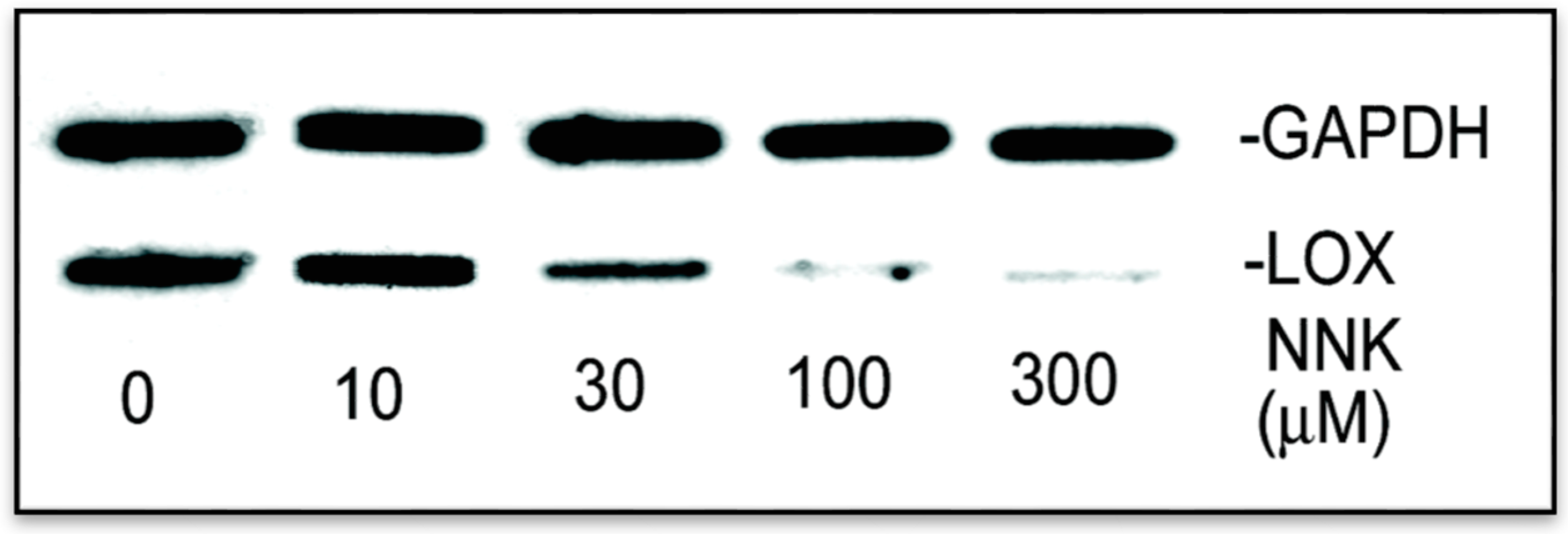
3.3. NNK Effects on the LOX Promoter Activation
3.3.1. Inhibition of LOX Promoter Activities in NNK Treated Cells
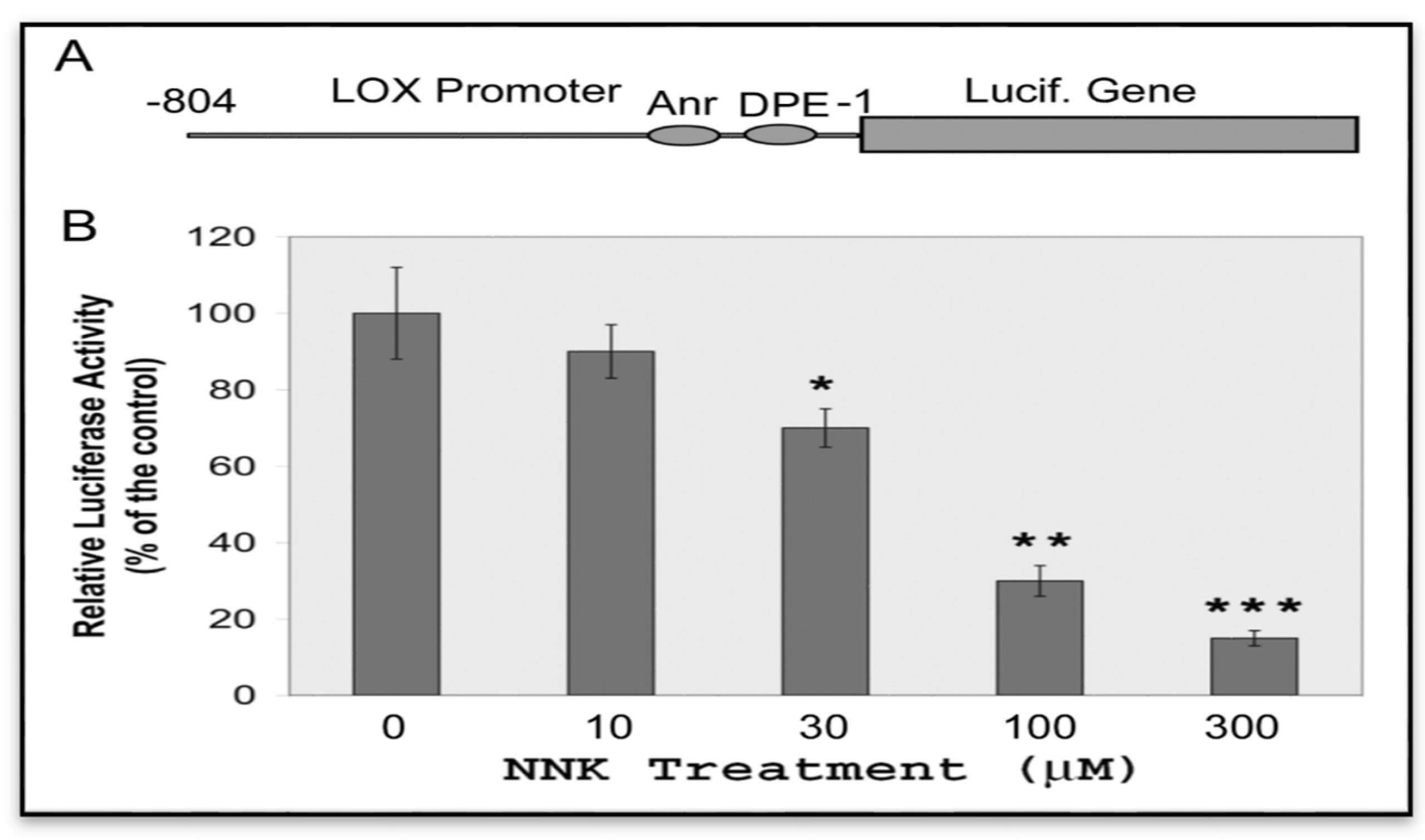
3.3.2. Inactivation of the Core Promoter of the LOX Gene in NNK-Treated Cells

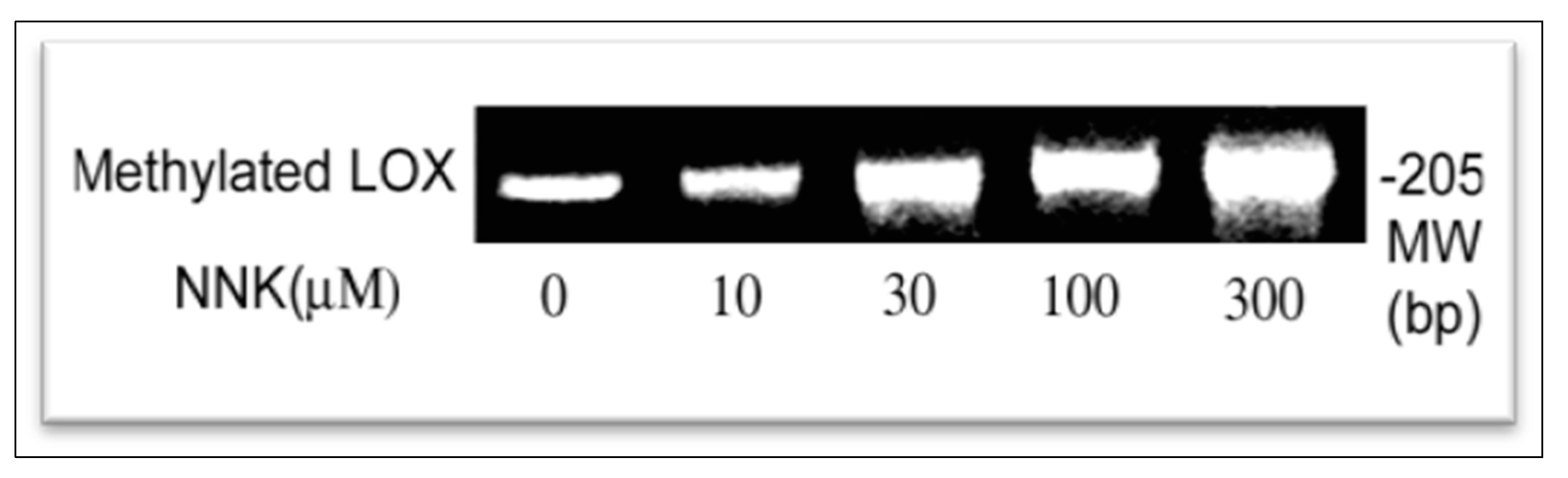
3.4. NNK Effects on Methylation of the LOX Promoter
NNK Enhancement of LOX Promoter Methylation in Treated Cells
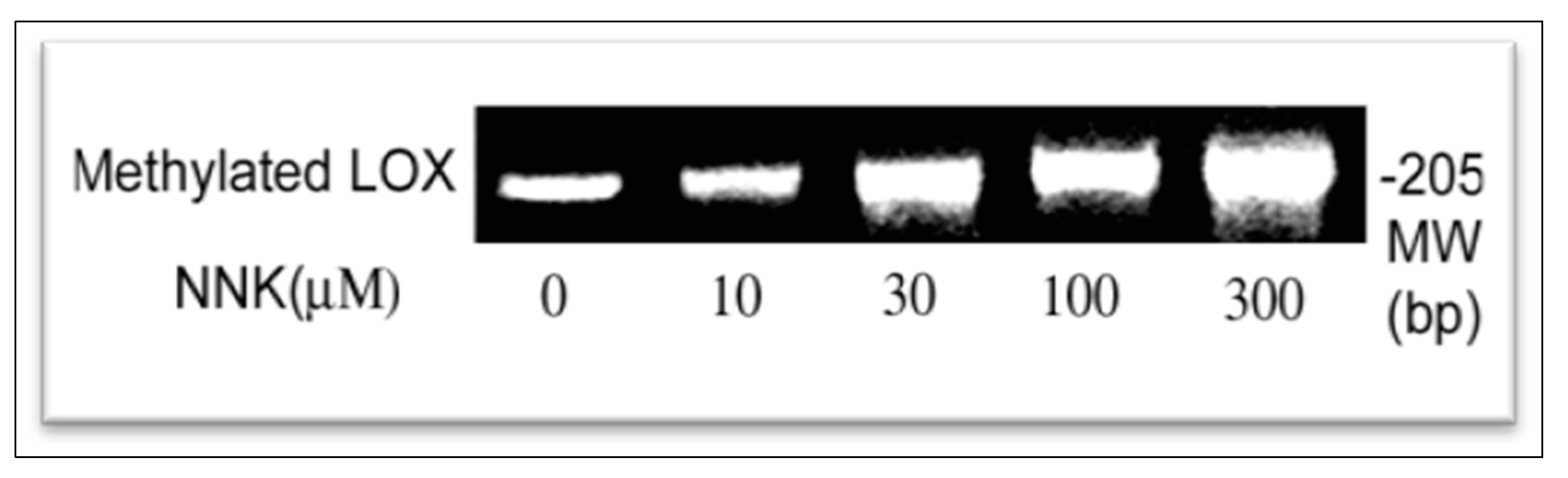
3.5. LOX, a Tumor Suppressor
3.6. NNK, a Genetic and Epigenetic Carcinogen
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kagan, H.M.; Li, W. Lysyl oxidase: Properties, specificity and biological roles inside of the cell. J. Cell. Biochem. 2003, 88, 660–672. [Google Scholar]
- Giampuzzi, M.; Oleggini, R.; Di Donato, A. Demonstration of in vitro interaction between tumor suppressor lysyl oxidase and histone H1 and H2: Definition of the regions involved. Biochim. Biophys. Acta 2003, 1647, 245–251. [Google Scholar]
- Kagan, H.M.; Williams, M.A.; Calaman, S.D.; Berkowitz, E.M. Histone H1 is a substrate for lysyl oxidase and contains endogenous sodium borotritide reducible residues. Biochem. Biophys. Res. Commun. 1983, 115, 186–192. [Google Scholar]
- Li, W.; Nugent, M.A.; Zhao, Y.; Chau, A.N.; Li, S.J.; Chou, I.N.; Liu, G.; Kagan, H.M. Lysyl oxidase oxidizes basic fibroblast growth factor and inactivates its mitogenic potential. J. Cell. Biochem. 2003, 88, 152–164. [Google Scholar]
- Li, W.; Nellaiappan, K.; Strassmaier, T.; Graham, L.; Thomas, K.M.; Kagan, H.M. Localization and activity of lysyl oxidase within nuclei of fibrogenic cells. Proc. Natl. Acad. Sci. USA. 1997, 94, 12817–12822. [Google Scholar]
- Mello, M.L.S.; Contente, S.; Vidal, B.C.; Planding, W.; Schenck, U. Modulation of ras transformation affecting chromatin supraorganization as assessed by image analysis. Exp. Cell. Res. 1995, 220, 374–382. [Google Scholar]
- Kenyon, K.; Contente, S.; Trackman, P.C.; Tang, J.; Kagan, H.M.; Friedman, R.M. Lysyl oxidase and rrg messenger RNA. Science 1991, 253. [Google Scholar] [CrossRef]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhofer, N.; Kong, C.; Le, Q.T.; Chi, J.T.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar]
- Hecht, S.S. Biochemistry, biology and carcinogenicity of tobacco-specific N-nitrosamines. Chem. Res. Toxicol. 1998, 11, 559–603. [Google Scholar]
- Belinsky, S.A.; Foley, J.F.; White, C.M.; Anderson, M.W.; Maronpot, R.R. Dose-response relationship between O6-methylguanine formation in Clara cells and induction of pulmonary neoplasia in rat by 4-(methyl nitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res. 1990, 50, 3772–3780. [Google Scholar]
- Jalas, J.R.; Ding, X.; Murphy, S.E. Comparative metabolism of the tobacco-specific nitrosamines 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol by rat cytochrome P450 2A3 and human cytochrome P450 2A13. Drug Metab. Dispos. 2003, 31, 1199–1202. [Google Scholar]
- Pfeifer, G.P.; Denissenko, M.F.; Olivier, M.; Tretyakova, N.; Hecht, S.S.; Hainaut, P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–7451. [Google Scholar]
- Zhao, Y.; Gao, S.; Chou, I.-N.; Toselli, P.; Stone, P.; Li, W. Inhibition of the expression of lysyl oxidase and its substrates in cadmium-resistant rat fetal lung fibroblasts. Toxicol. Sci. 2006, 90, 478–489. [Google Scholar]
- Chuang, C.H.; Hu, M.L. Synergistic DNA damage and lipid peroxidation in cultured human white blood cells exposed to 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone and ultraviolet A. Environ. Mol. Mutagen. 2006, 47, 73–81. [Google Scholar]
- Gupta, P.; Ghergherehchi, L.; Kulp, GA.; Tirgan, N.; Godley, B.F. Effect of tobacco specific N-nitrosamines, NNN and NNK on cultured retinal pigment epithelial cells: Implications to age related macular degeneration. In Proceedings of ARVO 2011 Visionary Genomics, Fort Lauderdale, FL, USA, 1–5 May ; 2011. [Google Scholar]
- Chou, D.K.; Zhao, Y.; Gao, S.; Chou, I.-N.; Toselli, P.; Stone, P.; Li, W. Perturbation of copper (Cu) homeostasis and expression of Cu-binding proteins in cadmium-resistant lung fibroblasts. Toxicol. Sci. 2007, 99, 267–276. [Google Scholar]
- Chen, L.-J.; Zhao, Y.; Gao, S.; Chou, I.-N.; Toselli, P.; Stone, P.; Li, W. Downregulation of lysyl oxidase and upregulation of cellular thiols in rat fetal lung fibroblasts treated with cigarette smoke condensate. Toxicol. Sci. 2005, 83, 372–379. [Google Scholar]
- Gao, S.; Chen, K.; Zhao, Y.; Rich, C.B.; Chen, L.; Li, S.J.; Toselli, P.; Stone, P.; Li, W. Transcriptional and posttranscriptional inhibition of lysyl oxidase expression by cigarette smoke condensate in cultured rat fetal lung fibroblasts. Toxicol. Sci. 2005, 87, 197–203. [Google Scholar]
- Gao, S.; Zhao, Y.; Kong, L.; Toselli, P.; Chou, I.-N.; Stone, P.; Li, W. Cloning andcharacterization of the rat lysyl oxidase gene promoter: Identification of core promoter elements and functional nuclear factor I binding sites. J. Biol. Chem. 2007, 282, 25322–25337. [Google Scholar]
- Mishra, D.K.; Chen, Z.; Wu, Y.; Sarkissyan, M.; Koeffler, H.P.; Vadgama, J.V. Global methylation pattern of genes in androgen sensitive and androgen independent prostate cancer cells. Mol. Cancer Ther. 2010, 9, 33–45. [Google Scholar]
- Li, W.; Chou, I.-N.; Boak, A.; Kagan, H.M. Down-regulation of lysyl oxidase in cadmium-resistant fibroblasts. Am. J. Respir. Cell. Mol. Biol. 1995, 13, 418–425. [Google Scholar]
- Lee, T.I.; Young, R.A. Transcription of eukaryotic protein-coding genes. Ann. Rev. Genet. 2000, 34, 77–137. [Google Scholar]
- Mullar, F.; Demeny, M.A.; Tora, L. New problems in RNA polymerase II transcription initiation: Matching the diversity of core promoters with a variety of promoter recognition factors. J. Biol. Chem. 2007, 282, 14685–14689. [Google Scholar]
- Smale, S.T.; Kadonaga, J.T. The RNA polymerase II core promoter. Ann. Rev. Biochem. 2003, 72, 449–479. [Google Scholar]
- Zhou, T.; Chiang, C-M. The intronless and TATAless human TAFII 55 gene contains a functional initiator and a downstream promoter element. J. Biol. Chem. 2001, 276, 25503–25511. [Google Scholar]
- Narlikar, G.J.; Fan, H.Y.; Kingston, R.E. Cooperation between complexes that regulate chromatin structure and transcription. Cell 2002, 108, 475–487. [Google Scholar]
- Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998, 12, 599–606. [Google Scholar]
- Bogdanović, O.; Veenstra, G.J.C. DNA methylation and methyl-CpG binding proteins: Developmental requirements and function. Chromosoma 2009, 118, 549–565. [Google Scholar]
- Bestor, T.H. The DNA methyltransferase of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar]
- Ng, H.H.; Adrian, B. DNA methylation and chromatin modification. Curr. Opin. Genet. Dev. 1999, 9, 158–163. [Google Scholar]
- Lin, R.K.; Hsieh, Y.S.; Lin, P.; Hsu, H.S.; Chen, C.Y.; Tang, Y.A.; Lee, C.F.; Wang, Y.C. The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J. Clin. Invest. 2010, 120, 521–532. [Google Scholar]
- Castonguay, A.; Tharp, R.; Hecht, S.S. Kinetics of DNA methylation by the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in F344 rats. IARC Sci. Publ. 1984, 57, 805–810. [Google Scholar]
- Kaneda, A.; Wakazono, K.; Tsukamoto, T.; Watanabe, N.; Yagi, Y.; Tatematsu, M.; Kaminishi, M.; Sugimura, T.; Ushijima, T. Lysyl oxidase is a tumor suppressor gene inactivated by methylation and loss of heterozygosity in human gastric cancers. Cancer Res. 2004, 64, 6410–6415. [Google Scholar]
- Wu, M.; Min, C.; Wang, X.; Yu, Z.; Kirsch, K.H.; Trackman, P.C.; Sonenshein, G.E. Repression of BCL2 by the tumor suppressor activity of the lysyl oxidase propeptide inhibits transformed phenotype of lung and pancreatic cancer cells. Cancer Res. 2007, 67, 6278–6285. [Google Scholar]
- Giampuzzi, M.; Botti, G.; Gilli, M.; Gusmano, R.; Borel, A.; Sommer, P.; Di Donato, A. Down-regulation of lysyl oxidase-induced tumorigenic transformation in NRK-49F cells characterized by constitutive activation of ras-proto-oncogene. J. Biol. Chem. 2001, 276, 29226–29232. [Google Scholar]
- Hamalainen, E.R.; Kemppainen, R.; Kuivaniemi, H.; Tromp, G.; Vaheri, A.; Pihlajaniemi, T.; Kivirikko, K.I. Quantitative polymerase chain reaction of lysyl oxidase mRNA in malignantly transformed human cell lines demonstrates that their low lysyl oxidase activity is due to low quantities of its mRNA and low levels of transcription of the respective gene. J. Biol. Chem. 1995, 270, 21590–21593. [Google Scholar]
- Woznick, A.R.; Braddock, A.L.; Dulai, M.; Seymour, M.L.; Callahan, R.E.; Welsh, R.J.; Chmielewski, G.W.; Zelenock, G.B.; Shanley, C.J. Lysyl oxidase expression in bronchogenic carcinoma. Am. J. Surg. 2005, 189, 297–301. [Google Scholar]
- Rost, T.; Pyritz, V.; Rathcke, I.O.; Gorogh, T.; Dunne, A.A.; Werner, J.A. Reduction of LOX- and LOXL2-mRNA expression in head and neck squamous cell carcinomas. Anticancer. Res 2003, 23, 1565–1573. [Google Scholar]
- Ren, C.; Yang, G.; Timme, T.L.; Wheeler, T.M.; Thompson, T.C. Reduced lysyl oxidase messenger RNA levels in experimental and human prostate cancer. Cancer Res. 1998, 58, 1285–1290. [Google Scholar]
- Decitre, M.; Gleyzal, C.; Raccurt, M.; Peyrol, S.; Aubert-Foucher, E.; Csiszar, K.; Sommer, P. Lysyl oxidase-like protein localizes to sites of de novo fibrinogenesis and in the early stromal reaction of ductal breast carcinomas. Lab. Invest. 1998, 78, 143–151. [Google Scholar]
- Peyrol, S.; Raccurt, M.; Gerard, F.; Gleyzal, C.; Grimaud, J.A.; Sommer, P. Lysyl oxidase gene expression in the stromal reaction to in situ and invasive ductal breast carcinoma. Am. J. Pathol. 1997, 150, 497–507. [Google Scholar]
- Bouez, C.; Reynaud, C.; Noblesse, E.; Thépot, A.; Gleyzal, C.; Kanitakis, J.; Perrier, E.; Damour, O.; Sommer, P. The lysyl oxidase LOX is absent in basal and squamous cell carcinomas and its knockdown induces an invading phenotype in a skin equivalent model. Clin. Cancer Res. 2006, 12, 1463–1469. [Google Scholar]
- Csiszar, K.; Fong, F.S.T.; Ujfalusi, A.; Krawetz, S.A.; Salvati, E.P.; Mackenzie, J.W.; Boyd, C.D. Somatic mutations of the lysyl oxidase gene on chromosome 5q23.1 in colorectal tumors. Int. J. Cancer 2002, 97, 636–642. [Google Scholar]
- Hashimoto, K.; Ohsawa, K.; Kimura, M. Mutations induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in the lacZ and cII genes of muta mouse. Mutat. Res. 2004, 560, 119–131. [Google Scholar]
- Matzinger, S.A.; Crist, K.A.; Stoner, G.D.; Anderson, M.W.; Pereira, M.A.; Steele, V.E.; Kelloff, G.J.; Lubet, R.A.; You, M. K-ras mutations in lung tumors from A/J and A/J×TSG-p53 F1 mice treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and phenethylisothiocyanate. Carcinogenesis 1995, 16, 2487–2492. [Google Scholar]
- Herzog, C.R.; Desai, D.; Amin, S. Array CGH analysis reveals chromosomal aberrations in mouse lung adenocarcinomas induced by the human lung carcinogen 4-(methylnitrosamino)-1-(3- pyridyl)-1-butanone. Biochem. Biophys. Res. Commun. 2006, 341, 856–863. [Google Scholar]
- Herzog, C.R.; Bodon, N.; Pittman, B.; Maronpot, R.R.; Massey, T.E.; Anderson, M.W.; You, M.; Devereux, T.R. Carcinogen-specific targeting of chromosome 12 for loss of heterozygosity in mouse lung adenocarcinomas: implications for chromosome instability and tumor progression. Oncogene 2004, 23, 3033–3039. [Google Scholar]
- Xue, J.; Yang, S.; Seng, S. Mechanisms of cancer induction by tobacco-specific NNK and NNN. Cancers 2014, 6, 1138–1156. [Google Scholar]
- Wei, P.L.; Chang, Y.J.; Ho, Y.S.; Lee, C.H.; Yang, Y.Y.; An, J.; Lin, S.Y. Tobacco-specific carcinogen enhances colon cancer cell migration through α7-nicotinic acetylcholine receptor. Ann. Surg. 2009, 249, 978–985. [Google Scholar]
- Schuller, H.M.; Orloff, M. Tobacco-specific carcinogenic nitrosamines: Ligands for nicotinic acetylcholine receptors in human lung cancer cells. Biochem. Pharmacol. 1998, 55, 1377–1384. [Google Scholar]
- Jull, B.; Plummer, H.; Schuller, H. Nicotinic receptor mediated activation by the tobacco-specific nitrosamine NNK of a Raf-1/MAP kinase pathway, resulting in phosphorylation of c-myc in human small cell lung carcinoma cells and pulmonary neuroendocrine cells. J. Cancer Res. Clin. Oncol. 2001, 127, 707–717. [Google Scholar]
- West, K.A.; Brognard, J.; Clark, A.S.; Linnoila, I.R.; Yang, X.; Swain, S.M.; Harris, C.; Belinsky, S.; Dennis, P.A. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J. Clin. Invest. 2003, 111, 81–90. [Google Scholar]
- Tsurutani, J.; Castillo, S.S.; Brognard, J.; Granville, C.A.; Zhang, C.; Gills, J.J.; Sayyah, J.; Dennis, P.A. Tobacco components stimulate Akt-dependent proliferation and NFκB dependent survival in lung cancer cells. Carcinogenesis 2005, 26, 1182–1195. [Google Scholar]
- Wu, W.K.; Wong, H.P.; Luo, S.W.; Chan, K.; Huang, F.Y.; Hui, M.K.; Lam, E.K.; Shin, V.Y.; Ye, Y.N.; Yang, Y.H.; Cho, C.H. 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone from cigarette smoke stimulates colon cancer growth via β-adrenoceptors. Cancer Res. 2005, 65, 5272–5277. [Google Scholar]
- Schuller, H.M.; Porter, B.; Riechert, A. Beta-adrenergic modulation of NNK-induced lung carcinogenesis in hamsters. J. Cancer Res. Clin. Oncol. 2000, 126, 624–630. [Google Scholar]
- Schuller, H.M.; Cekanova, M. NNK-induced hamster lung adenocarcinomas over-express β2-adrenergic and EGFR signaling pathways. Lung Cancer 2005, 49, 35–45. [Google Scholar]
- Ho, Y.S.; Chen, C.H.; Wang, Y.J.; Pestell, R.G.; Albanese, C.; Chen, R.J.; Chang, M.C.; Jeng, J.H.; Lin, S.Y.; Liang, Y.C.; et al. Tobaccospecific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induces cell proliferation in normal human bronchial epithelial cells through NFκB activation and cyclinD1 up-regulation. Toxicol. Appl. Pharmacol. 2005, 205, 133–148. [Google Scholar]
- Jin, Z.; Gao, F.; Flagg, T.; Deng, X. Tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone promotes functional cooperation of Bcl2 and c-Myc through phosphorylation in regulating cell survival and proliferation. J. Biol. Chem. 2004, 279, 40209–40219. [Google Scholar]
- Belinsky, S.A.; Nikula, K.J.; Palmisano, W.A.; Michels, R.; Saccomanno, G.; Gabrielson, E.; Baylin, S.B.; Herman, J.G. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc. Natl. Acad. Sci. USA. 1998, 95, 11891–11896. [Google Scholar]
- Pulling, L.C.; Vuillemenot, B.R.; Hutt, J.A.; Devereux, T.R.; Belinsky, S.A. Aberrant promoter hypermethylation of the death-associated protein kinase gene is early and frequent in murine lung tumors induced by cigarette smoke and tobacco carcinogens. Cancer Res. 2004, 64, 3844–3848. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, G.; Li, J.; Zheng, M.; Zhao, Y.; Zhou, J.; Li, W. NNK, a Tobacco-Specific Carcinogen, Inhibits the Expression of Lysyl Oxidase, a Tumor Suppressor. Int. J. Environ. Res. Public Health 2015, 12, 64-82. https://doi.org/10.3390/ijerph120100064
Cheng G, Li J, Zheng M, Zhao Y, Zhou J, Li W. NNK, a Tobacco-Specific Carcinogen, Inhibits the Expression of Lysyl Oxidase, a Tumor Suppressor. International Journal of Environmental Research and Public Health. 2015; 12(1):64-82. https://doi.org/10.3390/ijerph120100064
Chicago/Turabian StyleCheng, Guang, Jianmin Li, Maoguen Zheng, Yinzhi Zhao, Jing Zhou, and Wande Li. 2015. "NNK, a Tobacco-Specific Carcinogen, Inhibits the Expression of Lysyl Oxidase, a Tumor Suppressor" International Journal of Environmental Research and Public Health 12, no. 1: 64-82. https://doi.org/10.3390/ijerph120100064
APA StyleCheng, G., Li, J., Zheng, M., Zhao, Y., Zhou, J., & Li, W. (2015). NNK, a Tobacco-Specific Carcinogen, Inhibits the Expression of Lysyl Oxidase, a Tumor Suppressor. International Journal of Environmental Research and Public Health, 12(1), 64-82. https://doi.org/10.3390/ijerph120100064