Arylesterase Phenotype-Specific Positive Association Between Arylesterase Activity and Cholinesterase Specific Activity in Human Serum

Abstract
:
1. Introduction
2. Experimental Section
2.1. Design
2.2. Subjects
2.3. Assays
2.4. Statistical Analysis
3. Results and Discussion
3.1. Results
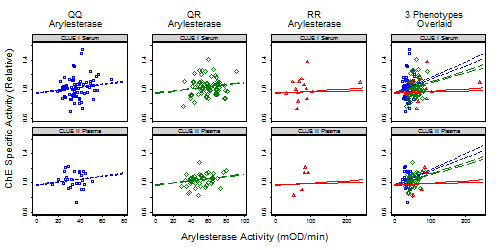
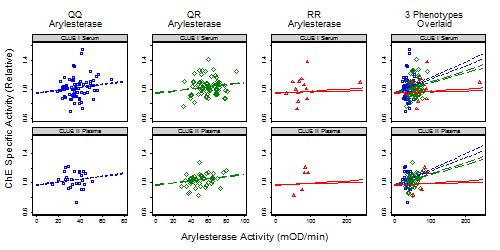
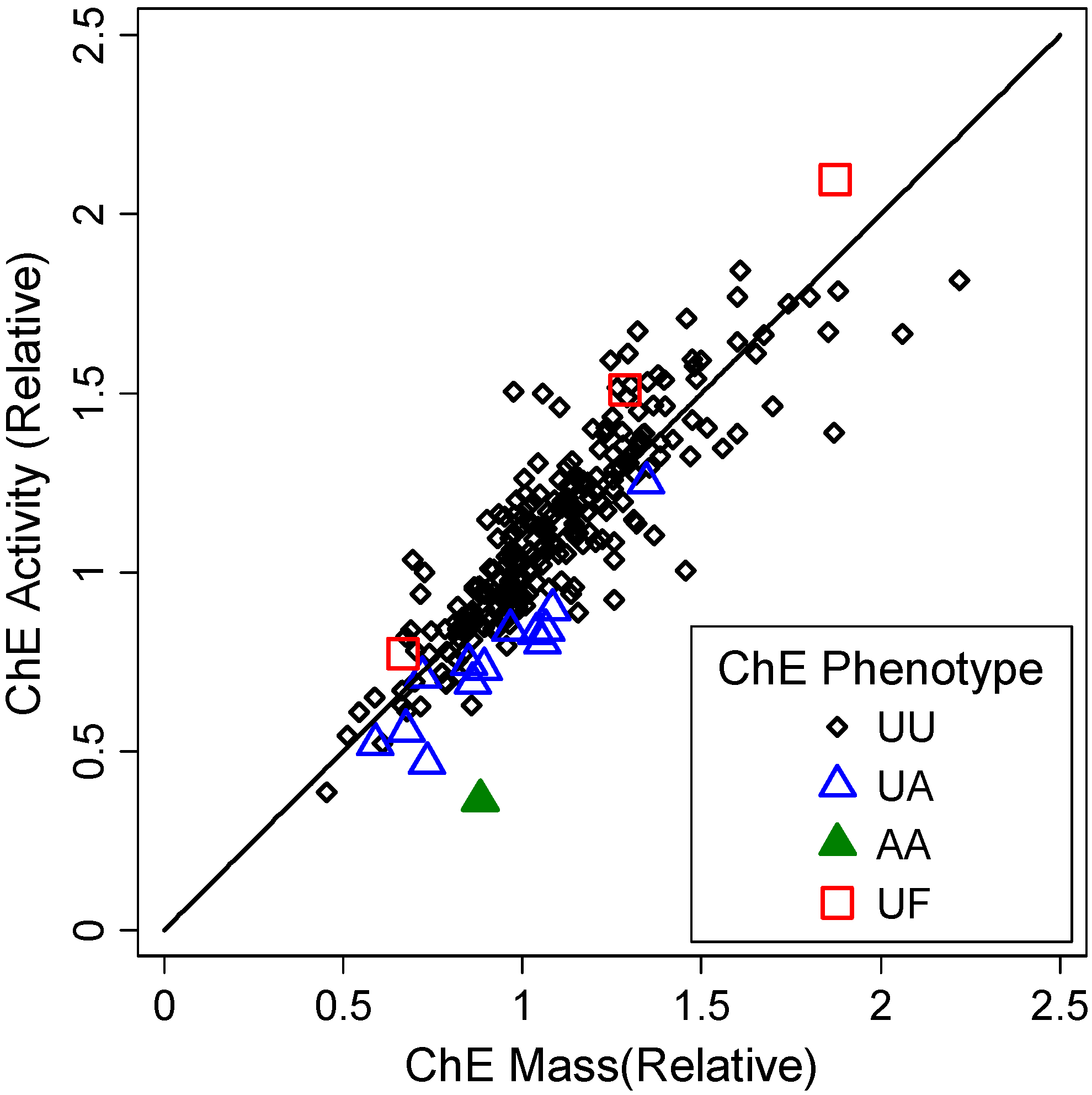
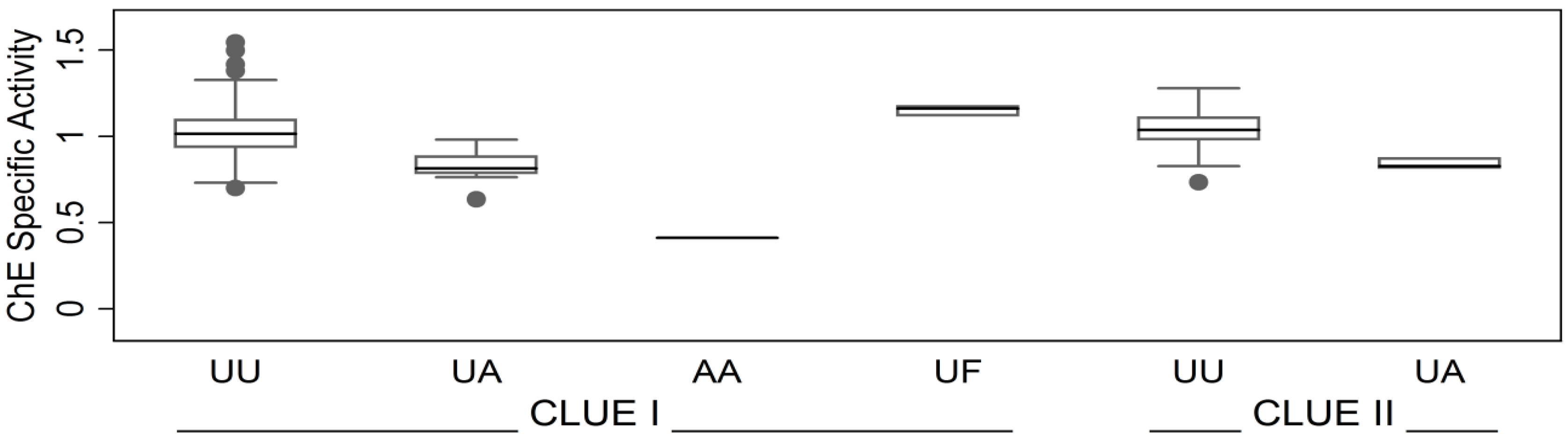
3.1.1. Subject and Sample Description

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | CLUE I | CLUE II | ||
|---|---|---|---|---|
| % | (n a) | % | (n) | |
| Sex: Male | 50 | (86) | 49 | (40) |
| Female | 50 | (85) | 51 | (42) |
| Race: White | 98 | (168) | 99 | (81) |
| Black | 2 | (3) | 1 | (1) |
| Smoking status:Never | 40 | (69) | 37 | (30) |
| Former | 29 | (49) | 51 | (42) |
| Current | 31 | (53) | 12 | (10) |
| Education: <12 years | 50 | (85) | 40 | (33) |
| ≥12 years | 50 | (86) | 60 | (49) |
| Mean | (SD) | Mean | (SD) | |
| Age at blood donation (years) | 52.7 | 13.3 | 63.1 | 11.8 |
| ChE Raw activity (unitless b) | 1.11 | 0.31 | 1.10 | 0.25 |
| ChE Mass (unitless) | 1.10 | 0.30 | 1.07 | 0.21 |
| ChE Specific activity (unitless) | 1.01 | 0.15 | 1.03 | 0.10 |
| Arylesterase activity (mOD/min): | ||||
| QQ phenotype | 37 | 10 | 34 | 8 |
| QR phenotype | 59 | 13 | 51 | 12 |
| RR phenotype | 84 | 44 c | 77 | 16 |
| Albumin (g/dL) | 5.1 | 0.7 | 4.6 | 0.48 |
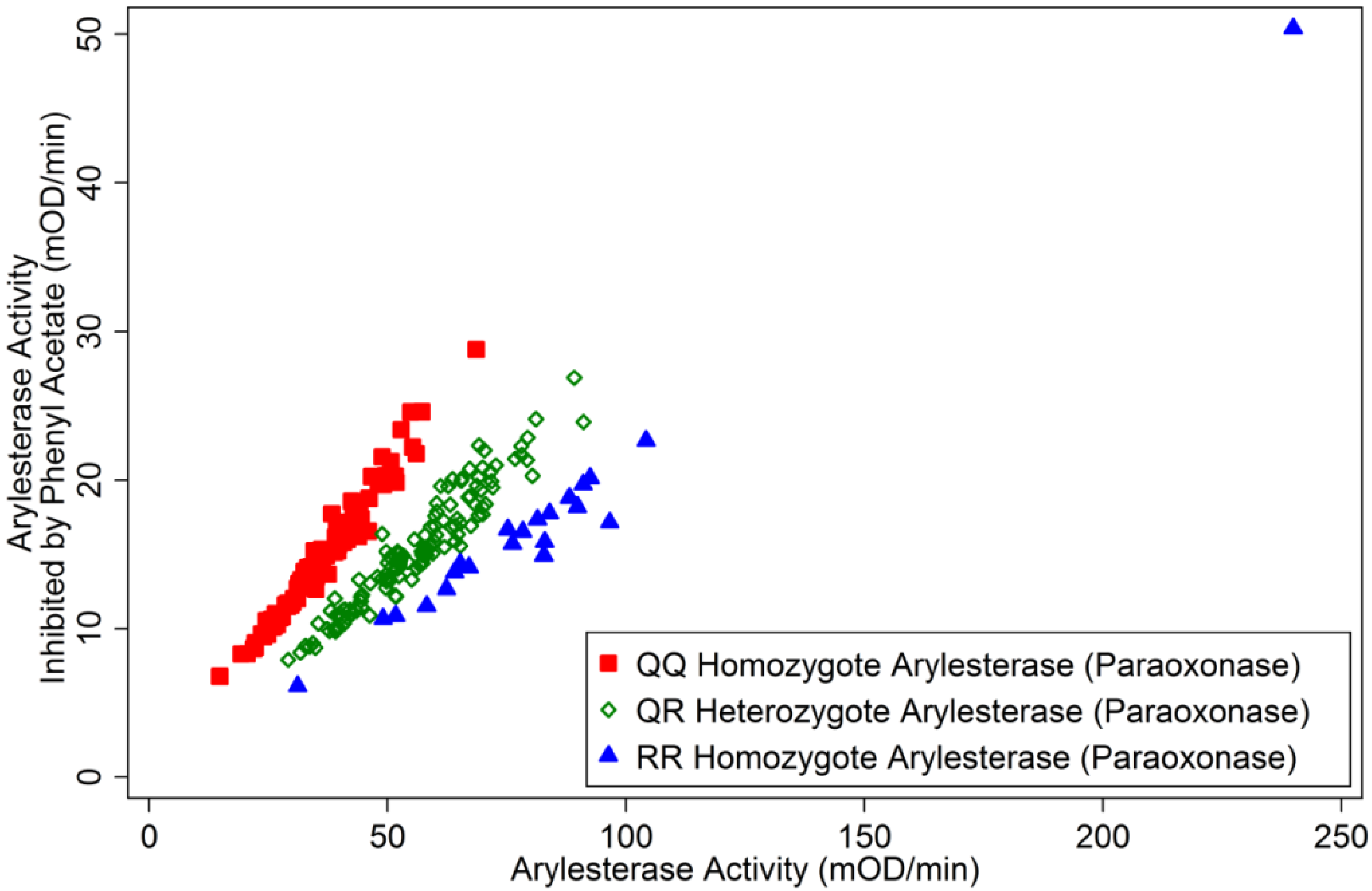
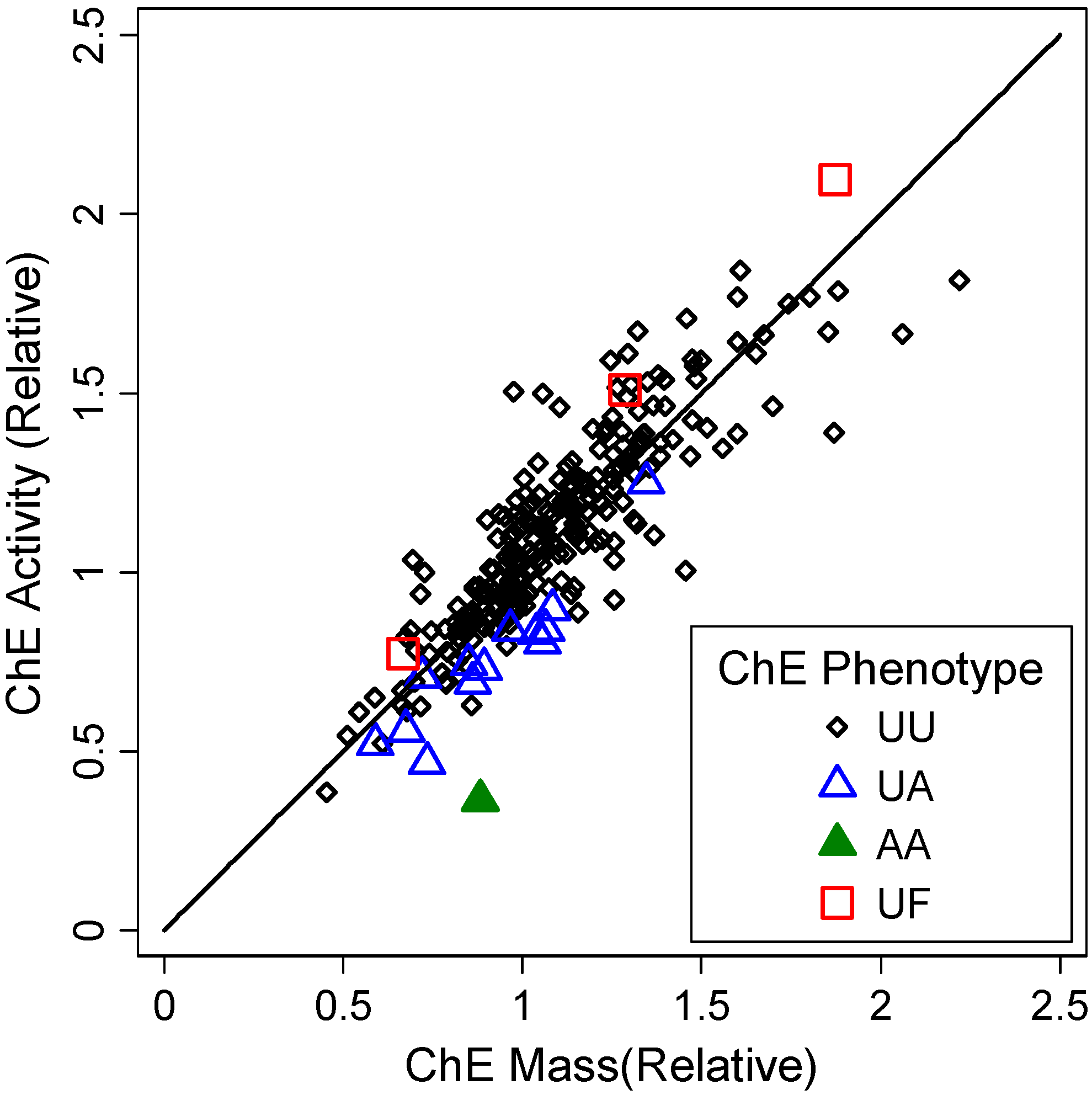
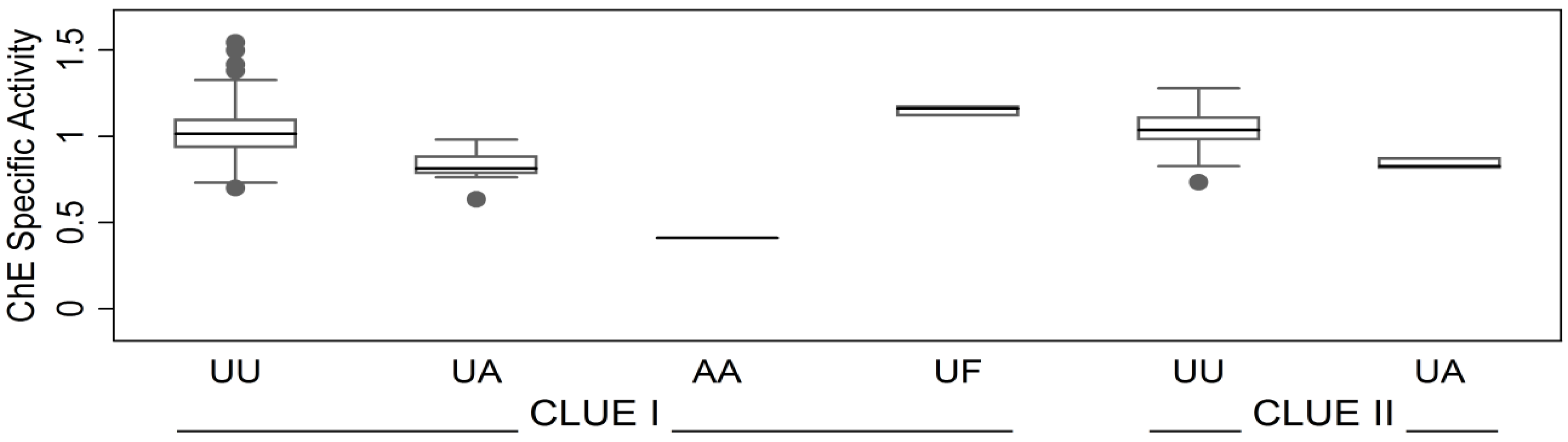
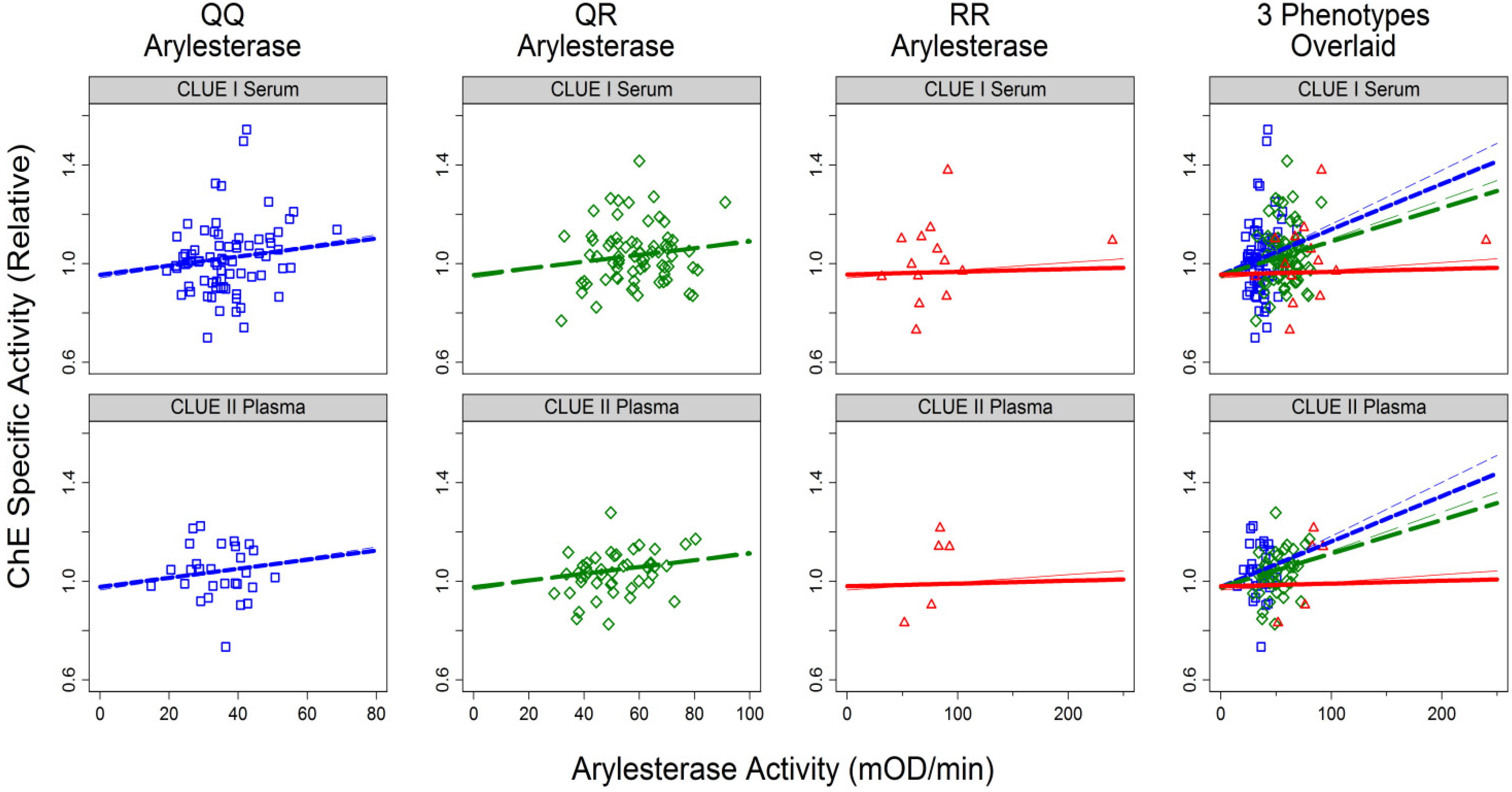
3.1.2. Correlates of ChE Specific Activity
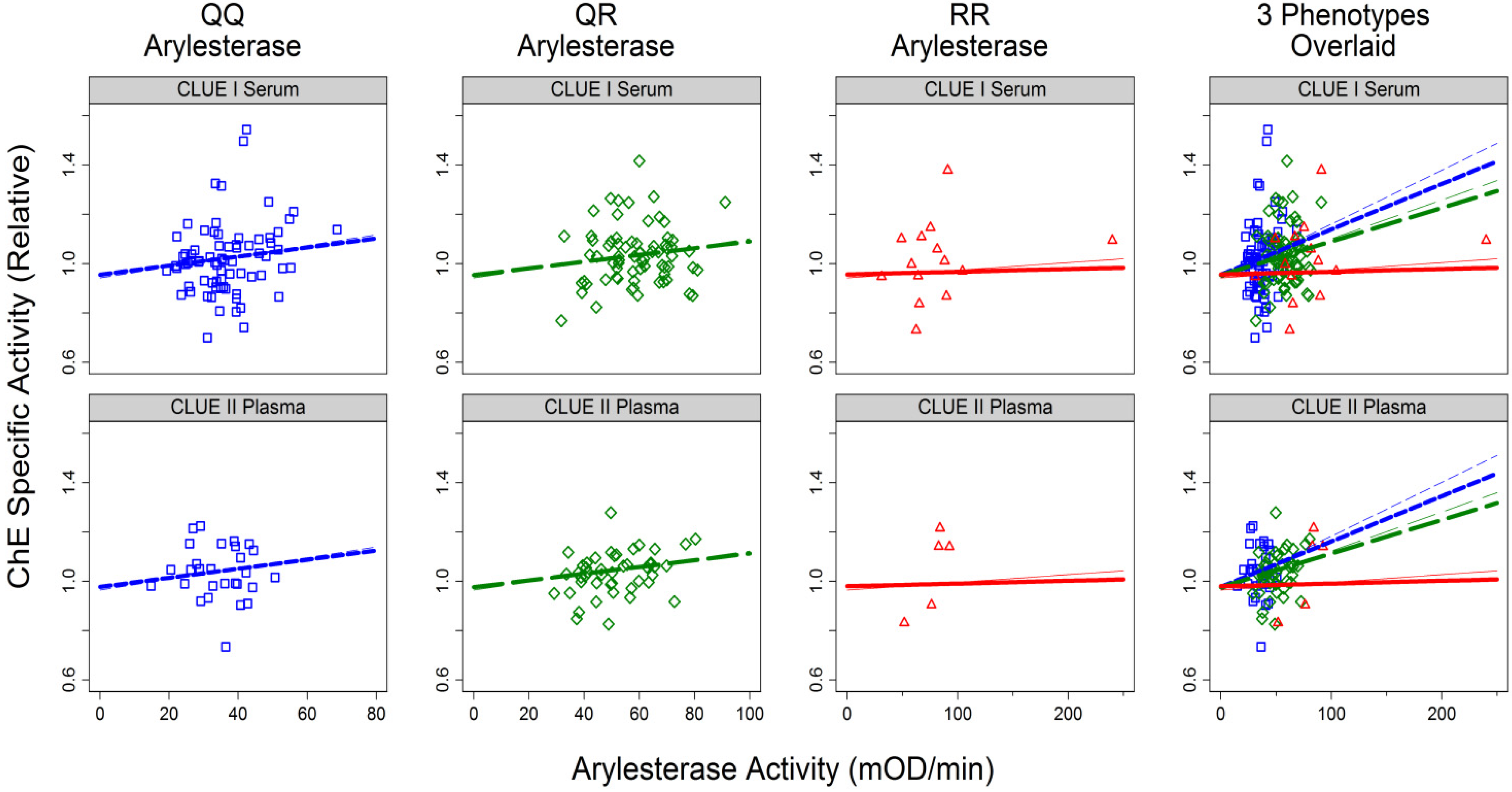

| Variable | Description (unit) | Partially-Adjusted Model a | Fully-Adjusted Model b | ||||
|---|---|---|---|---|---|---|---|
| β | (95% CI) | p c | β | (95% CI) | p | ||
| Arylesterase Activity | Continuous (mOD/min) | 0.004 | 0.007 | ||||
| For QQ phenotype | 0.0022 | (0.0007, 0.0037) | 0.004 | 0.0018 | (0.007, 0.068) | 0.01 | |
| For QR phenotype | 0.0016 | (0.0006, 0.003) | 0.001 | 0.0014 | (0.0004, 0.0023) | 0.005 | |
| For RR phenotype | 0.0003 | (−0.0004, 0.0010) | 0.4 | 0.0001 | (−0.0006, 0.0009) | 0.76 | |
| Interaction | 0.002 | 0.002 | |||||
| Albumin | Continuos (g/dL) | 0.033 | (0.010, 0.055) | 0.004 | 0.029 | (0.007, 0.051) | 0.01 |
| ChE Phenotype | Indicator (unitless) | 2 × 10−16 | 9 × 10−16 | ||||
| UA vs. UU (reference) | −0.21 | (−0.27, −0.15) | 4 × 10−11 | −0.19 | (−0.25, −0.13) | 5 × 10−10 | |
| AA vs. UU (reference) | −0.59 | (−0.81, −0.37) | 0.02 | −0.52 | (−0.73, −0.31) | 1 × 10−6 | |
| UF vs. UU (reference) | 0.15 | (0.023, 0.28) | 1 × 10−7 | 0.07 | (−0.05, 0.20) | 0.26 | |
| Sample Source | Indicator (unitless) | 0.35 | 0.02 | ||||
| CLUE II vs. I (reference) | 0.017 | (-0.017, 0.051) | 0.037 | (0.007, 0.068) | |||
| Intercept | Indicator (unitless) | 1.02 | (1.00, 1.05) | 0.81 | (0.69, 0.92) | ||
3.2. Discussion
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Wilson, B.W.; Henderson, J.D. Blood esterase determinations as markers of exposure. Rev. Environ. Contam Toxicol. 1992, 128, 55–69. [Google Scholar]
- Whittaker, M. Cholinesterase; Karger: Basel, Switzerland, 1986; Volume 11. [Google Scholar]
- Altland, K.; Goedde, H.W.; Held, K.; Jensen, M.; Munsch, H.; Solem, E. New biochemical and immunological data on quantitative and qualitative variability of human pseudocholinesterase. Humangenetik 1971, 14, 56–60. [Google Scholar]
- Brock, A. Inter and intraindividual variations in plasma cholinesterase activity and substance concentration in employees of an organophosphorus insecticide factory. Br. J. Ind. Med. 1991, 48, 562–567. [Google Scholar]
- Kotani, K.; Maekawa, M.; Hara, K.; Kanno, T. Clinical significance of serum cholinesterase concentrations determined by enzyme-linked immunosorbent assay (ELISA). Rinsho Byori 1996, 44, 965–969. [Google Scholar]
- Center for Disease Control and Prevention (CDC). Fourth National Report on Human Exposure to Environmental Chemicals; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2009.
- Stone, D.L.; Sudakin, D.L.; Jenkins, J.J. Longitudinal trends in organophosphate incidents reported to the National Pesticide Information Center, 1995–2007. Environ. Health 2009, 8, 18. [Google Scholar] [CrossRef]
- United States Environmental Protection Agency (EPA). Pesticide Industry Sales and Usage—2000 and 2001 Market Estimates; Environmental Protection Agency: Washington, DC, USA, 2004.
- Furlong, C.E.; Cole, T.B.; Jarvik, G.P.; Pettan-Brewer, C.; Geiss, G.K.; Richter, R.J.; Shih, D.M.; Tward, A.D.; Lusis, A.J.; Costa, L.G. Role of paraoxonase (PON1) status in pesticide sensitivity: Genetic and temporal determinants. Neurotoxicology 2005, 26, 651–659. [Google Scholar] [CrossRef]
- Costa, L.G.; Cole, T.B.; Furlong, C.E. Polymorphisms of paraoxonase (PON1) and their significance in clinical toxicology of organophosphates. J. Toxicol. Clin. Toxicol. 2003, 41, 37–45. [Google Scholar] [CrossRef]
- Aoki, Y. Exposure to Cholinesterase-Inhibiting Agents as a Risk Factor for Development of Non-Hodgkin’s Lymphoma; Johns Hopkins Bloomberg School of Public Health: Baltimore, MD, USA, 2004. [Google Scholar]
- Bush, D. The Interaction of Serum Micronutrients with Other Potential Risk Factors for the Development of Non-Hodgkin’s Lymphoma; The Johns Hopkins University School of Hygiene and Public Health: Baltimore, MD, USA, 1995. [Google Scholar]
- Rothman, N.; Cantor, K.P.; Blair, A.; Bush, D.; Brock, J.W.; Helzlsouer, K.; Zahm, S.H.; Needham, L.L.; Pearson, G.R.; Hoover, R.N.; et al. A nested case-control study of non-Hodgkin lymphoma and serum organochlorine residues. Lancet 1997, 350, 240–244. [Google Scholar] [CrossRef]
- Willig, S.; Hunter, D.L.; Dass, P.D.; Padilla, S. Validation of the use of 6,6'-dithiodinicotinic acid as a chromogen in the Ellman method for cholinesterase determinations. Vet. Hum. Toxicol. 1996, 38, 249–253. [Google Scholar]
- Dass, P.D.; Offutt, D.M.; Mejia, M.B.; VanGoethem, D.; Christenson, W.R.; Landes, M.M.; Stuart, B.P.; Sangha, G.K.; Thyssen, J.H. Comparative kinetic analysis of cholinesterase methods in rat and human erythrocytes and plasma. Vet. Hum. Toxicol. 1997, 39, 11–17. [Google Scholar]
- Whittaker, M.; Britten, J.J. Differential inhibition of plasma cholinesterase variants using the dibutyrate analogue of pancuronium bromide. Hum. Hered. 1981, 31, 242–247. [Google Scholar] [CrossRef]
- Hanel, H.K.; Viby-Mogensen, J. The inhibition of serum cholinesterase by urea. Mechanism of action and application in the typing of abnormal genes. Br. J. Anaesth. 1977, 49, 1251–1257. [Google Scholar] [CrossRef]
- Turner, J.M.; Hall, R.A.; Whittaker, M.; Holder, R.L.; Kricka, L.J. Application of stepwise discriminant analysis in the phenotyping of plasma cholinesterase variants. Ann. Clin. Biochem. 1985, 22 (Pt. 2), 175–178. [Google Scholar]
- Haagen, L.; Brock, A. A new automated method for phenotyping arylesterase (EC 3.1.1.2) based upon inhibition of enzymatic hydrolysis of 4-nitrophenyl acetate by phenyl acetate. Eur. J. Clin. Chem. Clin. Biochem. 1992, 30, 391–395. [Google Scholar]
- Hill, P.G.; Wells, T.N. Bromocresol purple and the measurement of albumin. Falsely high plasma albumin concentrations eliminated by increased reagent ionic strength. Ann. Clin. Biochem. 1983, 20 (Pt. 5), 264–270. [Google Scholar]
- Zheng, B. Summarizing the goodness of fit of generalized linear models for longitudinal data. Stat. Med. 2000, 19, 1265–1275. [Google Scholar] [CrossRef]
- StataCorp. Stata Statistical Software, Release 8.0, StataCorp LP: College Station, TX, USA, 2003.
- Furlong, C.E.; Richter, R.J.; Seidel, S.L.; Costa, L.G.; Motulsky, A.G. Spectrophotometric assays for the enzymatic hydrolysis of the active metabolites of chlorpyrifos and parathion by plasma paraoxonase/arylesterase. Anal. Biochem. 1989, 180, 242–247. [Google Scholar] [CrossRef]
- Davies, H.G.; Richter, R.J.; Keifer, M.; Broomfield, C.A.; Sowalla, J.; Furlong, C.E. The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin. Nat. Genet. 1996, 14, 334–336. [Google Scholar]
- Li, W.F.; Costa, L.G.; Richter, R.J.; Hagen, T.; Shih, D.M.; Tward, A.; Lusis, A.J.; Furlong, C.E. Catalytic efficiency determines the in-vivo efficacy of PON1 for detoxifying organophosphorus compounds. Pharmacogenetics 2000, 10, 767–779. [Google Scholar] [CrossRef]
- Hofmann, J.N.; Keifer, M.C.; Furlong, C.E.; de Roos, A.J.; Farin, F.M.; Fenske, R.A.; van, B.G.; Checkoway, H. Serum cholinesterase inhibition in relation to paraoxonase-1 (PON1) status among organophosphate-exposed agricultural pesticide handlers. Environ. Health Perspect. 2009, 117, 1402–1408. [Google Scholar]
- Mackness, B.; Durrington, P.; Povey, A.; Thomson, S.; Dippnall, M.; Mackness, M.; Smith, T.; Cherry, N. Paraoxonase and susceptibility to organophosphorus poisoning in farmers dipping sheep. Pharmacogenetics 2003, 13, 81–88. [Google Scholar]
- Richter, R.J.; Jarvik, G.P.; Furlong, C.E. Paraoxonase 1 (PON1) status and substrate hydrolysis. Toxicol. Appl. Pharmacol. 2009, 235, 1–9. [Google Scholar] [CrossRef]
- Payne-Sturges, D.; Cohen, J.; Castorina, R.; Axelrad, D.A.; Woodruff, T.J. Evaluating cumulative organophosphorus pesticide body burden of children: A national case study. Environ. Sci. Technol. 2009, 43, 7924–7930. [Google Scholar] [CrossRef]
- Nguyen, S.D.; Sok, D.E. Preferable stimulation of PON1 arylesterase activity by phosphatidylcholines with unsaturated acyl chains or oxidized acyl chains at sn-2 position. Biochim. Biophys. Acta 2006, 1758, 499–508. [Google Scholar]
- Schumacher, I.; Arad, A.; Margalit, R. Butyrylcholinesterase formulated in liposomes. Biotechnol. Appl. Biochem. 1999, 30 (Pt. 3), 225–230. [Google Scholar]
- Gaidukov, L.; Rosenblat, M.; Aviram, M.; Tawfik, D.S. The 192R/Q polymorphs of serum paraoxonase PON1 differ in HDL binding, lipolactonase stimulation, and cholesterol efflux. J. Lipid Res. 2006, 47, 2492–2502. [Google Scholar] [CrossRef]
- Gaidukov, L.; Tawfik, D.S. High affinity, stability, and lactonase activity of serum paraoxonase PON1 anchored on HDL with ApoA-I. Biochemistry 2005, 44, 11843–11854. [Google Scholar] [CrossRef]
- Paragh, G.; Harangi, M.; Seres, I. Effect of Lipid Lowering Medications on PON1. In The Paraoxonases: Their Role in Disease Development and Xenobiotic Metabolism; Mackness, B., Mackness, M., Aviram, M., Paragh, G., Eds.; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar]
- Kunec-Vajic, E.; Bernat, N.; Muacevic-Katanec, D. Effect of hypolipidemic drugs on cholinesterase activity in the rat. Gen. Pharmacol. 1992, 23, 217–219. [Google Scholar] [CrossRef]
- Butler, E.G.; England, P.J.; Williams, G.M. Effect of peroxisome proliferating hypolipidemic agents on serum activity levels of arylesterase and cholinesterase in rats and mice. Res. Commun. Chem. Pathol. Pharmacol. 1988, 60, 125–128. [Google Scholar]
- Kwon, E.Y.; Do, G.M.; Cho, Y.Y.; Park, Y.B.; Jeon, S.M.; Choi, M.S. Anti-atherogenic property of ferulic acid in apolipoprotein E-deficient mice fed Western diet: Comparison with clofibrate. Food Chem. Toxicol. 2010, 48, 2298–2303. [Google Scholar] [CrossRef]
- Downing, N.S.; Ross, J.S.; Jackevicius, C.A.; Krumholz, H.M. Avoidance of generic competition by Abbott Laboratories’ fenofibrate franchise. Arch. Intern. Med. 2012, 172, 724–730. [Google Scholar] [CrossRef]
- Saha, N.; Roy, A.C.; Teo, S.H.; Tay, J.S.; Ratnam, S.S. Influence of serum paraoxonase polymorphism on serum lipids and apolipoproteins. Clin. Genet. 1991, 40, 277–282. [Google Scholar]
- Hegele, R.A.; Brunt, J.H.; Connelly, P.W. A polymorphism of the paraoxonase gene associated with variation in plasma lipoproteins in a genetic isolate. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 89–95. [Google Scholar] [CrossRef]
- Nevin, D.N.; Zambon, A.; Furlong, C.E.; Richter, R.J.; Humbert, R.; Hokanson, J.E.; Brunzell, J.D. Paraoxonase genotypes, lipoprotein lipase activity, and HDL. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1243–1249. [Google Scholar] [CrossRef]
- Rozek, L.S.; Hatsukami, T.S.; Richter, R.J.; Ranchalis, J.; Nakayama, K.; McKinstry, L.A.; Gortner, D.A.; Boyko, E.; Schellenberg, G.D.; Furlong, C.E.; Jarvik, G.P.; et al. The correlation of paraoxonase (PON1) activity with lipid and lipoprotein levels differs with vascular disease status. J. Lipid Res. 2005, 46, 1888–1895. [Google Scholar] [CrossRef]
- Kutty, K.M.; Payne, R.H. Serum pseudocholinesterase and very-low-density lipoprotein metabolism. J. Clin. Lab Anal. 1994, 8, 247–250. [Google Scholar] [CrossRef]
- Mackness, B.; Mackness, M. Anti-inflammatory properties of paraoxonase-1 in atherosclerosis. Adv. Exp. Med. Biol. 2010, 660, 143–151. [Google Scholar] [CrossRef]
- Pavkovic, E.; Simeon, V.; Reiner, E.; Sucic, M.; Lipovac, V. Serum paraoxonase and cholinesterase activities in individuals with lipid and glucose metabolism disorders. Chem.-Biol. Interact. 1993, 87, 179–182. [Google Scholar] [CrossRef]
- Paragh, G.; Seres, I.; Balogh, Z.; Varga, Z.; Karpati, I.; Matyus, J.; Ujhelyi, L.; Kakuk, G. The serum paraoxonase activity in patients with chronic renal failure and hyperlipidemia. Nephron 1998, 80, 166–170. [Google Scholar] [CrossRef]
- Cucuianu, M.; Popescu, T.A.; Haragus, S. Pseudocholinesterase in obese and hyperlipemic subjects. Clin. Chim. Acta 1968, 22, 151–155. [Google Scholar] [CrossRef]
- Kleemola, P.; Freese, R.; Jauhiainen, M.; Pahlman, R.; Alfthan, G.; Mutanen, M. Dietary determinants of serum paraoxonase activity in healthy humans. Atherosclerosis 2002, 160, 425–432. [Google Scholar] [CrossRef]
- Ferre, N.; Camps, J.; Fernandez-Ballart, J.; Arija, V.; Murphy, M.M.; Ceruelo, S.; Biarnes, E.; Vilella, E.; Tous, M.; Joven, J. Regulation of serum paraoxonase activity by genetic, nutritional, and lifestyle factors in the general population. Clin. Chem. 2003, 49, 1491–1497. [Google Scholar] [CrossRef]
- Jarvik, G.P.; Tsai, N.T.; McKinstry, L.A.; Wani, R.; Brophy, V.H.; Richter, R.J.; Schellenberg, G.D.; Heagerty, P.J.; Hatsukami, T.S.; Furlong, C.E. Vitamin C and E intake is associated with increased paraoxonase activity. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1329–1333. [Google Scholar] [CrossRef]
- Rantala, M.; Silaste, M.L.; Tuominen, A.; Kaikkonen, J.; Salonen, J.T.; Alfthan, G.; Aro, A.; Kesaniemi, Y.A. Dietary modifications and gene polymorphisms alter serum paraoxonase activity in healthy women. J. Nutr. 2002, 132, 3012–3017. [Google Scholar]
- Aviram, M.; Dornfeld, L.; Rosenblat, M.; Volkova, N.; Kaplan, M.; Coleman, R.; Hayek, T.; Presser, D.; Fuhrman, B. Pomegranate juice consumption reduces oxidative stress, atherogenic modifications to LDL, and platelet aggregation: Studies in humans and in atherosclerotic apolipoprotein E-deficient mice. Am. J. Clin. Nutr. 2000, 71, 1062–1076. [Google Scholar]
- McGehee, D.S.; Krasowski, M.D.; Fung, D.L.; Wilson, B.; Gronert, G.A.; Moss, J. Cholinesterase inhibition by potato glycoalkaloids slows mivacurium metabolism. Anesthesiology 2000, 93, 510–519. [Google Scholar] [CrossRef]
- Masson, P. A naturally occurring molecular form of human plasma cholinesterase is an albumin conjugate. Biochim. Biophys. Acta 1989, 998, 258–266. [Google Scholar] [CrossRef]
- Sakoguchi, T.; Kobayashi, K.; Kimura, M.; Hase, A.; Shimozawa, M.; Matsuoka, A. Influence of blood proteins on biomedical analysis. VII. Electrophoretic analysis of the interaction of pseudocholinesterase with fatty acid and/or human serum albumin. Chem. Pharm. Bull. (Tokyo) 1984, 32, 3273–3276. [Google Scholar] [CrossRef]
- Hostmark, A.T.; Glattre, E.; Jellum, E. Effect of long-term storage on the concentration of albumin and free fatty acids in human sera. Scand. J. Clin. Lab Invest. 2001, 61, 443–447. [Google Scholar] [CrossRef]
- Burtis, C.A.; Ashwood, E.R.; Tietz, N.W. Tietz Textbook of Clinical Chemistry, 3rd ed.; W.B. Saunders: Philadelphia, PA, USA, 1999. [Google Scholar]
- Propert, D.N.; Brackenridge, C.J. The relation of sex, age, smoking status, birth rank and parental ages to pseudocholinesterase activity and phenotypes in a sample of Australian Caucasian adults. Hum. Genet. 1976, 32, 181–188. [Google Scholar] [CrossRef]
- Brock, A.; Brock, V. Factors affecting inter-individual variation in human plasma cholinesterase activity: Body weight, height, sex, genetic polymorphism and age. Arch. Environ. Contam. Toxicol. 1993, 24, 93–99. [Google Scholar] [CrossRef]
- Ratner, D.; Bar, S.P.; Schneeyour, A.; Kardontchik, A.; Eshel, E. Seasonal variation in blood cholinesterase activity. Isr. J. Med. Sci. 1989, 25, 247–250. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Aoki, Y.; Helzlsouer, K.J.; Strickland, P.T. Arylesterase Phenotype-Specific Positive Association Between Arylesterase Activity and Cholinesterase Specific Activity in Human Serum. Int. J. Environ. Res. Public Health 2014, 11, 1422-1443. https://doi.org/10.3390/ijerph110201422
Aoki Y, Helzlsouer KJ, Strickland PT. Arylesterase Phenotype-Specific Positive Association Between Arylesterase Activity and Cholinesterase Specific Activity in Human Serum. International Journal of Environmental Research and Public Health. 2014; 11(2):1422-1443. https://doi.org/10.3390/ijerph110201422
Chicago/Turabian StyleAoki, Yutaka, Kathy J. Helzlsouer, and Paul T. Strickland. 2014. "Arylesterase Phenotype-Specific Positive Association Between Arylesterase Activity and Cholinesterase Specific Activity in Human Serum" International Journal of Environmental Research and Public Health 11, no. 2: 1422-1443. https://doi.org/10.3390/ijerph110201422
APA StyleAoki, Y., Helzlsouer, K. J., & Strickland, P. T. (2014). Arylesterase Phenotype-Specific Positive Association Between Arylesterase Activity and Cholinesterase Specific Activity in Human Serum. International Journal of Environmental Research and Public Health, 11(2), 1422-1443. https://doi.org/10.3390/ijerph110201422