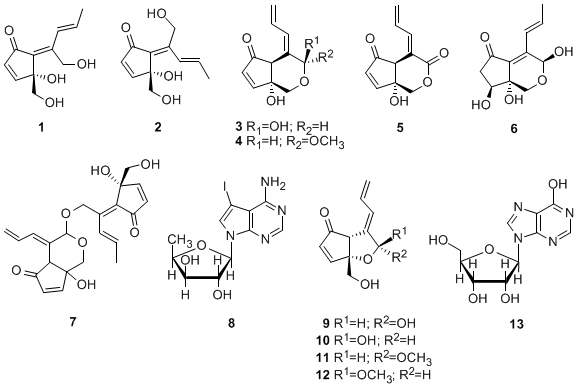
2. Results and Discussion
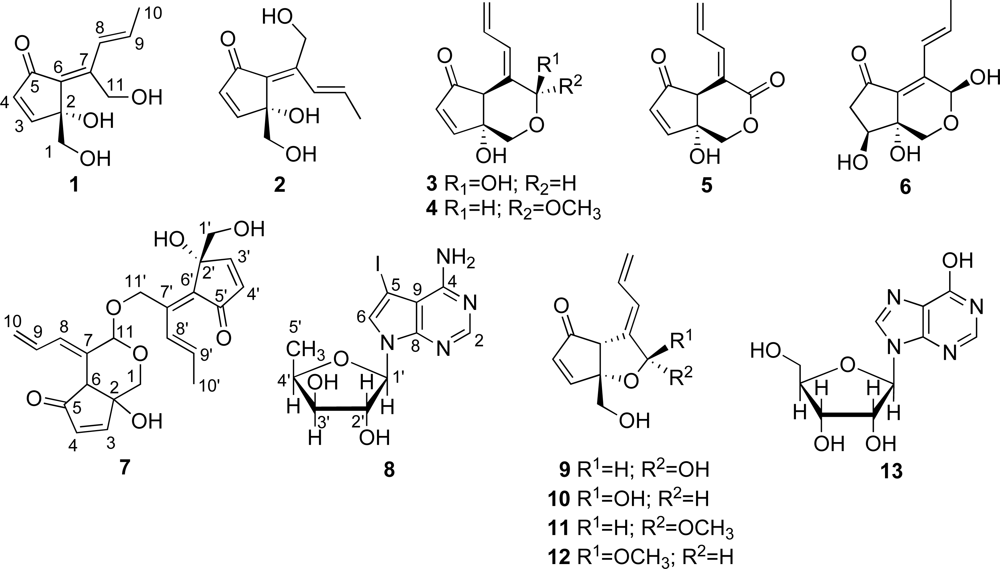
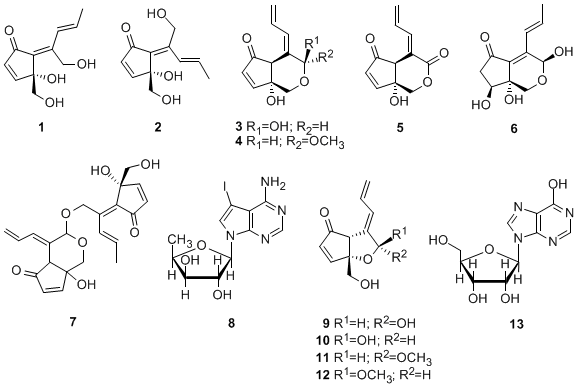
Specimens of colonial ascidian Lissoclinum sp. were collected off the coast of Tarama island, Okinawa, Japan. The specimens were extracted with acetone and the extract was partitioned between EtOAc and H2O. The aqueous layer was further partitioned between 1-BuOH and H2O. The H2O-soluble part was fractionated using RP-MPLC (reversed-phase MPLC) and eluted with a combination of H2O and MeOH. Further purification of the obtained fractions using RP-HPLC led to isolation of 1 (0.0059% of wet weight) and 13 (0.00067%). The EtOAc extract was suspended in aqueous MeOH and then successively extracted with hexane, CHCl3 and 1-BuOH. The resultant BuOH-soluble material was separated by a series of chromatographic steps [RP-OCC (reversed-phase open column chromatography), RP-MPLC and RP-HPLC] to afford 3 (0.0020%), 4 (0.00058%), 6 (0.0029%), 2 (0.011%), 1 (0.0049%), 12 (0.000014%), 5 (0.00030%), 7 (0.000019%) and an inseparable mixture of didemnenones A/B (9/10, 1:1, 0.00033%).
The colonial ascidian Diplosoma sp. was collected off the coast of Hateruma island, Okinawa, Japan. The specimens were extracted with acetone and the extract was partitioned between EtOAc and H2O. The EtOAc extract was suspended in aqueous MeOH and then successively extracted with hexane, CHCl3 and 1-BuOH. The CHCl3-soluble material was subjected to RP-OCC and eluted with H2O/MeOH, MeOH and MeOH/EtOAc. Further separation of the H2O/MeOH fraction by RP-HPLC eluted with a combination of H2O, MeOH and MeCN, led to the isolation of 1 (0.0021%), 2 (0.00066%), 5 (0.00047%), 8 (0.018%), 9/10 (1:1 mixture, 0.00069%), 11 (0.00098%) and 12 (0.00033%).
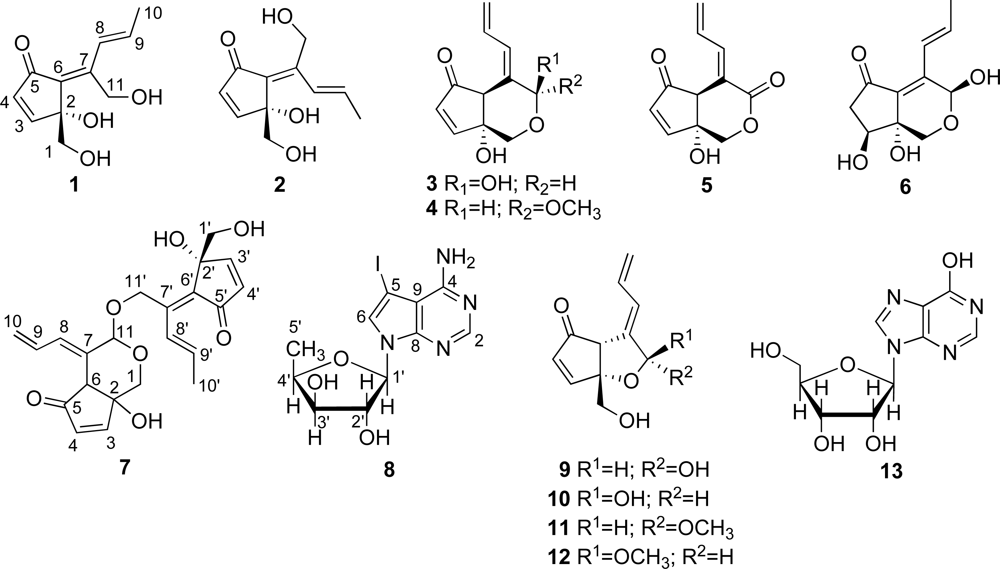
Analysis of
1 by NMR (
Tables 1 and
2) and HR-ESIMS [
m/z 233.0786 (M + Na)
+, calcd. for C
11H
14O
4Na, 233.0784] provided a molecular formula of C
11H
14O
4. The carbon resonating at δ
C 197.1 (s) suggested the presence of a carbonyl carbon in
1 and the IR absorption band at
vmax 1675 cm
−1 further supported the presence of the carbonyl group. Extensive analysis of
1H- and
13C-NMR data (
Tables 1 and
2) supported by
1H-
1H COSY data indicated the presence of a
cis double bond [δ
C 161.4 (d), 134.3 (d); δ
H 7.35 (d), 6.25 (d)], a
trans double bond [δ
C 127.2 (d), 134.8 (d); δ
H 7.73 (dd,
J = 1.6, 16.0 Hz), 6.43 (dq,
J = 16.0, 6.8 Hz)], a tetrasubstituted double bond [δ
C 133.5 (s), 144.5 (s)], a methyl group [δ
C 19.1 (t); δ
H 1.83 (dd)], two oxygenated methylenes [δ
C 66.8 (t); δ
H 3.74 (dd), 3.51 (dd) and δ
C 56.6 (t); δ
H 4.64 (dd), 4.36 (dd)] and an oxygenated quaternary carbon [δ
C 80.5 (s)] in
1. Degrees of unsaturation for these partial structures amount to four. Thus,
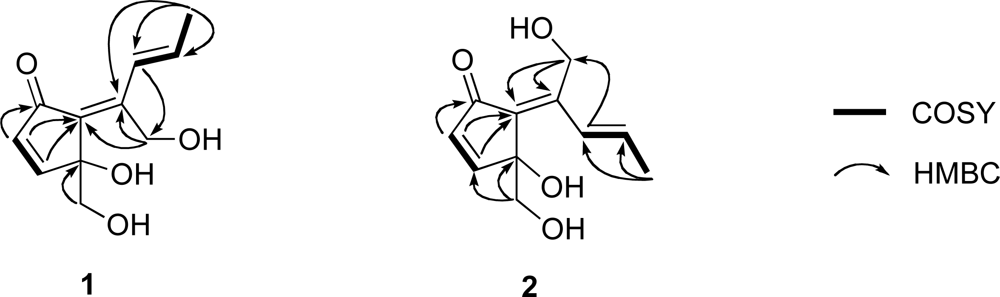
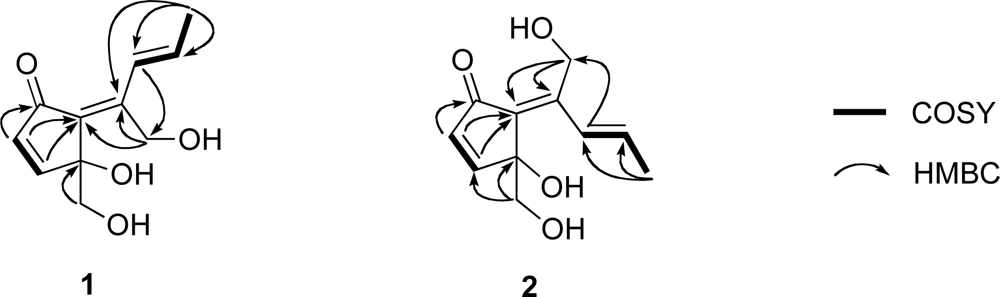
1 must be monocyclic to account for the five degrees of unsaturation required by the molecular formula. The connectivity of the aforementioned partial structures was established from the HMBC correlations of H
2-1/C-2, H-3/C-6, H-4/C-5, H-4/C-6, H-8/C-11, H
3-10/C-7, H
3-10/C-8, H
3-10/C-9, H
2-11/C-6 and H
2-11/C-7, as shown in
Figure 3, to describe the entire carbon framework of
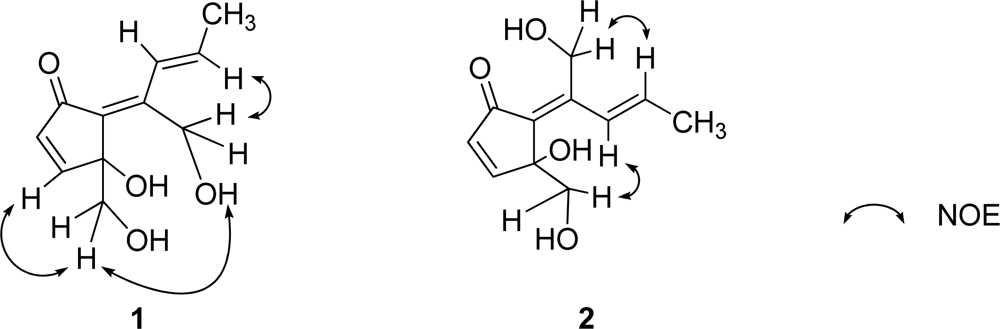
1. Geometric configuration of two olefins in
1 at C-6/C-7 and C-8/C-9 were assigned to be
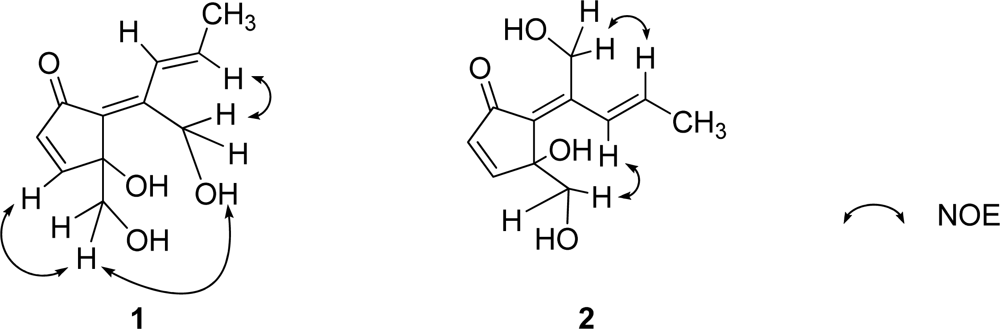
E by NOEDS experiments (
Figure 4), in which irradiation of H-9 caused enhancement of H-11 and irradiation of H-1 resulted in enhancement of the H-3 and OH-11 proton signals. Therefore, the planar structure of
1 was established as a class of didemnenone as shown in
1.
Compound
2 had the same molecular formula as
1, C
11H
14O
4, as established by HR-ESIMS [
m/z 233.0786 (M + Na)
+, calcd. for C
11H
14O
4Na, 233.0784]. The IR absorption bands at
vmax 1695 and 3360 cm
−1 indicated the presence of carbonyl and hydroxyl groups. The NMR data (
Tables 1 and
2) of
2 showed close similarity to those of
1, except for chemical shifts of H-8. The chemical shift of H-8 in
2 (δ
H 6.91) was at higher field than in
1 (δ
H 7.73) due to the magnetic anisotropy effect of the carbonyl group, suggesting a
Z configuration for the C-6 olefin in
2. This was confirmed by NOEDS experiments (
Figure 4), in which irradiation of H-9 caused enhancement of H-11 and irradiation of H-1 resulted in enhancement of H-8.
Analysis of the
13C-NMR and HR-ESIMS [
m/z 231.0632, (M + Na)
+, calcd for C
11H
12O
4Na, 231.0628] for
3 provided a molecular formula C
11H
12O
4, which suggested six degrees of unsaturation. The carbon resonating at δ
C 203.8 (s) suggested the presence of a carbonyl carbon in
3 and the IR absorption band at
vmax 1710 cm
−1 further supported the presence of the carbonyl group. The
1H- and
13C-NMR analyses (
Tables 1 and
2) coupled with
1H-
1H COSY data indicated the presence of an α, β-unsaturated ketone moiety [δ
C 203.8 (s), 132.8 (d), 165.1 (d); δ
H 6.28 (d), 7.52 (d)], a conjugated diene moiety [δ
C 135.0 (s), 131.7 (d); δ
H 6.48 (d) and δ
C 132.3 (d), 119.9 (t); δ
H 6.58 (ddd), 5.23 (d), 5.38 (d)], an acetal [δ
C 91.7 (d); δ
H 5.10 (brd)], a methine [δ
C 55.5 (d); δ
H 3.45 (s)], an oxygenated methylene [δ
C 63.2 (t); δ
H 3.53 (d), 3.42 (d)] and an oxygenated quaternary carbon [δ
C 79.7 (s)] in
3. Extensive analysis of
1H-
1H COSY demonstrated two isolated spin systems, C-3–C-4 and C-8–C-10. The connectivity of the aforementioned partial structures was established from the HMBC correlations of H
2-1/C-2, H
2-1/C-3, H
2-1/C-6, H
2-1/C-11, H-3/C-2, H-3/C-4, H-3/C-5, H-3/C-6, H-6/C-2, H-6/C-4, H-6/C-5, H-6/C-7, H-6/C-11, H-8/C-6, H-8/C-11, H-9/C-7, H-11/C-1, H-11/C-6 and H-11/C-7, to describe the entire carbon framework of
3. Geometric configuration of the olefins in
3 at C-7/C-8 was assigned to be
E by NOEDS experiments, in which irradiation of H-6 caused enhancement of H-9 and irradiation of H-8 caused enhancement of H-11 and H-10. Therefore, the planar structure of
3 was concluded to be a class of didemnenone, as depicted in
3. The NOE observed between a hydroxyl proton (OH-2) and H-6 revealed a
cis fusion of two rings. The NOEs; OH-2/H-11; H-1b/H-11 allowed the assignment of the H-11 as α.
The molecular formula of
4 was deduced to be C
12H
14O
4 based on HR-ESIMS [
m/z 245.0794, (M + Na)
+, calcd for C
12H
14O
4Na, 245.0784]. The
1H- and
13C-NMR spectral data (
Tables 1 and
2) of
4 resembled those of
3, except for the presence of a proton signal at δ
H 3.31 (s) and a carbon signal at δ
C 54.9 (q) in
4. Geometric configuration of the double bond at C-7/C-8 in
4 was assigned to be
E by NOEDS experiments, in which irradiation of H-6 caused enhancement of H-9 and irradiation of H-8 caused enhancement of H-11 and H-10. Therefore, the planar structure of
4 was elucidated to be a methylacetal of
3. The NOE between a hydroxyl proton (OH-2) and H-6 also indicated the ring junction to have the
cis-geometry. The NOEs; OH-2/ H-1b; H-1a /H-11 allowed the assignment of the H-11 as β. We cannot affirm that
4 is a natural product, because it is conceivable it arises from
3 in the isolation process.
The molecular formula of
5 was determined to be C
11H
10O
4, based on HR-ESIMS [
m/z 207.0654 (M + H)
+, calcd for C
11H
11O
4, 207.0652]. The carbons resonating at δ
C 202.9 (s) and 166.9 (s) suggested the presence of a carbonyl carbon and an ester carbonyl carbon, respectively in
5 (
Table 2) and the IR absorption band at
vmax 1717 cm
−1 further supported the presence of the carbonyl groups. The
1H- and
13C-NMR (
Tables 1 and
2) and 2D NMR spectral data of
5 are similar to those of γ-lactone didemnenone [
16], except for the HMBC correlation observed between an oxymethylene proton H
2-1 at δ
H 4.13 and a carbonyl carbon C-11 at δ
C 166.9. Geometric configuration of the double bond at C-7/C-8 in
5 was assigned to be
E by NOEDS experiments, in which irradiation of H-6 caused enhancement of H-9. Therefore, the planar structure of
5 was concluded to be a class of didemnenone, as depicted in
5.The NOE between a hydroxyl proton (OH-2) and H-6 allowed the ring junction to be assigned as
cis.
Analysis of the
13C-NMR and HR-ESIMS [
m/z 249.0742, (M + Na)
+, calcd for C
11H
14O
5Na, 249.0733] for
6 provided a molecular formula C
11H
14O
5. The carbon resonating at δ
C 206.1 (s) suggested the presence of a carbonyl carbon in
6 (
Table 2) and the IR absorption band at
vmax 1720 cm
−1 further supported the presence of the carbonyl group. Extensive analysis of
1H- and
13C-NMR data (
Tables 1 and
2) supported with
1H-
1H COSY data indicated the presence of a
trans double bond [δ
C 124.4 (d), 135.3 (d); δ
H 7.27 (dd,
J = 1.4, 16.0 Hz), 6.31 (dq,
J = 16.0, 6.8 Hz)], a tetrasubstituted double bond [δ
C 130.4 (s), 142.3 (s)], an oxygenated methylene [δ
C 63.1 (t); δ
H 4.27 (d), 3.50 (d)], two oxygenated methines [δ
C 86.6 (d); δ
H 5.51 (d) and δ
C 70.5 (d); δ
H 3.99 (brt)], an oxygenated quaternary carbon [δ
C 73.0 (s)], a methylene [δ
C 46.3 (t); δ
H 2.74 (dd), 1.98 (brd)] and a methyl [δ
C 19.1 (q); δ
H 1.82 (dd)] in
6. These functionalities accounted for three of the five degrees of unsaturation, therefore
6 is bicyclic. The connectivity of the aforementioned partial structures was established from the HMBC correlations of H-1/C-2, H-1/C-6, H-1/C-11, H-4/C-5, H-4/C-6, H-8/C-11 and H-11/C-6, to describe the entire carbon framework of
6. The NOE between H-9 and H-11 also indicated the double bond at C-8 to have the
trans-geometry, and the NOEs; OH-2/H-1a; OH-2/H-3; H-1a/H-11 allowed the assignment of the 3-OH and the 11-OH both as β.
Analysis of
7 by
13C-NMR (
Table 3) and HR-ESIMS [
m/z 401.1602 (M + H)
+, calcd for C
22H
25O
7, 401.1595] provided a molecular formula of C
22H
24O
7. Extensive analysis of
1H- (
Table 3) and
13C-NMR data, supported with
1H-
1H COSY data, indicated the presence of two α, β-unsaturated ketone moieties [δ
C 203.6 (s), 132.8 (d), 165.1 (d); δ
H 6.31 (d), 7.55 (d) and δ
C 196.7 (s), 134.9 (d), 161.6 (d); δ
H 6.28 (d), 7.37(d)], a conjugated diene moiety [δ
C 132.6 (s), 132.8 (d); δ
H 6.37 (d) and δ
C 132.2 (d), 120.3 (t); δ
H 6.59 (ddd), 5.30 (d), 5.26 (d)], a
trans double bond [δ
C 126.9 (d), 134.7 (d); G
H 7.78 (dd,
J = 1.4, 16.0 Hz), 6.41 (m)], a tetrasubstituted double bond [δ
C 135.2 (s), 140.7 (s)], an acetal [δ
C 97.9 (d); δ
H 5.08 (s)], three oxygenated methylenes [δ
C 63.3 (t); δ
H 3.53 (brd), δ
C 66.6 (t); δ
H 3.70 (dd), 3.46 (dd) and δ
C 62.7 (t); δ
H 4.86 (d), 4.49 (d)], two oxygenated quaternary carbons [δ
C 79.1 (s), 80.4 (s)], a methine [δ
C 55.1 (d); δ
H 3.48 (s)] and a methyl [δ
C 19.1 (q); δ
H 1.88 (dd)] in
7. The structure of
7 was elucidated to be a dimeric didemnenone composed of
1 and
3, from the molecular formula, the NMR data and the HMBC correlations of H-11/C-11′ and H
2-11′/C-11. Geometric configuration of three olefins in
7 at C-7/C-8, C-6′/C-7′ and C-8′/C-9′ was assigned to be
E by NOEDS experiments, in which irradiation of H-6 caused enhancement of H-9, irradiation of H-8 caused enhancement of H-11 and H-10, irradiation of H-9′ caused enhancement of H-11′ and irradiation of H-3′ resulted in enhancement of the H-1′ proton signal. Therefore, the structure of
7 was established as a didemnenone dimer as shown in
7. We could not determine the C-2/C-6 ring junction stereochemistry owing to decomposition of
7.
The structure of marine metabolite
8 was determined to be 4-amino-7-(5’-deoxy-β-
d-xylofuranosyl) -5-iodopyrrolo[2,3-
d]pyrimidine by 1D and 2D NMR spectra for
8 and
23, and by CD spectra of compounds
8,
24 and
25 (
Figure 5), as previously described [
23]. The absolute stereochemistry of the new compounds was tentatively deduced to be as depicted in
1–
7 based on the assumption that there is a similar biogenetic relationship between these compounds and (+)-didemnenone A. (+)Didemnenones
9–
12 and inosine (
13) were unambiguously identified by comparison of their spectral data with those described in the literature [
16].
Compounds
1–13 were tested
in vitro for their cytotoxic activities against the HCT116, A431 and A549 cancer cell lines (
Table 4). Compounds
1,
2 and
8 were significantly cytotoxic against the HCT116, A431 and A549 cancer cell lines, and compounds
3,
4,
7,
9/
10 and
12 were significantly cytotoxic against two cell lines, HCT116 and A431. In contrast to
12 (a β-anomer), its isomer
11 (an α-anomer) was not cytotoxic against any of the three cell lines. Among the isolated compounds tested, the iodinated nucleoside
8 showed the strongest cytotoxic activity against the HCT116, A431 and A549 cancer cell lines, with IC
50 values of 1.8, 3.1 and 3.5 μ g/mL, respectively.
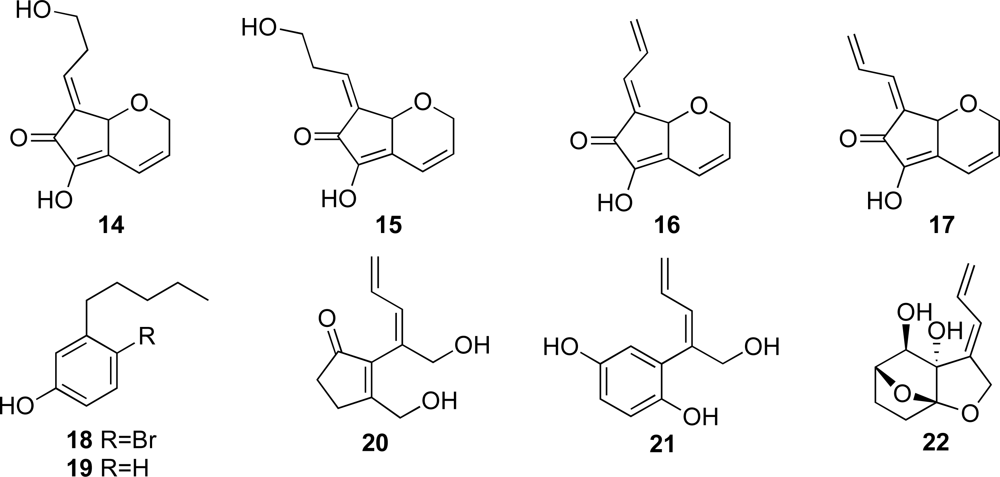
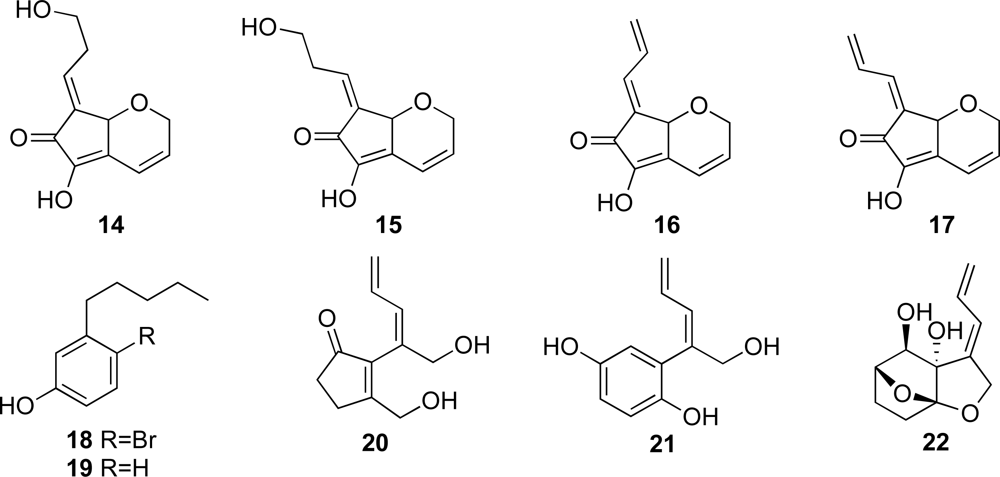
To date, a variety of C
11 compounds have been isolated from ascidians (compounds
9 and
10), cyanobacteria (compounds
20 and
21) and a sponge (compound
22) (
Figures 1 and
2) [
16,
35,
36]. Compounds
16 and
17 have been isolated from the ascidian
Diplosoma virens and a sponge
Ulosa sp. (
Figure 2) [
19,
20]. Isolation of a series of the C
11 compounds including compounds
18 and
19 (
Figure 2) from unrelated marine organisms supports the potential microbial origin of these compounds. From this viewpoint, we assume that the ascidian
Diplosoma sp. might not be the actual producer of the C
11 compounds, but suggest a possible microorganism source such as
Prochloron spp. We conducted, therefore, the following experiments. The
Prochloron spp., which are obligatory symbionts of ascidians, were separated from the body of the ascidians
Lissoclinum sp. and
Diplosoma spp. by squeezing through the plankton net.
1H-NMR spectra of the acetone extracts of the separated
Prochloron spp. showed the presence of the same peaks as present in those of didemnenones. This confirms our assumption that
Prochloron spp. are the actual producers of didemnenones.
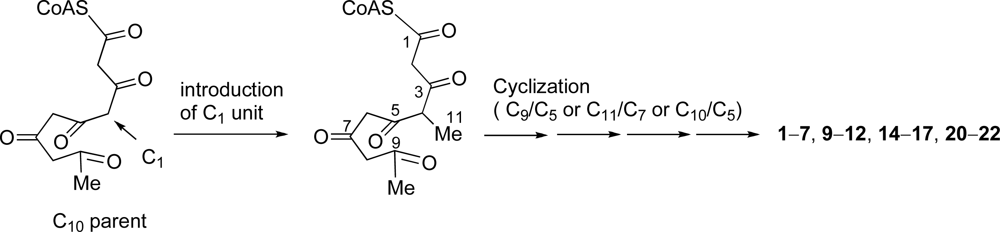
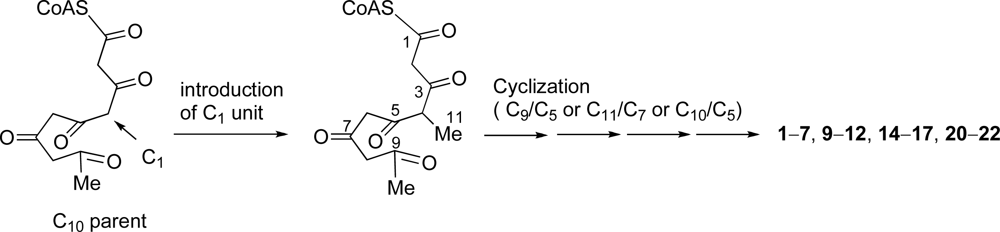
Most C
11 compounds are derived from polyketides (six acetates-C
1) or polyketides (five acetates + C
1) [
37]. Pentylphenols such as
18 and
19, and some compounds which have a carbon skeleton of 5-methyldecane are known to be derived from the former with a loss of CO
2 from C
12 parent (six acetates). Some C
11 metabolites are ascertained to be derived from a polyketide precursor (five acetates + C
1) which has a carbon skeleton of 4-methyldecane [
37]. We found that didemnenone-related compounds
1–
7,
9–
12,
14–
17 and
20–
22 have a common carbon skeleton of 4-methyldecane from a consideration of the carbon skeleton of these compounds. Consequently, We propose that these compounds should be derived from the polyketides (five acetates + C
1)
via cyclization between C-9/C-5, between C-11/C-7 or between C-10/C-5 (
Scheme 1).
3. Experimental Section
3.1. General experimental procedures
Optical rotations were measured on either a JASCO P-1020 or JASCO DIP-1000 polarimeter. Ultraviolet-visible spectra were obtained in methanol on a JASCO V-550 spectrophotometer. Infrared spectra were recorded as dry films on either JASCO FT/IR-300 or Spectrum 2000 Explorer (PERKIN ELMER). CD spectra were recorded on a JASCO J-720W Circular Dichroism Spectrometer. 1H- and 13C-NMR spectra were recorded on a JEOL JNM α-500 FT-NMR spectrometer or a JEOL JNM lambda 400 FT-NMR spectrometer, and chemical shifts were referenced to the solvent signals [δH 7.24 and δC 77.0 in chloroform-d, δH 2.49 and δC 39.5 in DMSO-d6, δH 3.30 and δC 49.0 in methanol-d4]. Inversed-detected heteronuclear correlations were measured using HMQC and HMBC pulse sequences with a pulse field gradient. HR-ESIMS data were obtained on a LTQ ORBITRAP (ThermoFisher Scientific, Germany), and HR-FABMS data were obtained on a JEOL JMS-700 mass spectrometer. LR-ESIMS data were measured on a Waters Quattro micro API triple quadruple mass analyzer. RP-OCC and RP-MPLC were performed on COSMOSIL® 140C18-OPN. Preparative RP-HPLC was run on a Waters 600 multi solvent system using ODS columns (YMC-Pack ODS-A, 150 × 20 mm I.D., YMC-Pack ODS-A, 250 × 20 mm I.D., YMC-Pack ODS-C8, 250 × 20 mm I.D., YMC-Pack ODS-AQ, 250 × 20 mm I.D., Develosil ODS-HG-5, 250 × 20 mm I.D. and COSMOSIL® -packed C18, 250 × 10 mm I.D.). All solvents used were reagent grade.
3.2. Animal material
The colonial brown ascidian was collected by hand at the tidal zone of Tarama island, Okinawa, Japan, and the colonial green ascidian was collected by hand from the coast of Hateruma island, Okinawa, Japan. The ascidians were stored at −15 °C until extraction. The brown ascidian and the green ascidian were identified as Lissoclinum sp. and Diplosoma sp., respectively, by Euichi Hirose, University of the Ryukyus, Japan. The voucher specimens were deposited at the University of the Ryukyus (Specimen no. URKU-801 for the brown ascidian and URKU-802).
3.3. Extraction and isolation
The ascidian Lissoclinum sp. (5.7 kg, wet weight) was initially extracted with acetone (18 L) and filtered to remove debris. The filtrate was concentrated in vacuo to remove acetone and the resultant mixture was partitioned between EtOAc (3.5 L) and H2O (3.5 L). The H2O layer was extracted with 1-BuOH to give the 1-BuOH extract (18.3 g) and the H2O extract (141.2 g). An aliquot of 27.8 g of the H2O-soluble material was subject to RP-MPLC on ODS (COSMOSIL® 140C18-OPN, 140 μm, 50 × 3 cm I.D.) with H2O (500 mL), H2O/MeOH (9:1, 500 mL 7:3, 500 mL; 5:5, 500 mL, 3:7, 500 mL; 1:9, 500 mL) and MeOH (500 mL) to give seven fractions. The second fraction (860 mg) was subjected to HPLC on ODS [YMC-Pack ODS-AQ, 250 × 20 mm I.D.; linear gradient elution, H2O/MeOH (3:1)-MeOH] to give 18 fractions. The 18th fraction contained pure 1 (66.0 mg). The 14th fraction (84.0 mg) was subjected to HPLC on ODS [YMC-Pack ODS-AQ, 250 × 20 mm I.D.; linear gradient elution, 1% HCOOH /H2O (1:9)-1% HCOOH/MeCN (1:9)] to give 13 (7.6 mg). The EtOAc layer was concentrated in vacuo to give a brown material (29.3 g). The material was subjected to a modified Kupchan’s partitioning procedure as follows. The material was suspended in a mixture of H2O/MeOH (1:1) and then successively extracted with hexane and CHCl3. The resultant aqueous phase was concentrated to remove MeOH and then extracted with 1-BuOH. An aliquot of 0.58 g of the BuOH-soluble material (3.93 g) was separated by RP-MPLC (COSMOSIL® 140C18-OPN, 140 μm, 50 × 3 cm I.D.) using H2O (500 mL), H2O/MeOH (9:1, 500 mL 7:3, 500 mL; 5:5, 500 mL, 3:7, 500 mL; 1:9, 500 mL) and MeOH (500 mL) to give seven fractions (fr. 1–7). The first fraction (fr. 1, 436 mg) was purified by RP-HPLC [YMC-Pack ODS-A, 250 × 20 mm I.D.; linear gradient elution, H2O/MeOH (4:1)-MeOH] to afford 3 (165 mg) and 4 (3.2 mg). The second fraction (fr. 2, 272 mg) was separated by RP-HPLC [YMC-Pack ODS-A, 150 × 20 mm I.D.; linear gradient elution, H2O/MeOH (4:1)-MeOH] to give 18 fractions (fr. 2-1–2-18). Purification of the second fraction (fr. 2-2, 170 mg) by RP-HPLC (Develosil ODS-HG-5, 250 × 20 mm I.D.) using H2O/MeOH (9:1) led to the isolation of 6 (24.6 mg), 2 (92.3 mg), 1 (92.7 mg) and an inseparable mixture of didemnenones A/B (9/10, 2.8 mg). HPLC separation [Develosil ODS-HG-5, 250 × 20 mm I.D., H2O/MeOH (9:1)] of seventh fraction (fr. 2–7, 6.8 mg) led to the isolation of 12 (0.8 mg). An aliquot of 3.35 g of the 1-BuOH extract (3.93 g) was chromatographed on ODS (COSMOSIL® 140C18-OPN, 140 μm, 100 g) with H2O/MeOH (3:7, 500 mL; 2:8, 100 mL; 1:9, 100 mL) and MeOH (100 mL) to give nine fractions. The second fraction (1.1 g) was purified by HPLC on ODS [YMC-Pack C8, 250 × 20 mm I.D.; linear gradient elution, H2O/MeCN (8:2–1:9)], to give 1 (186.8 mg), 5 (14.6 mg), 4 (29.7 mg) and 7 (0.9 mg).
The ascidian Diplosoma sp. (900 g, wet weight) was initially extracted with acetone (2.2 L). After filtration, the extracts were concentrated in vacuo to give an acetone extract. The acetone extract was partitioned between EtOAc and H2O. The H2O layer was further extracted with 1-BuOH. The 1-BuOH layer was concentrated in vacuo to give a BuOH-soluble material (2.14 g). The EtOAc layer was concentrated in vacuo to give an EtOAc extract (7 g). The EtOAc extract was suspended in H2O/MeOH (1:1, 400 mL) and then successively extracted with hexane and CHCl3 to give a hexane-soluble material (4.0 g), a CHCl3-soluble material (2.0 g) and an aqueous material (2.1 g). The aqueous material and the BuOH-soluble material (2.14 g) were combined. The combined polar fraction (4.24 g) was chromatographed on ODS (COSMOSIL® 140C18-OPN) with H2O/MeOH (1:1, 2:1), MeOH and MeOH/EtOAc (9:1) as eluent. The first fraction (2.0 g) contained more than 95% of 1 from its 1H NMR data. The CHCl3-soluble material (2.0 g) was subjected to OCC on ODS [COSMOSIL® 140C18-OPN, H2O/MeOH (1:4), MeOH and MeOH/EtOAc (7:3)]. The first fraction (fr. 1, 1.0 g) was further separated by OCC on ODS (COSMOSIL® 140C18-OPN) using H2O/MeOH (1:1, 2:1), MeOH and MeOH/EtOAc (9:1). The first aqueous methanol fraction (fr. 1-1, 902 mg) was subjected to HPLC on ODS (COSMOSIL® -packed C18, 250 × 10 mm I.D.) using H2O/MeOH (1:1) to give six fractions (fr. 1-1-1-fr. 1-1-6). The first fraction (fr. 1-1-1, 92.9 mg) was separated by HPLC on ODS [COSMOSIL® -packed C18, 250 × 10 mm I.D., H2O/MeCN (1:1)] first, and then by RP-HPLC [COSMOSIL®-packed C18, 250 × 10 mm I.D., H2O/MeOH/MeCN (5:2:2)] to yield 1 (18.7 mg) and 2 (5.9 mg). Further separation of the second fraction (fr. 1-1-2, 61.6 mg) by repeated HPLC [COSMOSIL® -packed C18, 250 × 10 mm I.D., H2O/MeOH/MeCN (50:31:19) and then H2O/MeCN (1:1)] to give 5 (4.2 mg). An inseparable mixture (6.2 mg) of didemnenones A and B (9 and 10) was obtained along with their methylacetals 11 (8.8 mg) and 12 (3.0 mg) from the fourth fraction (fr. 1-1-4, 70.4 mg) by repeated ODS HPLC [COSMOSIL® -packed C18, 250 × 10 mm I.D., H2O/MeOH/MeCN (50:31:19) and then H2O/MeCN (1:1)]. The fifth fraction (fr. 1-1-5, 40.0 mg) was purified by RP-HPLC (COSMOSIL®-packed C18, 250 × 10 mm I.D.) with H2O/MeOH/MeCN (50:31:19) to afford 8 (9.9 mg). The sixth fraction (fr. 1-1-6, 210.7 mg) was purified by HPLC on ODS (COSMOSIL®-packed C18, 250 × 10 mm I.D.) using MeOH/EtOAc (19:1) to yield 8 (164.4 mg).
3.3.1. Compound 1
Pale yellowish oil; [α]
27D +21.0° (
c 0.66, MeOH); UV (MeOH)
λmax (log
ɛ) 246 (3.39), 308 (3.69) nm; FT/IR (film)
vmax 3355, 1675, 1035 cm
−1;
1H-NMR and
13C-NMR (DMSO-
d6) see
Tables 1 and
2; LR-ESIMS
m/z 211 (M + H)
+, 209 (M − H)
− HR-ESIMS
m/z (M + Na)
+ 233.0786 (calcd. for C
11H
14O
4Na, 233.0784).
3.3.2. Compound 2
Pale yellowish oil; [α]
27D +22.5° (
c 0.43, MeOH); UV (MeOH)
λmax (log
ɛ) 245 (3.38), 307 (3.74) nm; FT/IR (film)
vmax 3359, 1712, 1379 cm
−1;
1H-NMR and
13C-NMR (DMSO-
d6) see
Tables 1 and
2; LR-ESIMS
m/z 233 (M + Na)
+, 211 (M + H)
+, 209 (M − H)
−; HR-ESIMS
m/z (M + Na)
+ 233.0786 (calcd. for C
11H
14O
4Na, 233.0784).
3.3.3. Compound 3
Pale yellowish oil; [α]
24D +147° (
c 0.62, H
2O); UV (H
2O)
λmax (log
ɛ) 246 (3.51), 274 (3.54); FT/IR (film)
vmax 3400, 1710, 1050 cm
−1;
1H-NMR and
13C-NMR (DMSO-
d6) see
Tables 1 and
2; LR-ESIMS
m/z 231 (M + Na)
+, 207 (M − H)
−; HR-ESIMS
m/z (M + Na)
+ 231.0632 (calcd. for C
11H
12O
4Na, 231.0628).
3.3.4. Compound 4
Pale yellowish oil; [α]
24D +459° (
c 0.015, MeOH); UV (MeOH)
λmax (log
ɛ) 249 (3.65); FT/IR (film)
vmax 3410, 1700, 1045 cm
−1;
1H-NMR and
13C-NMR (DMSO-
d6) see
Tables 1 and
2; LR-ESIMS
m/z 223 (M + H)
+; HR-ESIMS
m/z (M + Na)
+ 245.0794 (calcd. for C
12H
14O
4Na, 245.0784).
3.3.5. Compound 5
Pale yellowish oil; [α]
27D + 112° (
c 0.26, MeOH); UV (MeOH)
λmax (log
ɛ) 273 (3.83); FT/IR (film)
vmax 3419, 1717, 1049 cm
−1;
1H-NMR and
13C-NMR (DMSO-
d6) see
Tables 1 and
2; LR-ESIMS
m/z 207 (M + H)
+, 205 (M − H)
−; HR-ESIMS
m/z (M + H)
+ 207.0654 (calcd. for C
11H
11O
4, 207.0652).
3.3.6. Compound 6
Pale yellowish oil; [α]
24D +16.3° (
c 0.17, MeOH); UV (MeOH)
λmax (log
ɛ) 294 (3.91); FT/IR (film)
vmax 3390, 1720, 1050 cm
−1;
1H-NMR and
13C-NMR (DMSO-
d6) see
Tables 1 and
2; LR-ESIMS
m/z 249 (M + H)
+, 223 (M + H)
+; HR-ESIMS
m/z (M + Na)
+ 249.0742 (calcd. for C
11H
14O
5Na, 249.0733).
3.3.7. Compound 7
Pale yellowish oil; [λ]
24D +11.7° (
c 0.12, MeOH); UV (MeOH)
λmax (log
ɛ) 248 (3.78), 308 (3.83); FT/IR (film)
vmax 3400, 1580, 1050 cm
−1;
1H-NMR and
13C-NMR (DMSO-
d6) see
Table 3; LR-ESIMS
m/z 423 (M + Na)
+, 401 (M + H)
+, 399 (M − H)
−; HR-ESIMS
m/z (M + Na)
+ 423.1423 (calcd for C
22H
24O
7Na, 423.1414), (M + H)
+ 401.1602 (calcd. for C
22H
24O
7, 401.1595).
3.3.8. Compound 8
Pare yellowish oil: [α]
26D −69° (
c 0.1, MeOH); UV (MeOH)
λmax (log
ɛ) 283 nm (3.59); FT/IR (film)
vmax 3461, 3317, 3132, 1633, 1584, 1474, 1084, 755 cm
−1; NMR data were described in the previous paper [
13]. LR-EIMS
m/z (rel.%) 376 (M
+, 7), 303 (3), 289 (13), 261 (33), 260 (100), 233 (18). HR-FABMS
m/z (M)
+ 376.0016 (calcd. for C
11H
13IN
4O
3, 376.0027).
3.3.9. Acetal 23
To a solution of iodinated nucleoside
8, (10.0 mg, 26.6 μmol) in 2,2-dimethoxypropane (1 mL) and acetone (2 mL) was added a catalytic amount of camphorsulfonic acid. The mixture was stirred at rt for 24 h and at 45 °C for 4 h. The reaction mixture was diluted with ether, washed with saturated aqueous Na
2CO
3 and brine. The organic phase was dried (MgSO
4) and concentrated
in vacuo. The residual oil was purified by preparative TLC [CHCl
3-MeOH (3:0.2)] to give the acetal as colorless oil (
23, 3.0 mg, 25%). HR-FABMS and
1H NMR data for compound
23 were described in the earlier paper [
23].
3.3.10. Methyl 5-deoxy 2.3-di-O-(4-bromobenzoyl)-β-l-xylofuranoside (24)
The mixture of methyl 5-deoxy-β-
l-xylofuranoside [
38,
39] (56.5 mg, 0.11 mmol), 4-bromobenzoyl chloride (250.1 mg, 0.87 mmol), and DMAP (2.2 mg, 0.018 mmol), in pyridine (0.8 mL) was stirred at ambient temperature for 24 h, and water (0.5 mL) was added to the mixture. After being stirred at ambient temperature for 30 min, the mixture was concentrated. The residual solid was purified by HPLC [Develosil ODS-HG-5 (250 × 20 mm I.D.); flow rate 5 mL/min; detection UV 256 nm; solvent 85% MeOH] to give methyl 5-deoxy 2.3-di-
O-(4-bromobenzoyl)-β-
l-xylofuranoside (
24, 17.2 mg, 30%) and its corresponding α-anomer (15.3 mg, 27%), respectively. Spectral data for compound
24 were described in the previous paper [
23].
3.3.11. Dibenzoate 25
The mixture of iodinated nucleoside
8 (5.0 mg, 0.013 mmol), 4-bromobenzoyl chloride (25.2 mg, 0.115 mmol), and DMAP (2.2 mg, 0.018 mmol), in pyridine (0.5 mL) was stirred at ambient temperature for 24 h, and water (0.5 mL) was added to the mixture. After being stirred at ambient temperature for 30 min, the mixture was concentrated. The residual solid was purified by HPLC [Develosil ODS-HG-5 (250 × 20 mm I.D.); flow rate 5 mL/min; detection UV 256 nm; solvent 85% MeOH] to give dibenzoate
25 (2.0 mg, 21%). Spectral data for compound
25 were described in the earlier paper [
23].
3.3.12. Condition of cell cultures
Human colorectal carcinoma (HCT116), human epidermal carcinoma (A431) and human lung cancer (A549) cells were cultured in DMEM medium (including 10% FBS, 100 U/mL penicillin and 100 ng/mL streptomycin) at 37 °C in a 5% CO2 atmosphere.
3.3.13. Determination of cytotoxicity
Growth inhibition experiments were carried out in quadruplicate on 96-well flat-bottomed microplates, and the amount of viable cells at the end of incubation was determined with the MTT [3-(4,5-dimethylthiazol-2-yl) 2,5-diphenyltetrazolium bromide] dye reduction assay. Test compounds were dissolved in DMSO (4.0 mg/mL) and diluted with H2O such that the final DMSO concentration was 0.5%. Viable cells (HCT116, A431 and A549) in the growth medium were seeded on 96-well microplates (1.0 × 104 cells/well) and incubated at 37 °C in a 5% CO2 atmosphere, and continuously cultured without or with five concentrations (20, 10, 5, 2.5, 1.25 μg/mL, final concentration) of test compounds for 48 h from the next day. After incubation, 10 μL of MTT (5 mg/mL in phosphate-buffer saline) was added each well, the samples were again incubated. After standing for 3 h, the medium was removed, and the resulting formazan crystals were dissolved with DMSO (100 μL). The optical density (O.D.) was measured at 570 nm, provided the reference for reading at 655 nm with a microplate reader (Model 550, BIO-RAD, USA).

 and
and 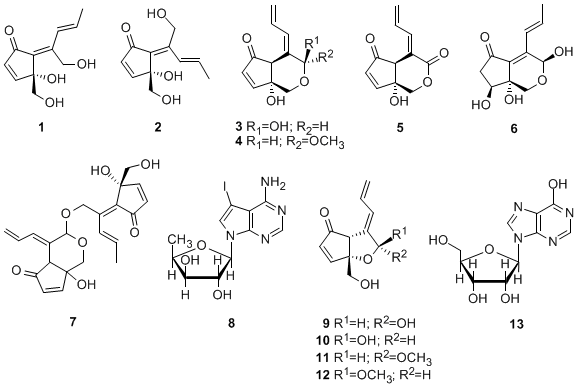
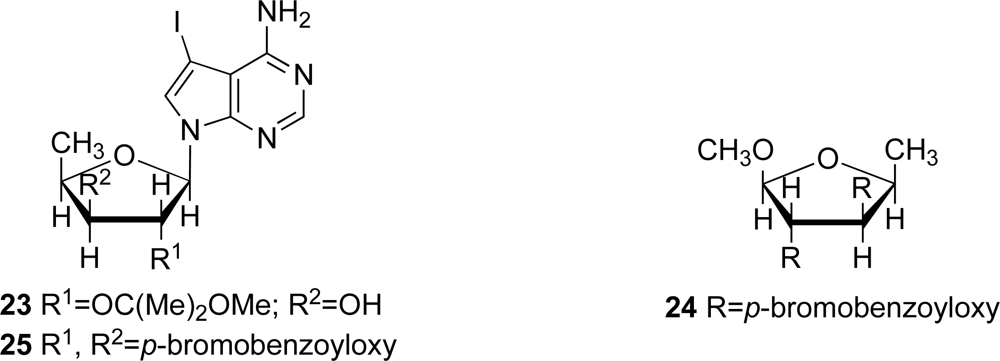

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
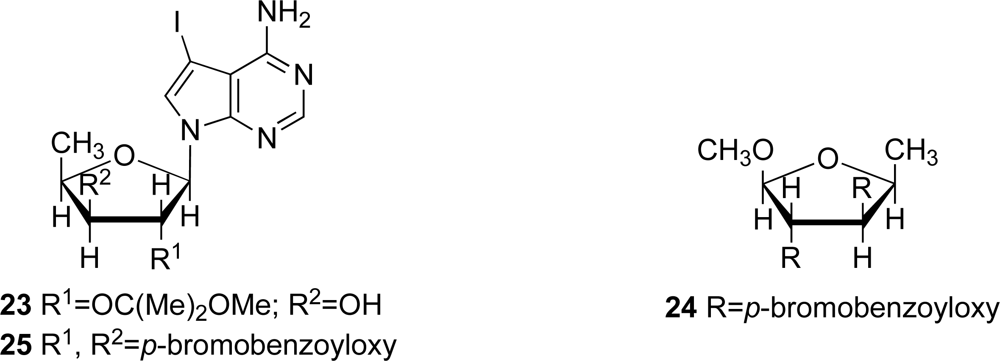{kind=link}
{kind=link}