Monitoring the Presence of 13 Active Compounds in Surface Water Collected from Rural Areas in Northwestern Spain



 ,
, 

Abstract
:1. Introduction
2. Experimental Section
2.1. Chemicals, Reagents and Stock Solutions

{kind=link}
{kind=link}
| Analyte | Therapeutic Class | CAS Number | Formula |
|---|---|---|---|
| Brilliant Green | Triphenylmethane dye | 633-03-4 | C27H34N2O4S |
| Diclofenac | Anti-inflammatory | 15307-79-6 | C14H11Cl2NO2 |
| Difloxacin | Antimicrobial | 98106-17-3 | C21H19F2N3O3 |
| Enrofloxacin | Antimicrobial | 93106-60-6 | C19H22FN3O3 |
| Malachite Green | Triphenylmethane dye | 2437-29-8 | C23H25ClN2 |
| Marbofloxacin | Antimicrobial | 115550-35-1 | C17H19FN4O4 |
| Oxolinic Acid | Antimicrobial | 14698-29-4 | C13H11NO5 |
| Propanolol | Anti-hypertensive | 525-66-6 | C16H21NO2 |
| Sarafloxacin | Antimicrobial | 98105-99-8 | C20H17F2N3O3 |
| Sulfamethoxazole | Antimicrobial | 723-46-6 | C10H11N303S |
| Tamoxifen | Anti-cancer | 10540-29-1 | C 26 H 29 NO |
| Triamcinolone | Glucocorticoid | 124-94-7 | C21H27FO6 |
| Trimethoprim | Antimicrobial | 738-70-5 | C14H18N4O3 |
2.2. Equipment
2.3. HPLC-MS/MS Conditions
| Total Time (min) | Mobile Phase A (%) | Mobile Phase B (%) |
|---|---|---|
| 0 | 90 | 10 |
| 2 | 90 | 10 |
| 7 | 50 | 50 |
| 15 | 25 | 75 |
| 24 | 0 | 100 |
| 28 | 0 | 100 |
| 33 | 75 | 25 |
| 36 | 90 | 10 |
| 40 | 90 | 10 |
| Analyte | tR (min) | Precursor (m/z) | Product 1 (m/z) | Product 2 (m/z) | Precursor > Product Ion 1 | |||
|---|---|---|---|---|---|---|---|---|
| DP | EP | CE | CXP | |||||
| Brilliant Green | 34.09 | 385 | 341 | 241 | 61 | 10 | 53 | 10 |
| Diclofenac | 20.29 | 296 | 214 | 250 | 51 | 10 | 43 | 14 |
| Difloxacin | 15.44 | 400 | 299 | 356 | 71 | 10 | 27 | 32 |
| Enrofloxacin | 14.91 | 360 | 316 | 342 | 76 | 10 | 28 | 8 |
| Malachite Green | 25.12 | 329 | 208 | 165 | 111 | 10 | 43 | 18 |
| Marbofloxacin | 13.74 | 363 | 72 | 320 | 76 | 10 | 51 | 4 |
| Oxolinic Acid | 16.62 | 262 | 216 | 160 | 36 | 10 | 39 | 14 |
| Propanolol | 15.97 | 260 | 116 | 183 | 106 | 10 | 23 | 10 |
| Sarafloxacin | 16.05 | 386 | 342 | 299 | 76 | 10 | 23 | 22 |
| Sulfamethoxazole | 13.20 | 254 | 156 | 92 | 81 | 10 | 25 | 14 |
| Tamoxifen | 21.2 | 372 | 340 | 235 | 106 | 10 | 73 | 14 |
| Triamcinolone | 15.34 | 395 | 375 | 357 | 66 | 10 | 15 | 6 |
| Trimethoprim | 12.05 | 291 | 230 | 123 | 56 | 10 | 35 | 20 |
| Sulfadoxine d3 | 13.70 | 314 | 156 | 108 | 41 | 10 | 27 | 12 |
| Malachite green d5 picrate | 25.8 | 334 | 213 | 170 | 126 | 10 | 47 | 24 |
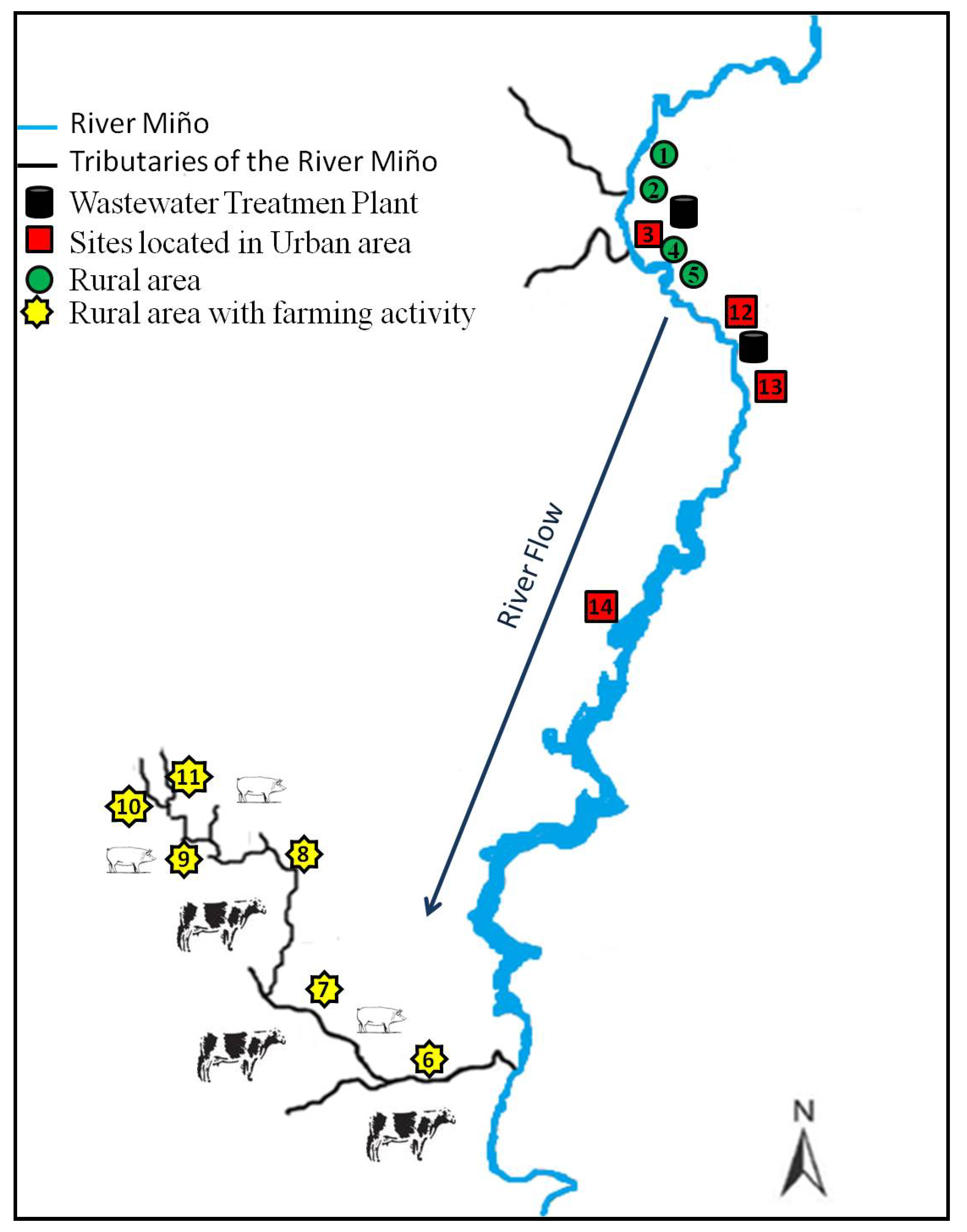
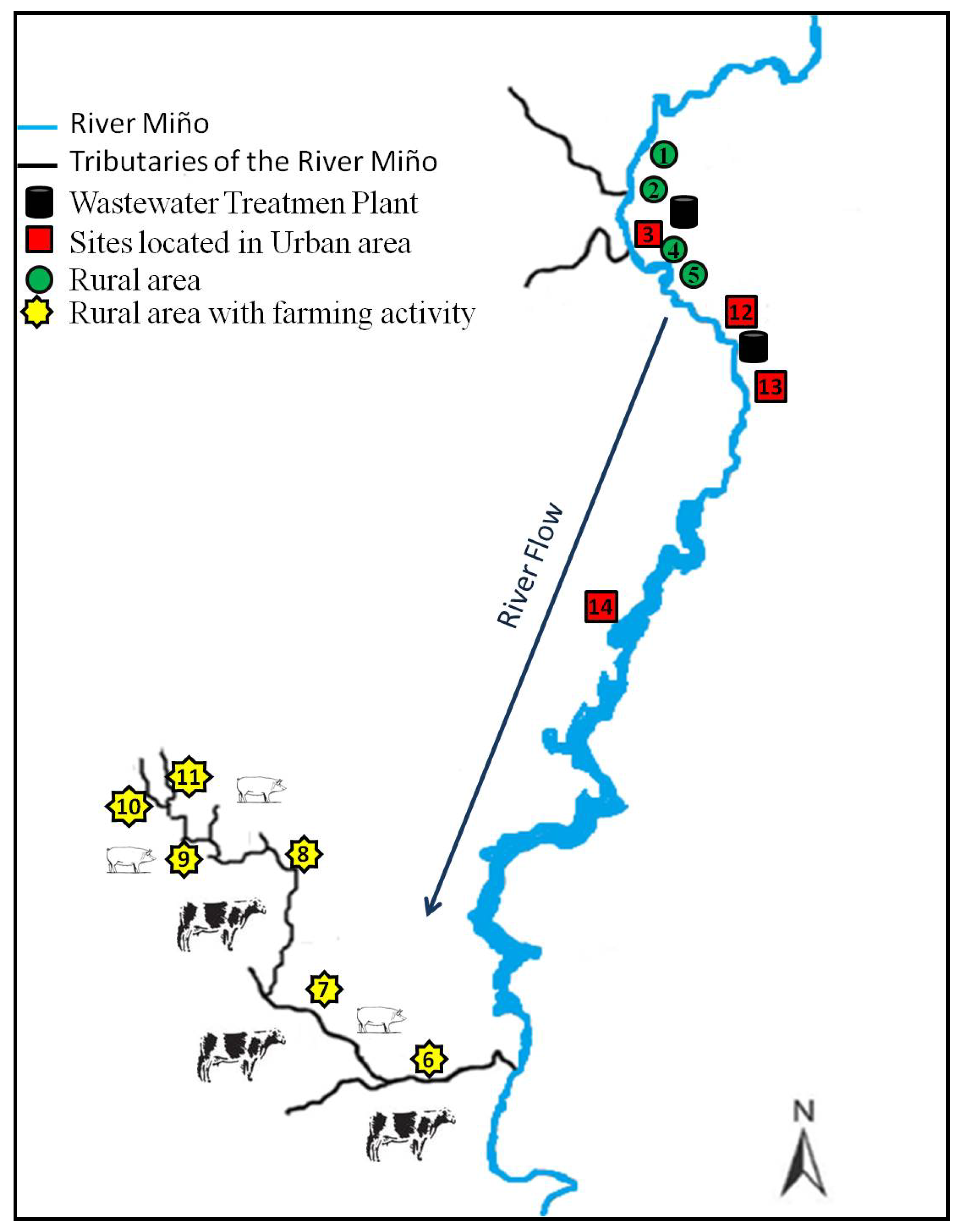
2.4. Study Site Description
2.5. Sampling Strategy, Method and Conservation
2.6. Analytical Procedure
2.7. Validation Procedure
2.8. Statistical Analysis
3. Results and Discussion
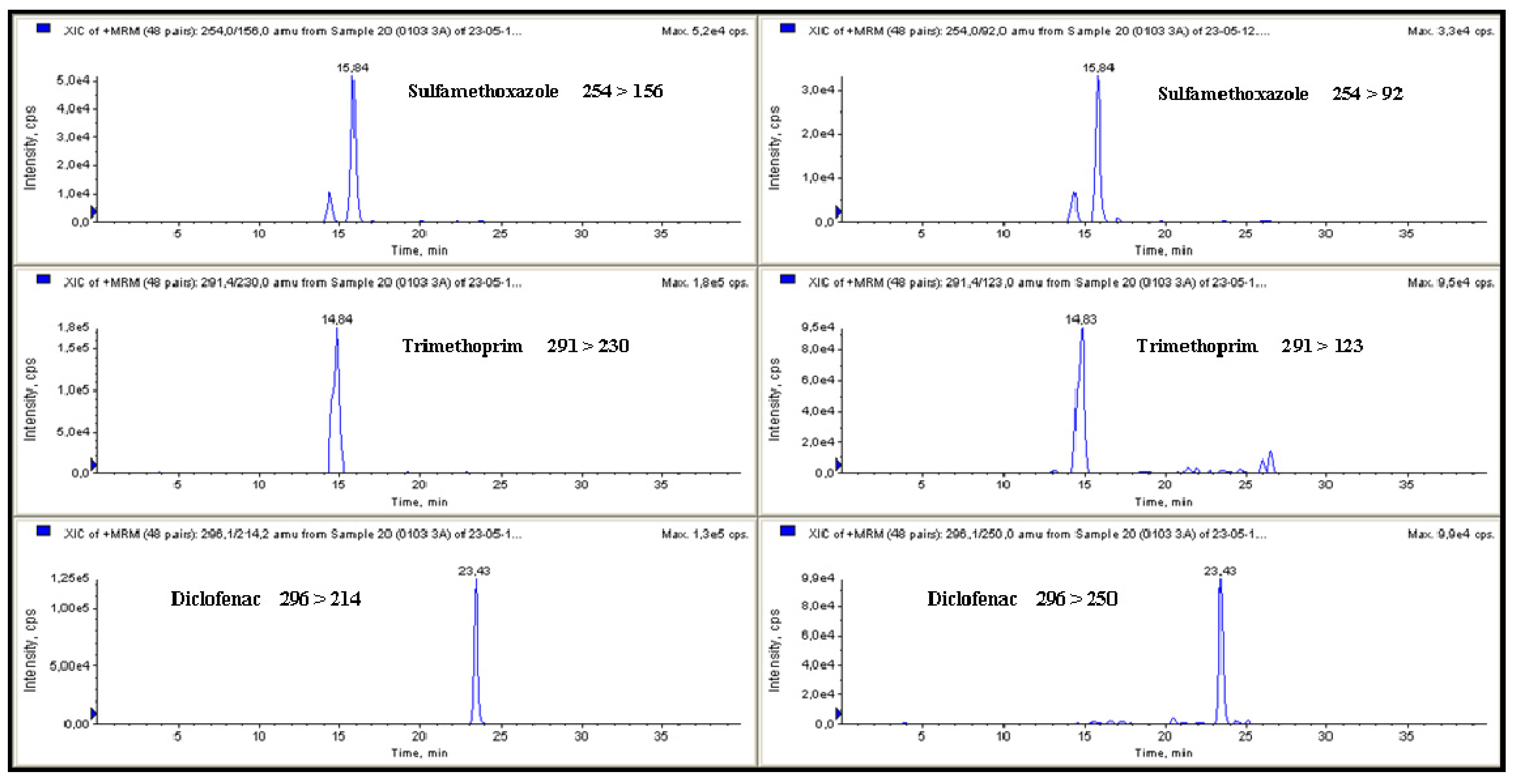
3.1. Optimisation of the HPLC-MS/MS and Extraction Protocols
3.2. Method Validation
| Analyte | IDL (ng·mL−1) | IQL (ng·mL−1) | LOD (ng·L−1) | LOQ (ng·L−1) | MDL (ng·L−1) |
|---|---|---|---|---|---|
| Brilliant Green | 0.2 | 0.3 | 2.6 | 3.0 | 7.4 |
| Diclofenac | 0.7 | 0.8 | 2.0 | 2.10 | 11.1 |
| Difloxacin | 0.3 | 0.5 | 1.0 | 3.0 | 8.8 |
| Enrofloxacin | 5.4 | 5.8 | 1.37 | 5.15 | 8.1 |
| Malachite Green | 0.2 | 0.3 | 1.5 | 3.3 | 10.9 |
| Marbofloxacin | 0.2 | 0.3 | 1.0 | 3.0 | 8.1 |
| Oxolinic Acid | 6.4 | 6.7 | 3.43 | 3.86 | 9.3 |
| Propanolol | 0.2 | 0.3 | 1.0 | 3.0 | 2.0 |
| Sarafloxacin | 4.6 | 4.8 | 2.1 | 5.2 | 11.9 |
| Sulfamethoxazole | 0.2 | 0.3 | 1.0 | 3.0 | 11.2 |
| Tamoxifen | 0.2 | 0.3 | 2.0 | 3.0 | 13.5 |
| Triamcinolone | 0.9 | 1.1 | 1.0 | 3.0 | 7.6 |
| Trimethoprim | 0.2 | 0.3 | 1.0 | 3.0 | 15.1 |
| Analyte | ICC R2 | SCC R2 | Mean Recovery (%) | RSD (%) |
|---|---|---|---|---|
| Brilliant Green | 0.996 | 0.989 | 80 | 20 |
| Diclofenac | 0.999 | 0.999 | 77 | 16 |
| Difloxacin | 0.993 | 0.995 | 60 | 26 |
| Enrofloxacin | 0.997 | 0.979 | 60 | 27 |
| Malachite Green | 0.999 | 0.997 | 79 | 25 |
| Marbofloxacin | 0.991 | 0.986 | 60 | 23 |
| Oxolinic Acid | 0.994 | 0.991 | 62 | 25 |
| Propanolol | 0.999 | 0.999 | 93 | 10 |
| Sarafloxacin | 0.995 | 0.990 | 72 | 20 |
| Sulfamethoxazole | 0.999 | 0.999 | 82 | 25 |
| Tamoxifen | 0.994 | 0.999 | 75 | 20 |
| Triamcinolone | 0.994 | 0.993 | 68 | 22 |
| Trimethoprim | 0.999 | 0.998 | 80 | 21 |
3.3. Monitoring the Presence of Drugs in Spanish Rivers
| Analyte | Maximum concentration (ng·L−1) | Minimum concentration (ng·L−1) | Mean concentration (ng·L−1) | Number of detections |
|---|---|---|---|---|
| Brilliant Green | 7.9 | 3.3 | 5.6 | 2 |
| Diclofenac | 46.0 | 2.8 | 13.6 | 69 |
| Difloxacin | 8.5 | 8.5 | 8.5 | 1 |
| Enrofloxacin | 164.5 | 60.0 | 119.9 | 3 |
| Malachite Green | 9.0 | 9.0 | 9.0 | 1 |
| Marbofloxacin | 20.1 | 3.6 | 8.8 | 10 |
| Oxolinic Acid | 39.1 | 39.1 | 39.1 | 1 |
| Propanolol | 62.6 | 4.2 | 11.5 | 14 |
| Sarafloxacin | 171.4 | 171.4 | 171.4 | 1 |
| Sulfamethoxazole | 40.1 | 3.0 | 10.7 | 42 |
| Tamoxifen | 11.7 | 3.5 | 7.5 | 6 |
| Triamcinolone | 8.4 | 8.4 | 8.4 | 1 |
| Trimethoprim | 110.4 | 3.5 | 16.1 | 46 |

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fent, K.; Anna, A.; Weston, A.A.; Caminada, D. Ecotoxicology of human pharmaceuticals. Aquat. Toxicol. 2006, 76, 122–159. [Google Scholar] [CrossRef]
- Kemper, N. Veterinary antibiotics in the aquatic and terrestrial environment. Ecol. Indic. 2008, 8, 1–13. [Google Scholar] [CrossRef]
- Crane, M.; Boxall, A.B.A.; Barrett, K. Veterinary Medicines in the Environment; Crane, M., Boxall, A.B.A., Barret, K., Eds.; Taylor & Francis: Boca Raton, FL, USA/CRC Press: London, UK, 2009; p. 224. [Google Scholar]
- Bergwerff, A.A.; Scherpenisse, P. Determination of residues of malachite green in aquatic animals. J. Chromatogr. B 2003, 788, 351–359. [Google Scholar] [CrossRef]
- Oplatowska, M.; Donnelly, R.F.; Majithiya, R.J.; Kennedy, D.G.; Elliott, C.T. The potential for human exposure, direct and indirect, to the suspected carcinogenic triphenylmethane dye Brilliant Green from green paper towels. Food Chem. Toxicol. 2011, 49, 1870–1876. [Google Scholar] [CrossRef]
- Srivastava, S.; Sinha, R.; Roy, D. Toxicological effects of malachite green. Aquat. Toxicol. 2004, 66, 319–329. [Google Scholar] [CrossRef]
- Daughton, C.G.; Ternes, T.A. Pharmaceuticals and personal care products in the environment, agents of subtle change? Environ. Health Perspect. 1999, 107, 907–938. [Google Scholar] [CrossRef]
- Jung, J.; Kim, Y.; Kim, J.; Jeong, D.H.; Choi, K. Environmental levels of ultraviolet light potentiate the toxicity of sulfonamide antibiotics in Daphnia magna. Ecotoxicology 2008, 17, 37–45. [Google Scholar] [CrossRef]
- Kim, J.W.; Ishibashi, H.; Yamauchi, R.; Ichikawa, N.; Takao, Y.; Hirano, M.; Koga, M.; Arizono, K. Acute toxicity of pharmaceutical and personal care products on freshwater crustacean (Thamnocephalus platyurus) and fish (Oryzias latipes). J. Toxicol. Sci. 2009, 34, 227–232. [Google Scholar] [CrossRef]
- Li, Z.H.; Randak, T. Residual pharmaceutically active compounds (PhACs) in aquatic environment—Status, toxicity and kinetics, a review. Vet. Med.-Czech. 2009, 54, 295–314. [Google Scholar]
- Madureira, T.V.; Cruzeiro, C.; Rochaa, M.J.; Rocha, E. The toxicity potential of pharmaceuticals found in the Douro River estuary (Portugal)—Experimental assessment using a zebrafish embryo test. Environ. Toxicol. Pharmacol. 2011, 32, 212–217. [Google Scholar]
- Heberer, T.; Fuhrmann, B.; Schmidt-Baumler, K.; Tsipi, D.; Koutsouba, V.; Hiskia, A. Occurrence of pharmaceutical residues in sewage, river, ground, and drinking water in Greece and Berlin (Germany). In Pharmaceuticals and Care Products in the Environment: Scientific and Regulatory Issues; Daughton, C.G., Jones-Lepp, T.L., Eds.; American Chemical Society: Washington, DC, USA, 2001; pp. 70–83. [Google Scholar]
- Alder, A.C.; Schaffner, C.; Majewsky, M.; Klasmeier, J.; Fenner, K. Fate of b-blocker human pharmaceuticals in surface water: Comparison of measured and simulated concentrations in the Glatt Valley Watershed, Switzerland. Water Res. 2010, 44, 936–948. [Google Scholar] [CrossRef]
- Dinh, Q.T.; Alliot, F.; Moreau-Guigon, E.; Eurin, J.; Chevreuil, M.; Labadie, P. Measurement of trace levels of antibiotics in river water using on-line enrichment and triple-quadrupole LC-MS/MS. Talanta 2011, 85, 1238–1245. [Google Scholar] [CrossRef]
- Ferrer, I.; Zweigenbaum, J.A.; Thurman, E.M. Analysis of 70 Environmental Protection Agency priority pharmaceuticals in water by EPA Method 1694. J. Chromatogr. A 2010, 1217, 5674–5686. [Google Scholar]
- Minh, T.B.; Leung, H.W.; Loi, I.H.; Chan, W.H.; Som, M.K.; Maob, J.Q.; Choi, D.; Lam, J.C.W.; Zheng, G.; Martín, M.; et al. Antibiotics in the Hong Kong metropolitan area: Ubiquitous distribution and fate in Victoria Harbour. Mar. Pollut. Bull. 2009, 58, 1052–1062. [Google Scholar] [CrossRef]
- Tamtam, F.; Mercier, F.; Eurin, J.; Chevreuil, M.; le Bot, B. Ultra performance liquid chromatography tandem mass spectrometry performance evaluation for analysis of antibiotics in natural waters. Anal. Bioanal. Chem. 2009, 393, 1709–1718. [Google Scholar] [CrossRef]
- Zuccato, E.; Castiglioni, S.; Bagnati, R.; Melis, M.; Fanelli, R. Source, occurrence and fate of antibiotics in the Italian aquatic environment. J. Hazard. Mater. 2010, 179, 1042–1048. [Google Scholar] [CrossRef]
- Carpinteiro, J.; Quintana, J.B.; Martínez, E.; Rodríguez, I.; Carro, A.M.; Lorenzo, R.A.; Cela, R. Application of strategic sample composition to the screening of anti-inflammatory drugs in water samples using solid-phase microextraction. Anal. Chim. Acta 2004, 524, 63–71. [Google Scholar] [CrossRef]
- Castro-Puyana, M.; Crego, A.L.; Marina, M.L. Recent advances in the analysis of antibiotics by CE and CEC. Electrophoresis 2010, 31, 229–250. [Google Scholar] [CrossRef]
- Herrera-Herrera, A.V.; Ravelo-Péreza, L.M.; Hernández-Borgesa, J.; Afonso, M.M.; Palenzuela, J.A.; Rodríguez-Delgado, M.A. Oxidized multi-walled carbon nanotubes for the dispersive solid-phase extraction of quinolone antibiotics from water samples using capillary electrophoresis and large volume sample stacking with polarity switching. J. Chromatogr. A 2011, 1218, 5352–5361. [Google Scholar]
- Lin, C.Y.; Huang, S.D. Application of liquid–liquid–liquid microextraction and high-performance liquid-chromatography for the determination of sulfonamides in water. Anal. Chim. Acta 2008, 612, 37–43. [Google Scholar] [CrossRef]
- Raich-Montiu, J.; Folch, J.; Compañó, R.; Granados, M.; Prat, M.D. Analysis of trace levels of sulfonamides in surface water and soil samples by liquid chromatography-fluorescence. J. Chromatogr. A 2007, 1172, 186–193. [Google Scholar]
- Serrano, J.M.; Silva, M. Rapid and sensitive determination of aminoglycoside antibiotics in water samples using a strong cation-exchange chromatography non-derivatisation method with chemiluminescence detection. J. Chromatogr. A 2006, 1117, 176–183. [Google Scholar] [CrossRef]
- Sun, L.; Chen, L.; Sun, X.; Du, X.; Yue, Y.; He, D.; Xu, H.; Zeng, Q.; Wang, H.; Ding, L. Analysis of sulfonamides in environmental water samples based on magnetic mixed hemimicelles solid-phase extraction coupled with HPLC-UV detection. Chemosphere 2009, 77, 1306–1312. [Google Scholar] [CrossRef]
- Díaz-Cruz, M.S.; García-Galán, M.J.; Barceló, D. Highly sensitive simultaneous determination of sulfonamide antibiotics and one metabolite in environmental waters by liquid chromatography-quadrupole linear ion trap-mass spectrometry. J. Chromatogr. A 2008, 1193, 50–59. [Google Scholar]
- García-Galán, M.J.; Díaz-Cruz, M.S.; Barceló, D. Determination of 19 sulfonamides in environmental water samples by automated on-line solid-phase extraction-liquid chromatography–tandem mass spectrometry (SPE-LC-MS/MS). Talanta 2010, 81, 355–366. [Google Scholar] [CrossRef]
- Huerta-Fontela, M.; Galceran, M.T.; Ventura, F. Ultraperformance liquid chromatography-tandem mass spectrometry analysis of stimulatory drugs of abuse in wastewater and surface waters. Anal. Chem. 2007, 79, 3821–3829. [Google Scholar] [CrossRef]
- Ibáñez, M.; Guerrero, C.; Sancho, J.V.; Hernández, F. Screening of antibiotics in surface and wastewater samples by ultra-high-pressure liquid chromatography coupled to hybrid quadrupole time-of-flight mass spectrometry. J. Chromatogr. A 2009, 1216, 2529–2539. [Google Scholar]
- Kuster, M.; López de Alda, M.J.; Hernando, M.D.; Petrovic, M.; Martín-Alonso, J.; Barceló, D. Analysis and occurrence of pharmaceuticals, estrogens, progestogens and polar pesticides in sewage treatment plant effluents, river water and drinking water in the Llobregat river basin (Barcelona, Spaiin). J. Hydrol. 2008, 358, 112–123. [Google Scholar] [CrossRef]
- Pedrouzo, M.; Borrull, F.; Marcé, R.M.; Pocurull, E. Simultaneous determination of macrolides, sulfonamides, and other pharmaceuticals in water samples by solid-phase extraction and LC-(ESI) MS. J. Sep. Sci. 2008, 31, 2182–2188. [Google Scholar] [CrossRef]
- Martín, J.; Camacho-Muñóz, D.; Santos, J.L.; Aparicio, I.; Alonso, E. Monitoring of pharmaceutically active compounds on the Guadalquivir River basin (Spain): Occurrence and risk assessment. J. Environ. Monit. 2011, 13, 2042–2049. [Google Scholar] [CrossRef]
- Valcárcel, Y.; González Alonso, S.; Rodríguez-Gil, J.L.; Gil, A.; Catalá, M. Detection of pharmaceutically active compounds in the rivers and tap water of the Madrid Region (Spain) and potential ecotoxicological risk. Chemosphere 2011, 84, 1336–1348. [Google Scholar] [CrossRef]
- González-Mariño, I.; Quintana, J.B.; Rodríguez, I.; Cela, R. Determination of drugs of abuse in water by solid-phase extraction, derivatisation and gas chromatography–ion trap-tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 1748–1760. [Google Scholar]
- Cancel-Barrio, J.J.; Fandiño, M. Management of irrigation water in Terra Chá: Indicators. IBADER 2009, 5, 49–57. (In Spanish) [Google Scholar]
- Nebot, C.; Gibb, S.W.; Boyd, K.G. Quantification of human pharmaceuticals in water samples by high performance liquid chromatography–tandem mass spectrometry. Anal. Chim. Acta 2007, 598, 87–94. [Google Scholar] [CrossRef]
- Environmental Protection Agency (EPA). Definition and Procedure for the Method Detection Limit-Revision 136, Appendix B; EPA—Environmental Protection Agency: Washington, DC, USA, 1984. [Google Scholar]
- Kim, S.C.; Carlson, K. Quantification of human and veterinary antibiotics in water and sediment using SPE/LC/MS/MS. Anal. Bioanal. Chem. 2007, 387, 1301–1315. [Google Scholar] [CrossRef]
- Senta, I.; Terzic, S.; Ahel, M. Simultaneous determination of sulfonamides, fluoroquinolones, macrolides and trimethoprim in wastewater and river water by LC-tandem-MS. Chromatographia 2008, 68, 747–758. [Google Scholar] [CrossRef]
- Pozo, O.J.; Guerrero, C.; Sancho, J.V.; Ibáñez, M.; Pitarch, E.; Hogendoorn, E.; Hernández, F. Efficient approach for the reliable quantification and confirmation of antibiotics in water using on-line solidphase extraction liquid chromatography/tandem mass spectrometry. J. Chromatogr. A 2006, 1103, 83–93. [Google Scholar] [CrossRef]
- Hao, C.; Lissemore, L.; Nguyen, B.; Kleywegt, S.; Yang, P.; Solomon, K. Determination of pharmaceuticals in environmental waters by liquid chromatography/electrospray ionization/tandem mass spectrometry. Anal. Bioanal. Chem. 2006, 384, 505–513. [Google Scholar]
- Hilton, M.; Thomas, K.V. Determination of selected human pharmaceutical compounds in effluent and surface water samples by high-performance liquid chromatography–Electrospray tandem mass spectrometry. J. Chromatogr. A 2003, 1015, 129–141. [Google Scholar] [CrossRef]
- Kasprzyk-Hordern, B.; Dinsdale, R.M.; Guwy, A.J. Multi-residue method for the determination of basic/neutral pharmaceuticals and illicit drugs in surface water by solid-phase extraction and ultra performance liquid chromatography-positive electrospray ionization tandem mass spectrometry. J. Chromatogr. A 2007, 1161, 132–145. [Google Scholar] [CrossRef]
- Tölgyesi, A.; Verebey, Z.; Sharma, V.K.; Kovacsics, L.; Fekete, J. Simultaneous determination of corticosteroids, androgens, and progesterone in river water by liquid chromatography-tandem mass spectrometry. Chemosphere 2010, 78, 972–979. [Google Scholar] [CrossRef]
- Batt, A.L.; Aga, D.S. Simultaneous analysis of multiple classes of antibiotics by ion trap LC/MS/MS for assessing surface water and groundwater contamination. Anal. Chem. 2005, 77, 2940–2947. [Google Scholar] [CrossRef]
- Batt, A.L.; Kostich, M.S.; Lazorchak, J.M. Analysis of ecologically relevant pharmaceuticals in wastewater and surface water using selective solid-phase extraction and UPLC-MS/MS. Anal. Chem. 2008, 80, 5021–5030. [Google Scholar] [CrossRef]
- Ashton, D.; Hilton, M.; Thomas, K.V. Investigating the environmental transport of human pharmaceuticals to streams in the United Kingdom. Sci. Total Environ. 2004, 333, 167–184. [Google Scholar] [CrossRef]
- Cahill, J.D.; Furlong, E.T.; Burkhardt, M.R.; Kolpin, D.; Anderson, L.G. Determination of pharmaceutical compounds in surface- and ground-water samples by solid-phase extraction and high-performance liquid chromatography–electrospray ionization mass spectrometry. J. Chromatogr. A 2004, 1041, 171–180. [Google Scholar] [CrossRef]
- Yang, S.; Cha, J.; Carlson, K.H. Quantitative determination of trace concentrations of tetracycline and sulfonamide antibiotics in surface water using solid-phase extraction and liquid chromatography/ion trap tandem mass spectrometry. Rapid Commun. Mass 2004, 18, 2131–2145. [Google Scholar] [CrossRef]
- Muñóz, I.; López-Doval, J.C.; Ricart, M.; Villagrasa, M.; Brix, R.; Geiszinger, A.; Ginebreda, A.; Guasch, H.; López de Alda, M.J.; Romaní, A.M.; et al. Bridging levels of pharmaceuticals in river water with biological community structure in the Llobregat river basin (northeast Spain). Environ. Toxicol. Chem. 2009, 28, 2706–2714. [Google Scholar] [CrossRef]
- Kasprzyk-Hordern, B.; Dinsdale, R.M.; Guwy, A.J. The occurrence of pharmaceuticals, personal care products, endocrine disruptors and illicit drugs in surface water in South Wales, UK. Water Res. 2008, 42, 3498–3518. [Google Scholar] [CrossRef]
- Benito-Peña, E.; Urraca, J.L.; Sellergren, B.; Moreno-Bondi, M.C. Solid-phase extraction of fluoroquinolones from aqueous samples using a water-compatible stochiometrically imprinted polymer. J. Chromatogr. A 2008, 1208, 62–70. [Google Scholar] [CrossRef]
- Rao, R.N.; Venkateswarlu, N.; Narsimha, R. Determination of antibiotics in aquatic environment by solid-phase extraction followed by liquid chromatography-electrospray ionization mass spectrometry. J. Chromatogr. A 2008, 1187, 151–164. [Google Scholar] [CrossRef]
- Forbes, B.; Sahm, D.F.; Weissfeld, A.S. Bailey & Scott’s Diagnostic Microbiology, 12th ed.; Médica Panamericana: Argentina, Spian, 2009; p. 1026. [Google Scholar]
- Pastor-Navarro, N.; Maquieira, Á.; Puchades, R. Review on immunoanalytical determination of tetracycline and sulfonamide residues in edible products. Anal. Bioanal. Chem. 2009, 395, 907–920. [Google Scholar] [CrossRef]
- Ferguson, P.J.; Bermot, M.J.; Doll, J.C.; Lauer, T.E. Detection of pharmaceuticals and personal care products (PPCPs) in near-shore habitats of southern Lake Michigan. Sci. Total. Environ. 2013, 458, 187–196. [Google Scholar]
- Gros, M.; Rodríguez-Mozaz, S.; Barceló, D. Fast and comprehensive multi-residue analysis of a broad range of human and veterinary pharmaceuticals and some of their metabolites in surface and treated waters by ultra-high-performance liquid chromatography coupled to quadrupole-linear ion trap tandem mass spectrometry. J. Chromatogr. A 2012, 1248, 104–121. [Google Scholar] [CrossRef]
- Park, J. Pharmaceuticals in the Environment and Management Approaches in Korea; Korea Environment Institute: Seoul, Korea, 2005. [Google Scholar]
- Yang, J.F.; Ying, G.G.; Zhao, J.L.; Tao, R.; Su, H.C.; Liu, Y.S. Spatial and seasonal distribution of selected antibiotics in surface waters of the Pearl Rivers, China. J. Environ. Sci. Health B 2011, 46, 272–280. [Google Scholar] [CrossRef]
- Huang, C.H.; Renew, J.E.; Smeby, K.L.; Pinkston, K.; Sedlak, D.L. Assessment of potential antibiotic contaminants in water and preliminary occurrence analysis. J. Contemp. Water Res. Educ. 2011, 120, 30–40. [Google Scholar]
- Jiang, L.; Hu, X.; Yin, D.; Zhang, H.; Yu, Z. Occurrence, distribution and seasonal variation of antibiotics in the Huangpu River, Shanghai, China. Chemosphere 2011, 82, 822–828. [Google Scholar] [CrossRef]
- Gros, M.; Petrovic, M.; Barceló, D. Development of a multi-residue analytical methodology based on liquid chromatography-tandem mass spectrometry (LC-MS/MS) for screening and trace level determination of pharmaceuticals in surface and wastewaters. Talanta 2006, 70, 678–690. [Google Scholar] [CrossRef]
- Kim, S.D.; Cho, J.; Kim, I.S.; Vanderford, B.J.; Snyder, S.A. Occurrence and removal of pharmaceuticals and endocrine disruptors in South Korean surface, drinking, and waste waters. Water Res. 2007, 41, 1013–1021. [Google Scholar] [CrossRef]
- Grujíc, S.; Vasiljevic, T.; Lausevic, M. Determination of multiple pharmaceutical classes in surface and ground waters by liquid chromatography–ion trap–tandem mass spectrometry. J. Chromatogr. A 2009, 1216, 4989–5000. [Google Scholar]
- Gros, M.; Petrović, M.; Ginebreda, A.; Barceló, D. Removal of pharmaceuticals during wastewater treatment and environmental risk assessment using hazard indexes. Environ. Int. 2010, 36, 15–26. [Google Scholar] [CrossRef]
- Bartels, P.; Tümpling, W.V. Solar radiation influence on the decomposition process of diclofenac in surface waters. Sci. Total Environ. 2007, 374, 143–155. [Google Scholar] [CrossRef]
- Farré, M.; Ferrer, I.; Ginebreda, A.; Figueras, M.; Olivella, L.; Tirapu, L.; Vilanova, M.; Barceló, D. Determination of drugs in surface water and wastewater samples by liquid chromatography-mass spectrometry: Methods and preliminary results including toxicity studies with Vibrio fischeri. J. Chromatogr. A 2001, 938, 187–197. [Google Scholar]
- Isidori, M.; Lavorgna, M.; Nardelli, A.; Pascarella, L.; Parrella, A. Toxic and genotoxic evaluation of six antibiotics on non-target organisms. Sci. Total Environ. 2005, 346, 87–98. [Google Scholar] [CrossRef]
- Schwaiger, J.; Ferling, H.; Mallow, U.; Wintermayr, H.; Negele, R.D. Toxic effects of the non-steroidal anti-inflammatory drug diclofenac. Part I: Histopathological alterations and bioaccumulation in rainbow trout. Aquat. Toxicol. 2004, 68, 141–150. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Iglesias, A.; Nebot, C.; Vázquez, B.I.; Coronel-Olivares, C.; Abuín, C.M.F.; Cepeda, A. Monitoring the Presence of 13 Active Compounds in Surface Water Collected from Rural Areas in Northwestern Spain. Int. J. Environ. Res. Public Health 2014, 11, 5251-5272. https://doi.org/10.3390/ijerph110505251
Iglesias A, Nebot C, Vázquez BI, Coronel-Olivares C, Abuín CMF, Cepeda A. Monitoring the Presence of 13 Active Compounds in Surface Water Collected from Rural Areas in Northwestern Spain. International Journal of Environmental Research and Public Health. 2014; 11(5):5251-5272. https://doi.org/10.3390/ijerph110505251
Chicago/Turabian StyleIglesias, Alejandra, Carolina Nebot, Beatriz I. Vázquez, Claudia Coronel-Olivares, Carlos M. Franco Abuín, and Alberto Cepeda. 2014. "Monitoring the Presence of 13 Active Compounds in Surface Water Collected from Rural Areas in Northwestern Spain" International Journal of Environmental Research and Public Health 11, no. 5: 5251-5272. https://doi.org/10.3390/ijerph110505251
APA StyleIglesias, A., Nebot, C., Vázquez, B. I., Coronel-Olivares, C., Abuín, C. M. F., & Cepeda, A. (2014). Monitoring the Presence of 13 Active Compounds in Surface Water Collected from Rural Areas in Northwestern Spain. International Journal of Environmental Research and Public Health, 11(5), 5251-5272. https://doi.org/10.3390/ijerph110505251