Microbial Community Structures and Dynamics in the O3/BAC Drinking Water Treatment Process




{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Sample Collection
2.2. Total DNA Extraction
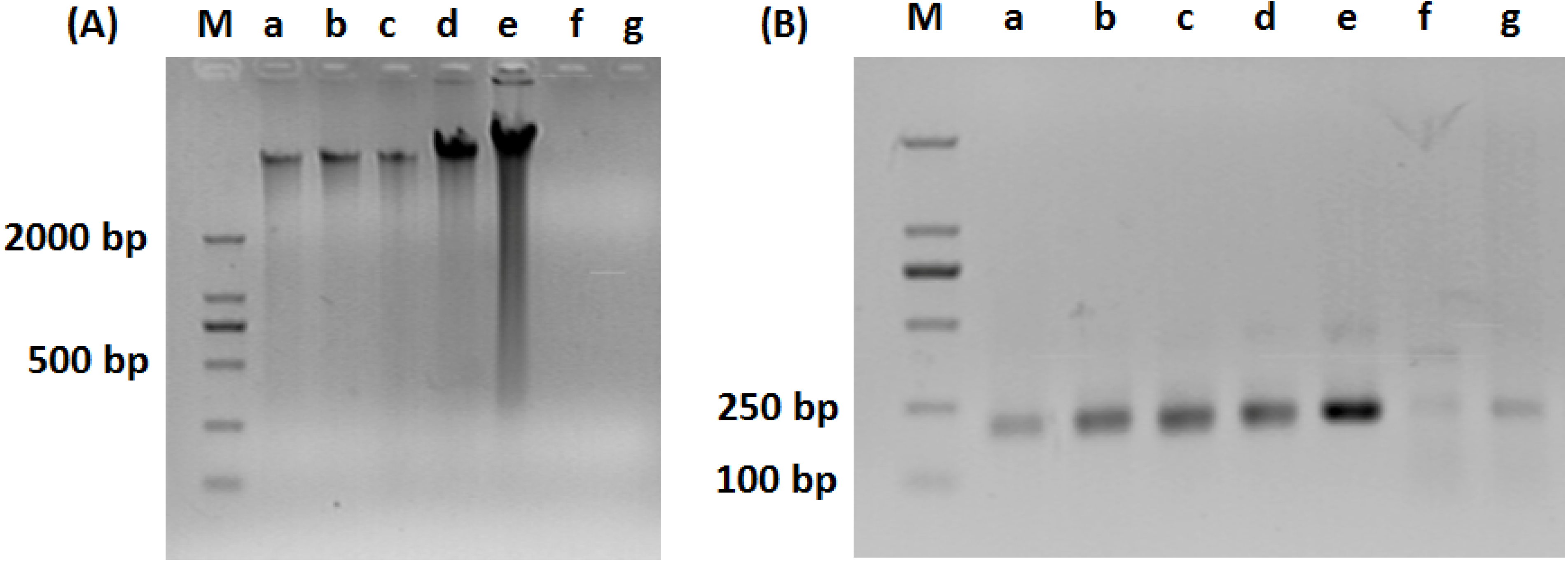
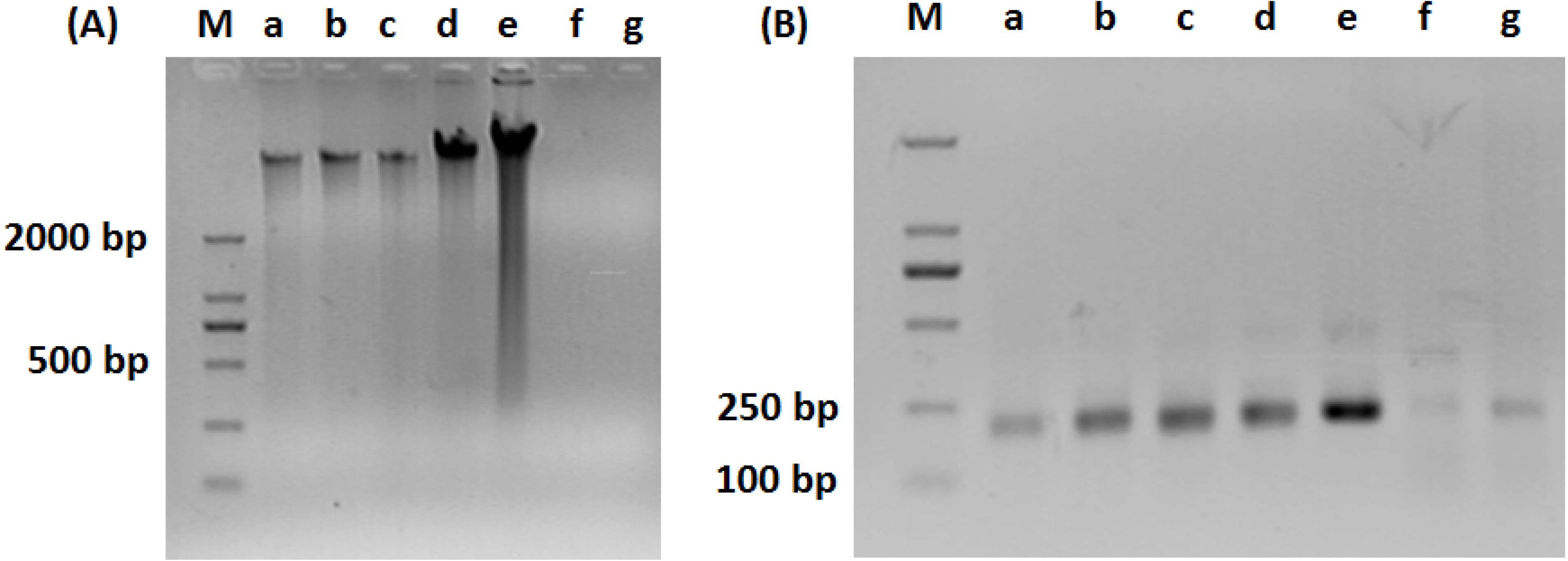
2.3. PCR-DGGE Analysis
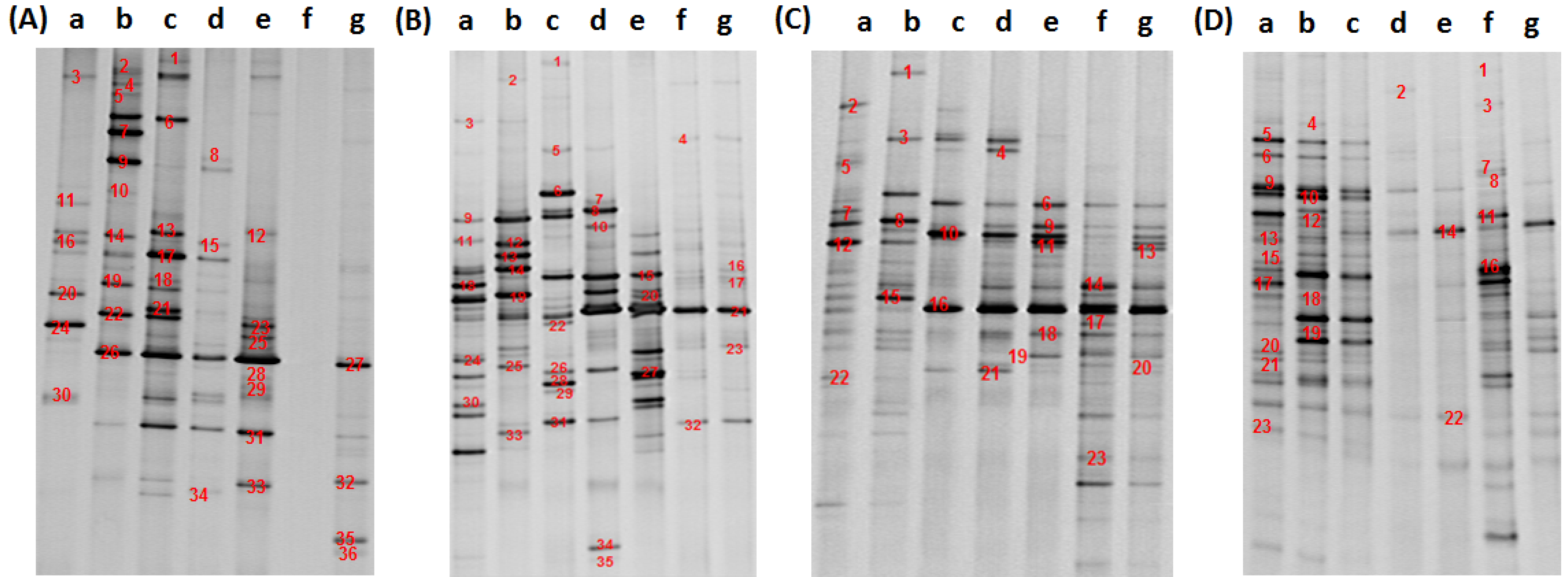
2.4. 16S rDNA Sequencing and Community Structure Analysis
3. Results and Discussion
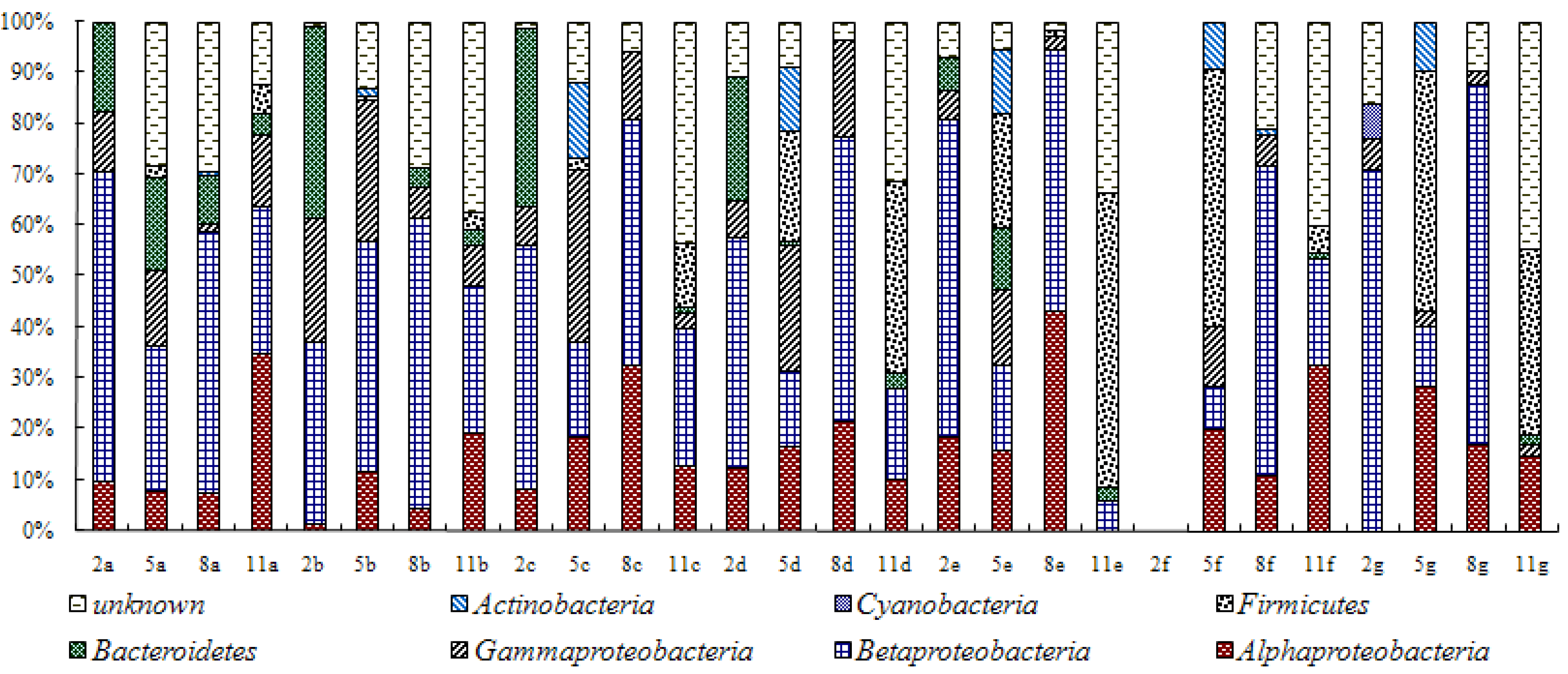
3.1. Composition of Bacteria Community at Different Stage of Water Treatment
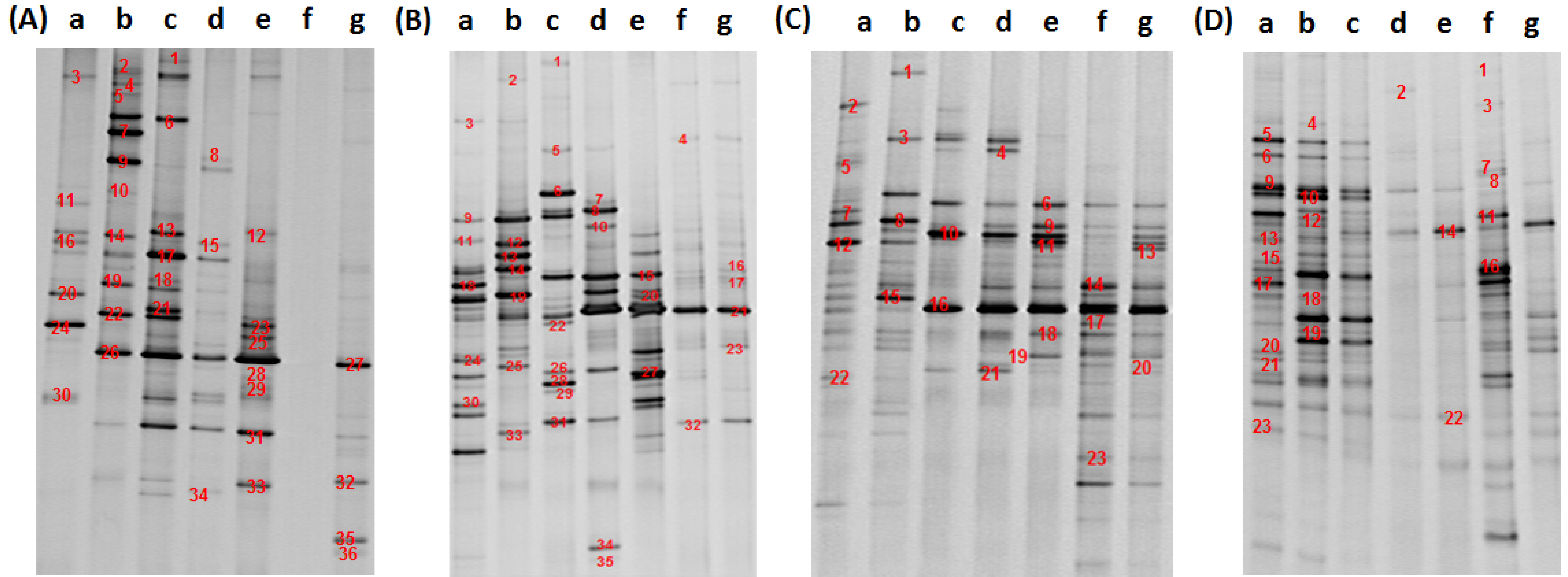
3.2. Analysis of Water Bacteria Community Dynamics at Various Process Stages
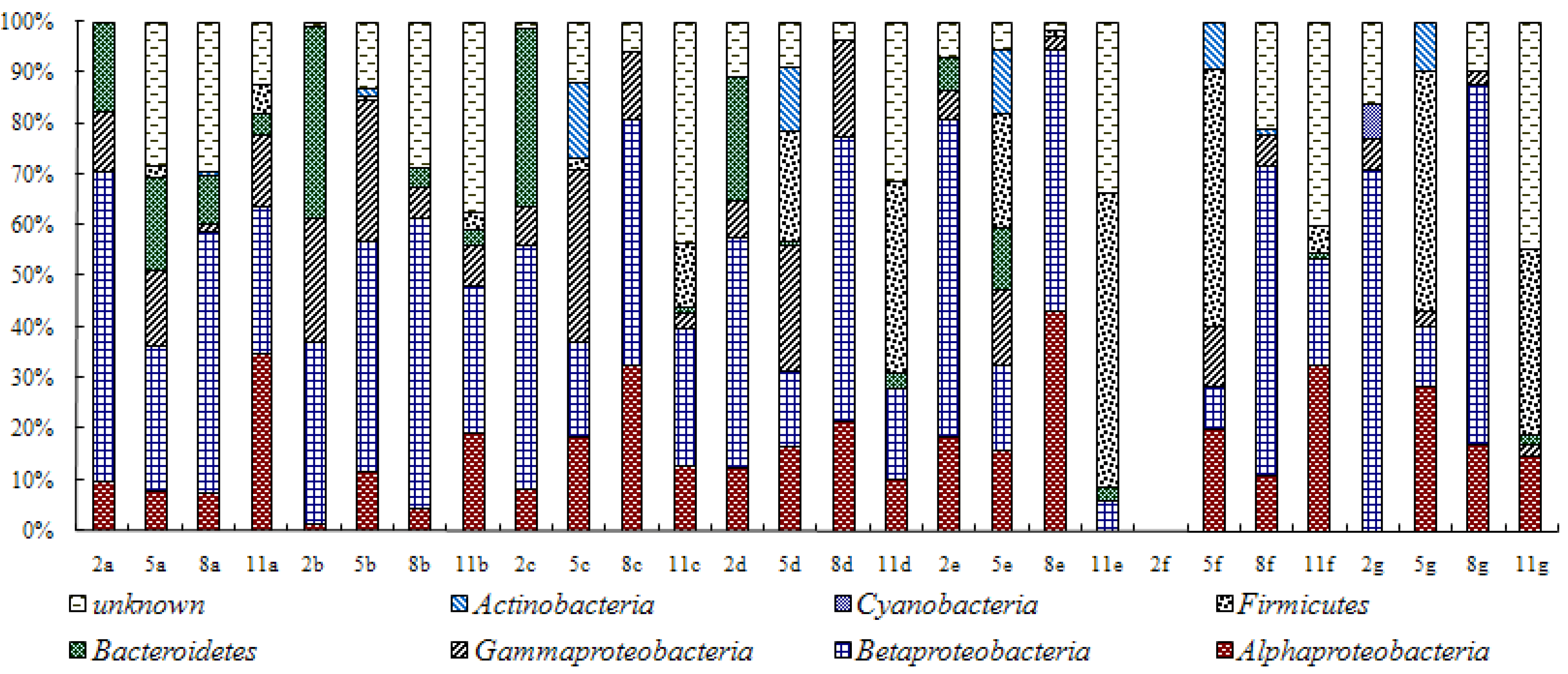
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Niquette, P.; Servais, P.; Savoir, R. Bacterial dynamics in the drinking water distribution system of Brussels. Water Res. 2001, 35, 675–682. [Google Scholar] [CrossRef]
- Ward, D.M.; Bateson, M.M.; Weller, R.; Ruff-Roberts, A.L. Ribosomal RNA analysis of microorganisms as they occur in nature. Adv. Microb. Ecol. 1992, 12, 219–286. [Google Scholar] [CrossRef]
- Colwell1, R.R.; Brayton, P.R.; Grimes, D.J.; Roszak, D.B.; Huq, S.A.; Palmer, L.M. Viable but non-culturable Vibrio cholerae in the environment: Implications for release of genetically engineered microorganisms. Nat. Biotechnol. 1985, 3, 817–820. [Google Scholar] [CrossRef]
- Rahman, I.; Shahamat, M.; Kirchman, P.A.; Russek-Cohen, E.; Colwell, R.R. Methionine uptake and cytopathogenicity of viable but nonculturable Shigella dysenteriae type 1. Appl. Environ. Microbiol. 1994, 60, 3573–3578. [Google Scholar]
- Pommepuy, M.; Butin, M.; Derrien, A.; Gourmelon, M.; Colwell, R.R.; Cormier, M. Retention of enteropathogenicity by viable but nonculturable Escherichia coli exposed to seawater and sunlight. Appl. Environ. Microbiol. 1996, 62, 4621–4626. [Google Scholar]
- Muyzer, G.; de Waal, E.C.; Uitterlinden, A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction amplified genes encoding for 16S rRNA. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar]
- Li, Z.Y.; He, L.M.; Miao, X.L. Cultivable Bacterial Community from South China Sea Sponge as Revealed by DGGE Fingerprinting and 16S rDNA Phylogenetic Analysis. Curr. Microbiol. 2007, 55, 465–472. [Google Scholar] [CrossRef]
- Kandeler, E.; Tscherko, D.; Bruce, K.D.; Stemmer, M.; Hobbs, P.J.; Bardgett, R.D.; Amelung, W. Structure and function of the soil microbial community in microhabitats of a heavy metal polluted soil. Biol. Fertil. Soils 2000, 32, 390–400. [Google Scholar]
- Paniagua-Michel, J.; Franco-Rivera, A.; Cantera, J.J.L.; Stein, L.Y. Activity of Nitrifying Biofilms Constructed on Low-Density Polyester Enhances Bioremediation of a Coastal Wastewater Effluent. World J. Microb. Biot. 2005, 1, 1371–1377. [Google Scholar]
- Wang, J.; Ma, T.; Zhao, L.; Lv, J.; Li, G.; Zhang, H.; Zhao, B.; Liang, F.; Liu, R. Monitoring exogenous and indigenous bacteria by PCR-DGGE technology during the process of microbial enhanced oil recovery. J. Ind. Microbiol. Biotechnol. 2008, 35, 619–628. [Google Scholar] [CrossRef]
- Pearce, D.A. Bacterioplankton Community Structure in a Maritime Antarctic Oligotrophic Lake during a Period of Holomixis, as Determined by Denaturing Gradient Gel Electrophoresis (DGGE) and Fluorescence in Situ Hybridization (FISH). Microb. Ecol. 2003, 46, 92–105. [Google Scholar] [CrossRef]
- Wang, H.Y.; Zhang, X.J.; Zhao, L.P. Analysis and comparison of the bacterial community in fermented grains during the fermentation for two different styles of Chinese liquor. J. Ind. Microbiol. Biotechnol. 2008, 35, 603–609. [Google Scholar] [CrossRef]
- Wu, Q.; Zhao, X.H.; Zhao, S.Y. Application of PCR-DGGE in Research of Bacterial Diversity in Drinking Water. Biomed. Environ. Sci. 2006, 19, 371–374. [Google Scholar]
- Molnár, O.; Wuczkowski, M.; Prillinger, H. Yeast biodiversity in the guts of several pests on maize;comparison of three methods: classical isolation, cloning and DGGE. Mycol. Progress 2008, 7, 111–123. [Google Scholar] [CrossRef]
- Garbeva, P.; Overbeek, L.S.; Vuurde, J.W.; Elsas, J.D. Analysis of endophytic bacterial communities of potato by plating and denaturing gradient gel electrophoresis (DGGE) of 16S rDNA based PCR fragments. Microb. Ecol. 2001, 41, 369–383. [Google Scholar]
- Wang, X.H.; Zhang, K.; Ren, N.Q.; Li, N.; Ren, L.J. Monitoring microbial community structure and succession of an A/O SBR during start-up period using PCR-DGGE. J. Environ. Sci. 2009, 21, 223–228. [Google Scholar] [CrossRef]
- Dewettinck, T.; Hulsbosch, W.; Van Hege, K.; Top, E.M.; Verstraete, W. Molecular fingerprinting of bacterial populations in groundwater and bottled mineral water. Appl. Microbiol. Biotechnol. 2001, 57, 412–418. [Google Scholar] [CrossRef]
- Eichler, S.; Christen, R.; Höltje, C.; Westphal, P.; Bötel, J.; Brettar, I.; Mehling, A.; Höfle, M.G. Composition and Dynamics of Bacterial Communities of a Drinking Water Supply System as Assessed by RNA-and DNA-Based 16S rRNA Gene Fingerprinting. Appl. Environ. Microbiol. 2006, 72, 1858–1872. [Google Scholar] [CrossRef]
- Schmeisser, C.; Stöckigt, C.; Raasch, C.; Wingender, J.; Timmis, K.N.; Wenderoth, D.F.; Flemming, H.C.; Liesegang, H.; Schmitz, R.A.; Jaeger, K.E.; Streit, W.R. Metagenome Survey of Biofilms in Drinking-Water Networks. Appl. Environ. Microbiol. 2003, 69, 7298–7309. [Google Scholar] [CrossRef]
- Liu, Z.P.; Wang, B.J.; Liu, Y.H.; Liu, S.J. Novosphingobium taihuense sp nov., a novel aromatic-compound-degrading bacterium isolated from Taihu Lake, China. Int. J. Syst. Evol. Microbiol. 2005, 55, 1229–1232. [Google Scholar] [CrossRef]
- Berg, K.A.; Lyra, C.; Sivonen, K.; Paulin, L.; Suomalainen, S.; Tuomi, P.; Rapala, J. High diversity of cultivable heterotrophic bacteria in association with cyanobacterial water blooms. ISME. J. 2009, 3, 314–325. [Google Scholar] [CrossRef]
- Qu, J.H.; Yuan, H.L.; Yang, J.S.; Li, H.F.; Chen, N. Lacibacter cauensis gen.nov., sp.nov., a novel member of the phylum Bacteroidetes isolated from sediment of a eutrophic lake. Int. J. Syst. Evol. Microbiol. 2009, 59, 1153–1157. [Google Scholar] [CrossRef]
- Muyzer, G.; Smalla, K. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. A. Van. Leeuw. 1998, 73, 127–141. [Google Scholar] [CrossRef]
- Yang, C.H.; Crowley, D.E. Rhizosphere microbial community structure in relation to root location and plant iron nutritional status. Appl. Environ. Microbiol. 2000, 66, 345–351. [Google Scholar] [CrossRef]
- Vaz-Moreira, I.; Egas, C.; Nunes, O.C.; Manaia, C.M. Bacterial diversity from the source to the tap: A comparative study based on 16S rRNA gene-DGGE and culture-dependent methods. FEMS. Microbiol. Ecol. 2013, 83, 361–374. [Google Scholar] [CrossRef]
- Delgado, S.; Rachid, C.T.; Fernández, E.; Rychlik, T.; Alegría, A.; Peixoto, R.S.; Mayo, B. Diversity of thermophilic bacteria in raw, pasteurized and selectively-cultured milk, as assessed by culturing, PCR-DGGE and pyrosequencing. Food Microbiol. 2013, 36, 103–111. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tian, J.; Lu, J.; Zhang, Y.; Li, J.-C.; Sun, L.-C.; Hu, Z.-L. Microbial Community Structures and Dynamics in the O3/BAC Drinking Water Treatment Process. Int. J. Environ. Res. Public Health 2014, 11, 6281-6290. https://doi.org/10.3390/ijerph110606281
Tian J, Lu J, Zhang Y, Li J-C, Sun L-C, Hu Z-L. Microbial Community Structures and Dynamics in the O3/BAC Drinking Water Treatment Process. International Journal of Environmental Research and Public Health. 2014; 11(6):6281-6290. https://doi.org/10.3390/ijerph110606281
Chicago/Turabian StyleTian, Jian, Jun Lu, Yu Zhang, Jian-Cheng Li, Li-Chen Sun, and Zhang-Li Hu. 2014. "Microbial Community Structures and Dynamics in the O3/BAC Drinking Water Treatment Process" International Journal of Environmental Research and Public Health 11, no. 6: 6281-6290. https://doi.org/10.3390/ijerph110606281
APA StyleTian, J., Lu, J., Zhang, Y., Li, J.-C., Sun, L.-C., & Hu, Z.-L. (2014). Microbial Community Structures and Dynamics in the O3/BAC Drinking Water Treatment Process. International Journal of Environmental Research and Public Health, 11(6), 6281-6290. https://doi.org/10.3390/ijerph110606281