Abstract
The pyridoacridine alkaloids kuanoniamines A and C were isolated together with 24 α-methylcholestanol, p-hydroxybenzaldehyde, p-hydroxybenzoic acid, phenylacetic acid and 3-formylindole from the ethyl acetate extract of the marine sponge Oceanapia sagittaria (Sollas), collected from the Gulf of Thailand. Kuanoniamines A and C were evaluated for their effect on the growth of five human tumour and a non-tumour cell lines, as well as on the proliferation of human lymphocytes. Kuanoniamine A was found to be a potent growth inhibitor of all the tumour and a non-tumour cell lines while kuanoniamine C was less potent but showed high selectivity toward the estrogen dependent (ER+) breast cancer cell line. Kuanoniamine A has shown to be a more potent inhibitor of DNA synthesis than kuanoniamine C. Kuanoniamine A was also found to cause an extensive reduction of the MCF-7 cells in G2/M phase as well as an increase in the apoptotic cells.
1. Introduction
Species of Oceanapia are common, with more than 50 nominal species recorded for the Indo-west Pacific region alone, relatively abundant in both soft and hard substrates, and widely distributed [1]. However, the chemistry of this genus seems to vary significantly. Though a majority of the compounds isolated from the marine sponges of this genus are alkaloids, several non alkaloidal constituents have also been isolated. Thus, while an antifungal bis α,ϖ amino alcohol glycoside, oceanapiside, was reported from O. phillipensis, collected from Southern Australia [2], Oceanapia sp., collected from the Northwest coast of Australia, furnished a hybrid α,ϖ-bifunctionalized sphingoid tetrahydroquinoline β-glucoside [3]. Capon et al. [4, 5] reported the isolation of nematocidal bisthiocyanates from the ethanol extract of Oceanapia sp., also collected from Australia. Matsugana et al. have reported isolation of a bioactive acetylenic acid from the lipophillic extract of the Japanese collection of Oceanapia sp. [6]. On the other hand, most of the alkaloids reported from the genus Oceanapia were pyridoacridine alkaloids except for oceanapamine, a sesquiterpene alkaloid from specimens of Oceanapia sp., collected from Palawan island, the Phillipines [7] and the phloeodictynes from O. fistulusa from the Eastern coast of New Caledonia [8].
The most studied pyridoacridine alkaloids are the kuanoniamines. So far, only kuanoniamines C and D have been reported from the sponges of the genus Oceanapia. While kuanoniamine C, together with sagitol, were found to be the major constituents of the marine sponge O. sagitaria from Palau [9], kuanoniamines C and D and their N-acetyl derivative were also isolated from the Micronesian sponge Oceanapia sp. [10]. Interestingly, kuanoniamine C was shown to be produced by the sponge cells and not by intracellular symbiotic organisms of O. sagittaria [11]. Schupp et al. [12] have found that amounts of kuanoniamines C and D, the major secondary metabolites of the Micronesian Oceanapia sp, increased sharply from the basal root to the capitum. These findings let them to conclude that these alkaloids had ecological implication to this sponge. Recently, the alkaloid petrosamine B, an inhibitor of the Helicobacter pylori enzyme aspartyl semialdehydedehydrogenase, was isolated from the methanol extract of the Australia sponge Oceanapia sp. [13].
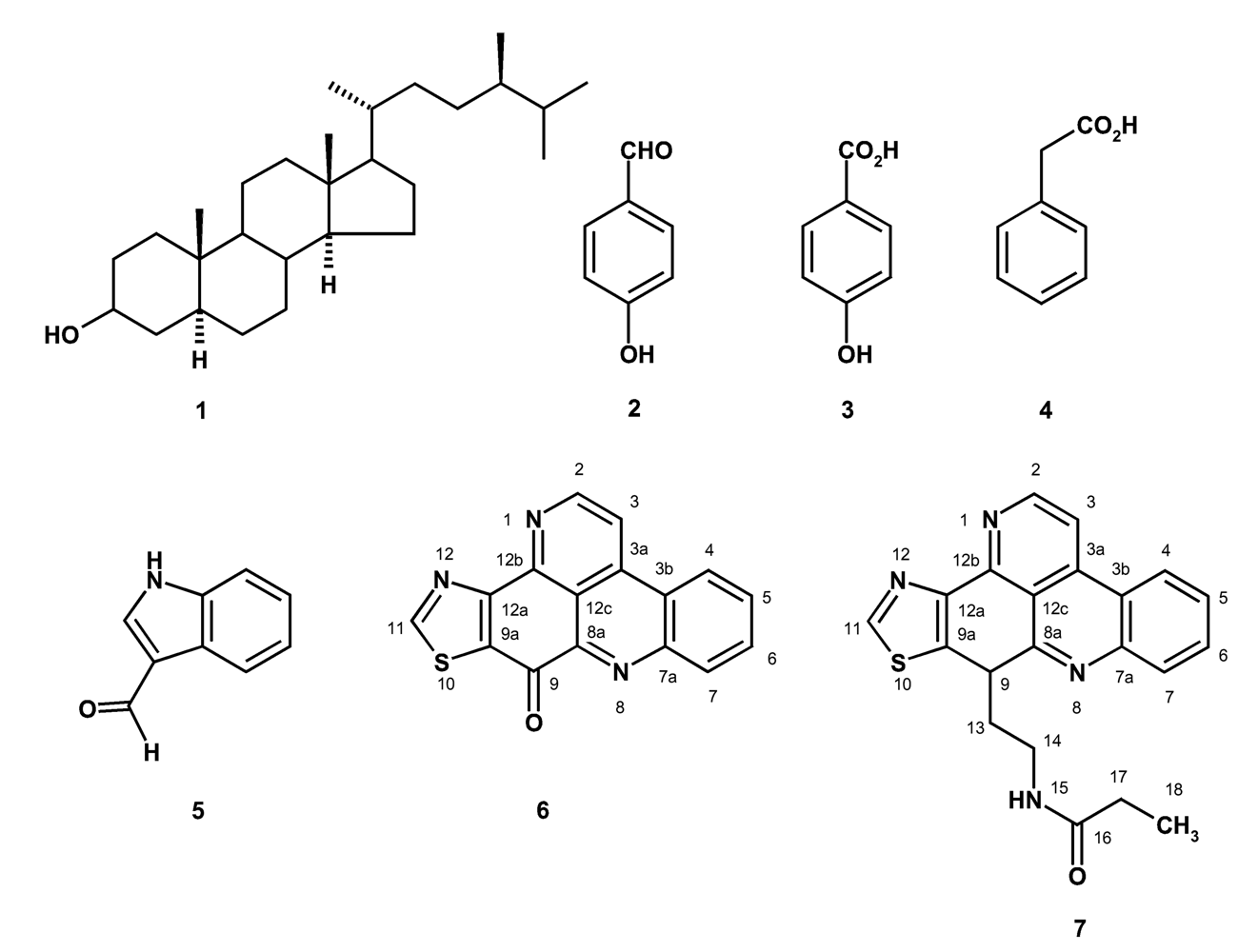
In the course of our investigation on bioactive compounds from the marine sponges from the Gulf of Thailand, we have isolated 24 α-methylcholestanol (1), p-hydroxybenzaldehyde (2), p-hydroxybenzoic acid (3), phenylacetic acid (4), 3-formylindole (5), kuanoniamine A (6) and kuanoniamine C (7) from the ethyl acetate extract of the marine sponge Oceanapia sagittaria (Sollas), collected near Koh Samed island in the Gulf of Thailand (Figure 1). We have also investigated the effect of kuanoniamines A (6) and C (7) on the growth of five human tumour cell lines: MCF-7 (breast carcinoma, estrogen–dependent ER+), MDA-MB-231 (breast carcinoma, estrogen–independent ER-), SF-268 (glioma), NCI-H460 (non small cell lung cancer), UACC-62 (melanoma), and a non-tumour human cell line MRC-5 (diploid embryonic lung fibroblast), as well as on the proliferation of peripheral human lymphocytes. As kuanoniamines A and C were found to be potent growth inhibitors of the MCF-7 cells, they were also evaluated for their effect on DNA biosynthesis and MTT-reducing capacity as well as on a cell cycle progression and apoptosis of these cells.
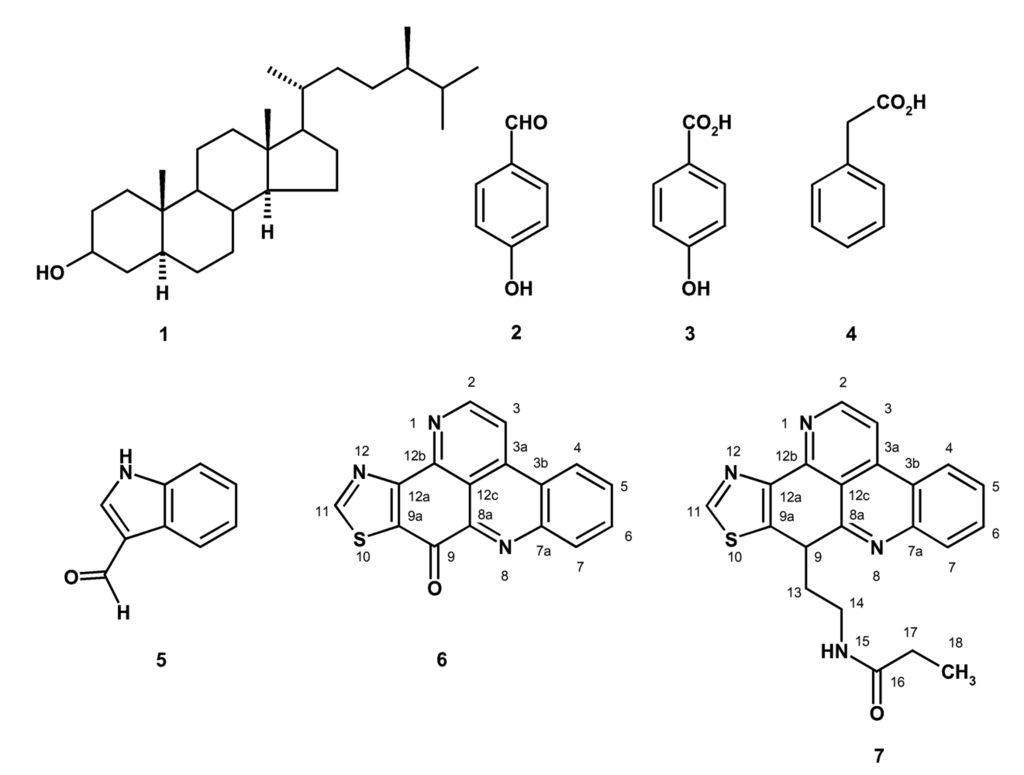
Figure 1.
Structure of compounds 1–7.
2. Results and Discussion
Kuanoniamine A (6) has been initially reported, together with kuanonamine C, from an unidentified purple colonial tunicate and its prosobranch mollusc predator Chelynotus semprei and was found to inhibit the proliferation of KB (human pharyngeal cancer) cell lines in vitro at an IC50 of 1 μg/ml [14]. Though a total synthesis of kuanoniamine A has been proposed [15], to our knowledge it has never been reported from the marine sponges of the genus Oceanapia. On the contrary, kuanoniamine C (7) has been reported later from Oceanapia sagittaria [9, 11] and from the Micronesian sponge Oceanapia sp. [12] and was found to exhibit significant in vitro cytotoxicity to Hela and MONO-MAC 6 tumour cells [10]. On the other hand, 24 α-methylcholestanol (1) is one of the common marine sterols while p-hydroxybenzaldehyde (2), p-hydroxybenzoic acid (3), phenylacetic acid (4) and 3-formylindole (5) are probably degradation products of the secondary metabolites and have been commonly found in many marine sponges.
The structures of 24 α-methylcholestanol (1), 3-formylindole (5), kuanoniamine A (6) and kuanoniamine C (7) were established by 1H, 13CNMR, DEPTs, COSY, HSQC, HMBC, NOESY spectroscopy and HRMS as well as by comparison of their proton and carbon chemical shifts and other physical data with those reported previously [14].
The effect of kuanoniamine A (6) and kuanoniamine C (7) on the in vitro growth of five human tumour cell lines: MCF-7 (ER+), MDA-MB-231 (ER-), SF-268, NCI-H460, UACC-62, and on the non-tumour cell line MRC-5 was evaluated after a continuous exposure of 48 h. Results given in concentrations that were able to cause 50% of cell growth inhibition (GI50) are summarized in Table 1. Kuanoniamine A (6) was found to potently inhibit the growth of all the five human tumour cell lines, as demonstrated by its low GI50 values (≤ 5 μM). This potent effect was also observed for the non-tumour fibroblast cell line MRC-5. Kuanoniamine C (7) also inhibited the growth of all these tumour cell lines. However, its effect was at least ten times less potent than that observed for kuanoniamine A for MDA-MB-231, SF-268, NCI-H460 and UACC-62. Interestingly, the low value of the GI50 of kuanoniamine C on MCF-7 (0.81 ± 0.11 μM) indicates a high selectivity of this estrogen dependent (ER +) breast cancer cell line to this compound. Since the growth inhibitory effect of kuanoniamine C was less intense on the estrogen independent (ER-) MDA-MB-231, it is possible that the estrogen receptors could be involved in this antiproliferative effect. However more studies are needed to confirm the possible antiestrogenic effect of kuanoniamine C.

Table 1.
Effect of kuanoniamine A (6) and kuanoniamine C (7) on the growth of human tumour and non-tumour cell lines *.
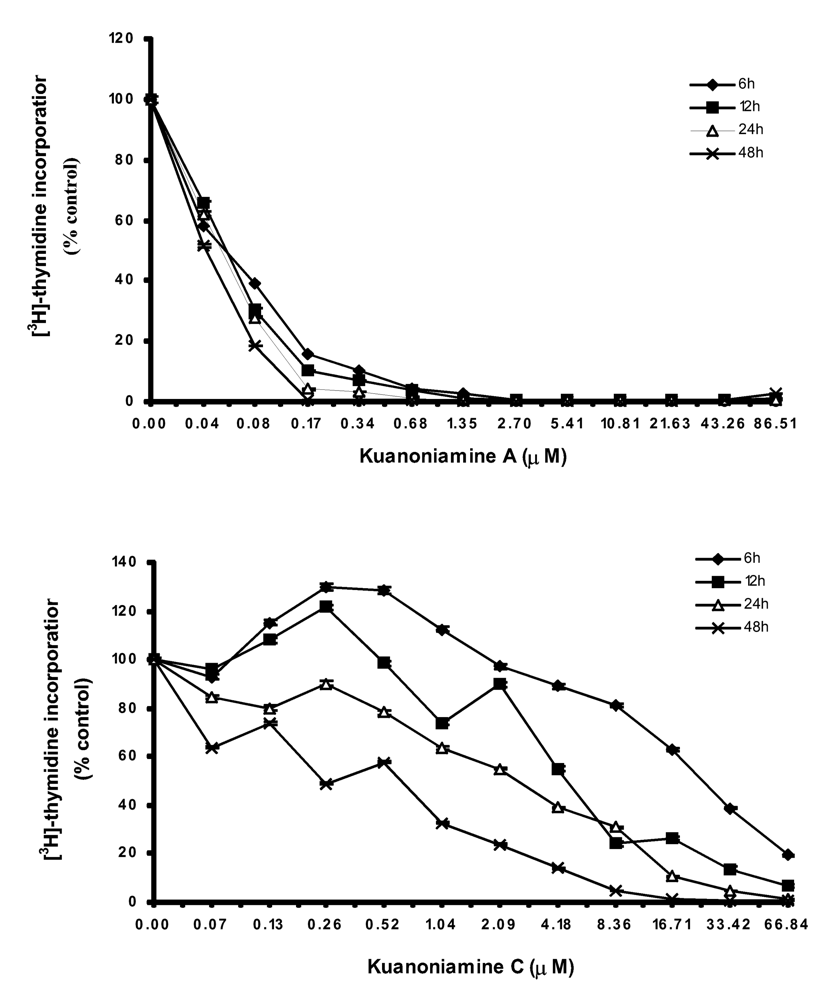
As both kuanoniamine A (6) and kuanoniamine C (7) were found to be potent inhibitors of the MCF-7 cells, we have selected this breast cancer cell line to further characterize the growth inhibitory effect of the two compounds. We first evaluated their capacity to interfere with the biosynthesis of DNA by exposing the exponential growing MCF-7 cells to a range of increasing concentrations of kuanoniamine A (6) and kuanoniamine C (7), below and above their GI50 (0.04 – 86.51 μM and 0.03 – 66.84 μM, respectively) for 6, 12, 24 and 48 h and the DNA synthesis was quantified by thymidine incorporation (Figure 2). The dose-response curve showed that kuanoniamine A (6) dramatically affected DNA synthesis, even at concentrations as low as 0.08 μM, which were associated with a decrease in thymidine incorporation of more than 60% when compared with untreatead control cells. It was also found that the effect on DNA synthesis was mainly dependent on the concentration of kuanoniamine A (6) rather than on the exposure time. On the contrary, the effect of kuanoniamine C (7) was found to be dependent either on the concentration or the exposure time. Although an inhibition of DNA synthesis was also observed with kuanoniamine C (7), an interesting stimulatory effect was detected at low concentrations and exposure periods of 6 and 12 h. A maximum stimulation of 130% was reached after 6 h of treatment with 0.26 and 0.52 μM. This biphasic effect in DNA synthesis of ER (+) MCF-7 cells, expressed by a stimulatory effect at low concentrations and an inhibitory effect at high concentrations, resembles the behaviour described for phytoestrogens on ER (+) breast cancer cells [16, 17].
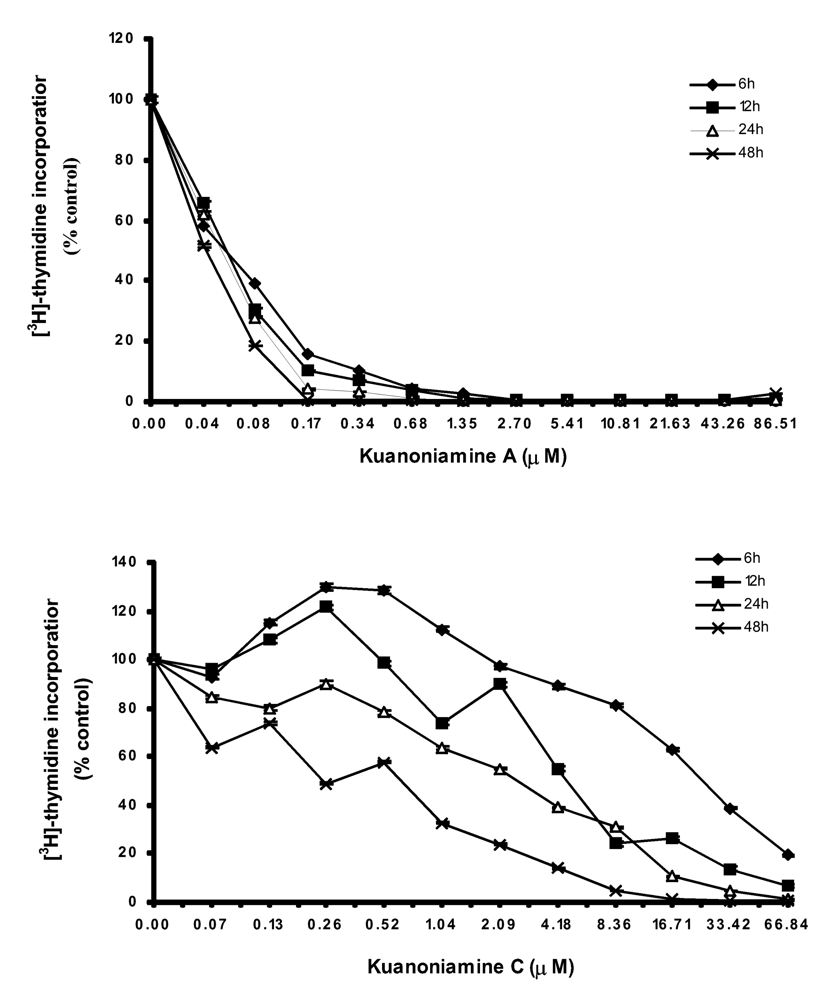
Figure 2.
Time and dose-dependent curves of kuanoniamine A (6) and kuanoniamine C (7) in DNA synthesis of MCF-7 cells determined by [3H]-thymidine incorporation assay. Results are the mean ± SEM of four replicates from a representative experiment.
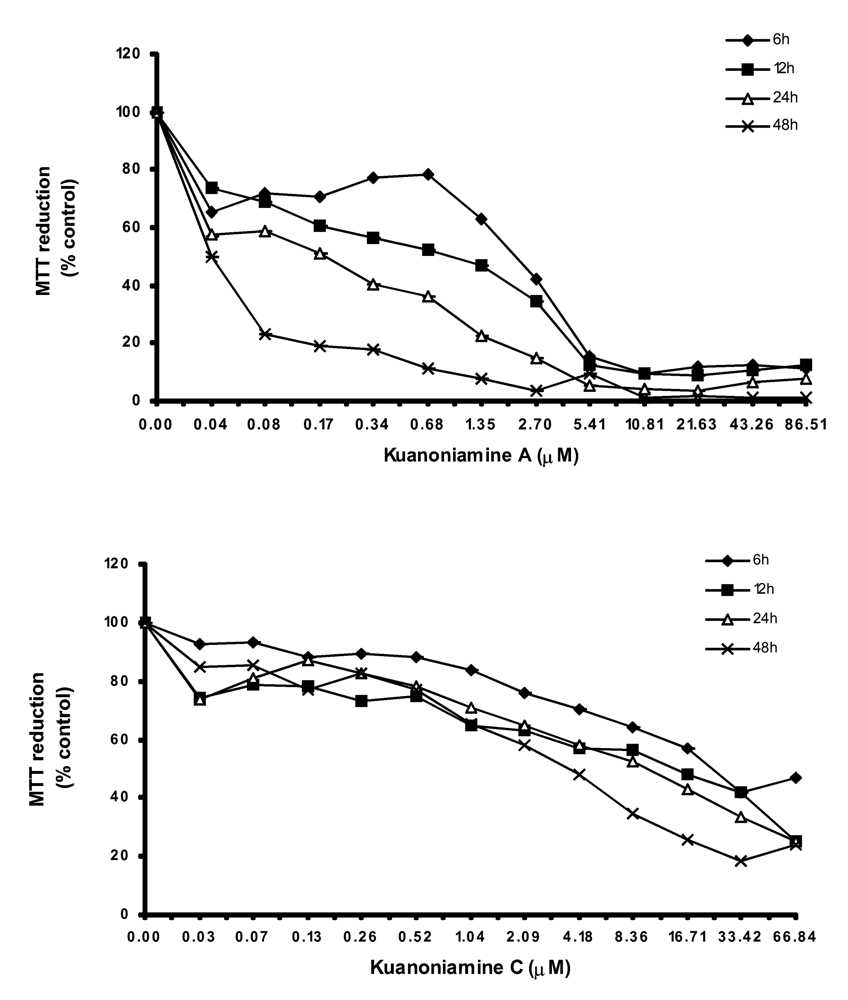
In order to get more insight on the mechanism of kuanoniamine A (6) and kuanoniamine C (7), both compounds were evaluated for their effect on the capacity of MCF-7 cells to reduce MTT (Figure 3).
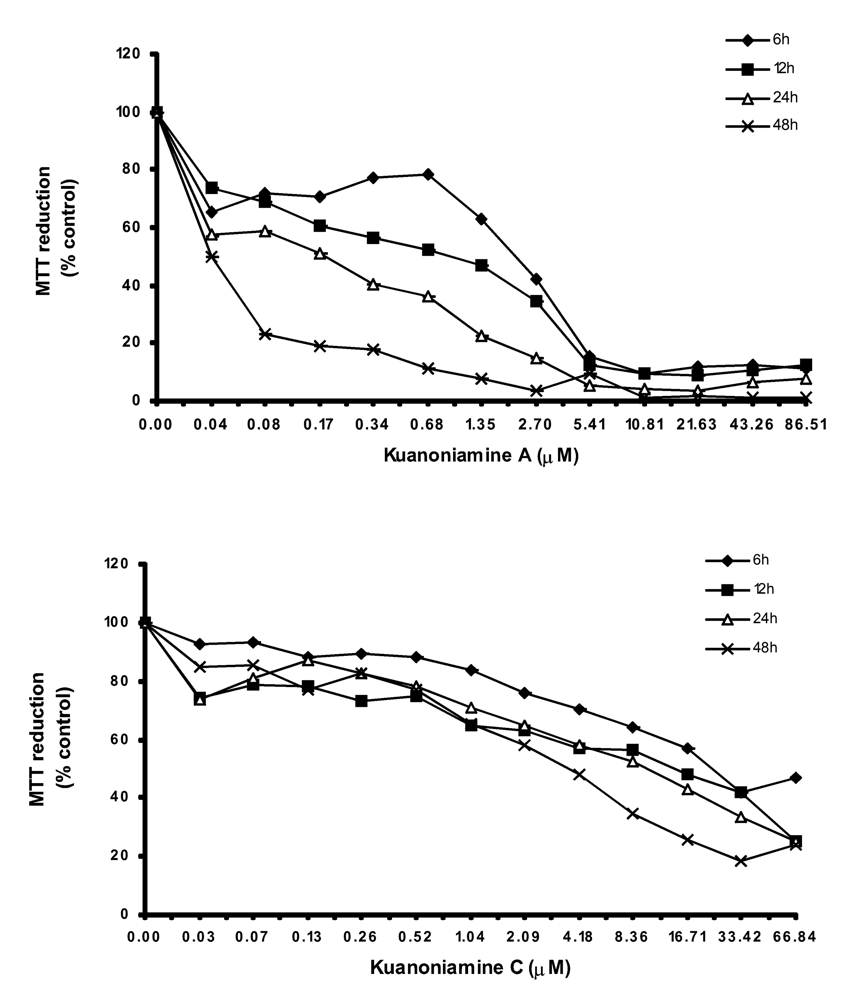
Figure 3.
Time and dose-dependent curves of kuanoniamine A (6) and kuanoniamine C (7) in MTT reduction capacity of MCF-7 cells. Results are the mean ± SEM of three replicates from a representative experiment.
Kuanoniamine A (6) was shown to be more drastic and more dependent on the exposure time than kuanoniamine C (7). Cell viability was dramatically affected even when treatments with low concentrations of kuanoniamine A (6) and in short periods of time. A decrease of viability to 70% was detected after 6 h of exposure at the lowest concentration studied (0.04 μM) and exposure of 24 h and 48 h were associated with a dramatic falls of the viability. This lost of viability is in accordance with the drastic effect observed in DNA synthesis after these treatments. However the cells whose DNA synthesis was dramatically affected are still viable and capable of reducing MTT, similar to those treated with concentrations of 0.06 μM. On the contrary, kuanoniamine C (7) did not affect the cells so drastically as kuanoniamine A (6) and 70% of the cells remained viable and with a capacity to reduce MTT, even after treatments with concentrations near the GI50 and exposure time of 24 and 48 h.
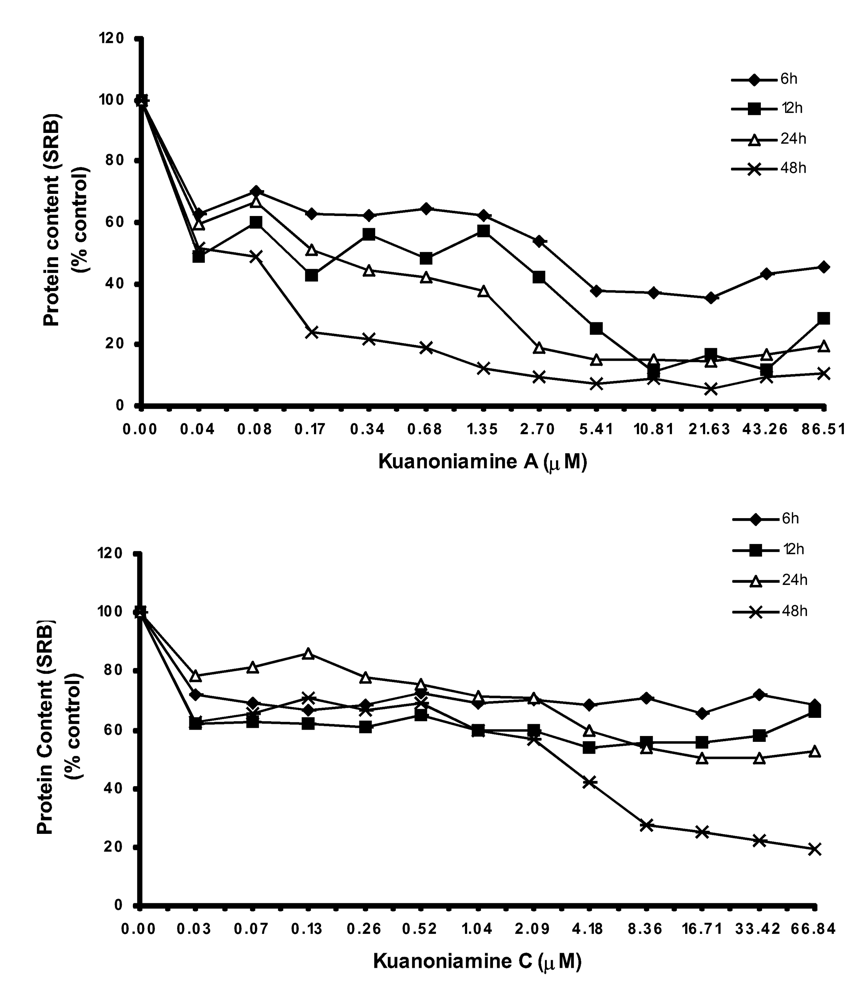
We have found also that the effect of kuanoniamine A (6) and kuanoniamine C (7) on the protein content of MCF-7 cells (Figure 4) was comparable to that observed with MTT, as can be concluded by the similarity between the curves slop.
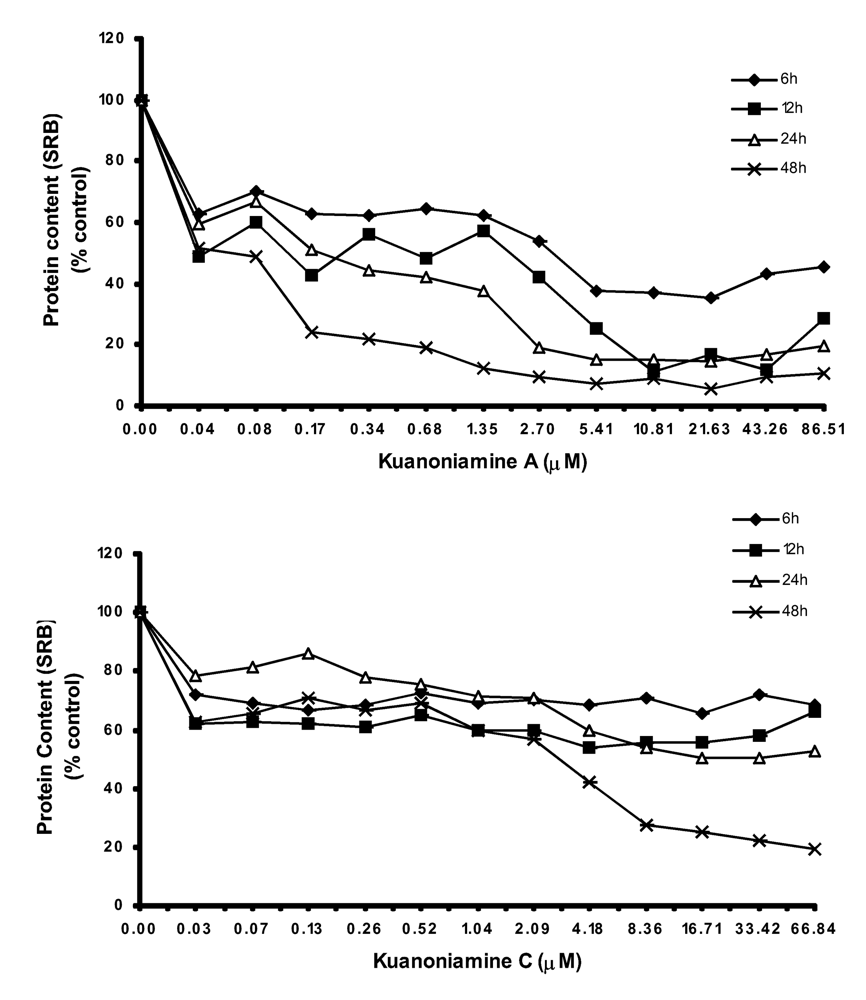
Figure 4.
Time and dose dependent curves of kuanoniamine A (6) and kuanoniamine C (7) on the cell growth of MCF-7 cells determined by protein content (SRB) assay. Results are the mean ± SEM of three replicates from a representative experiment.
With kuanoniamine A (6), the loss of cellular protein content falls immediately for the values below 70% after the exposure time as low as 6 h. This effect was found to be significantly dependent on the exposure time. On the contrary, the effect of kuanoniamine C (7) was found to be less intense and the cellular protein content was stable for almost all the treatments.
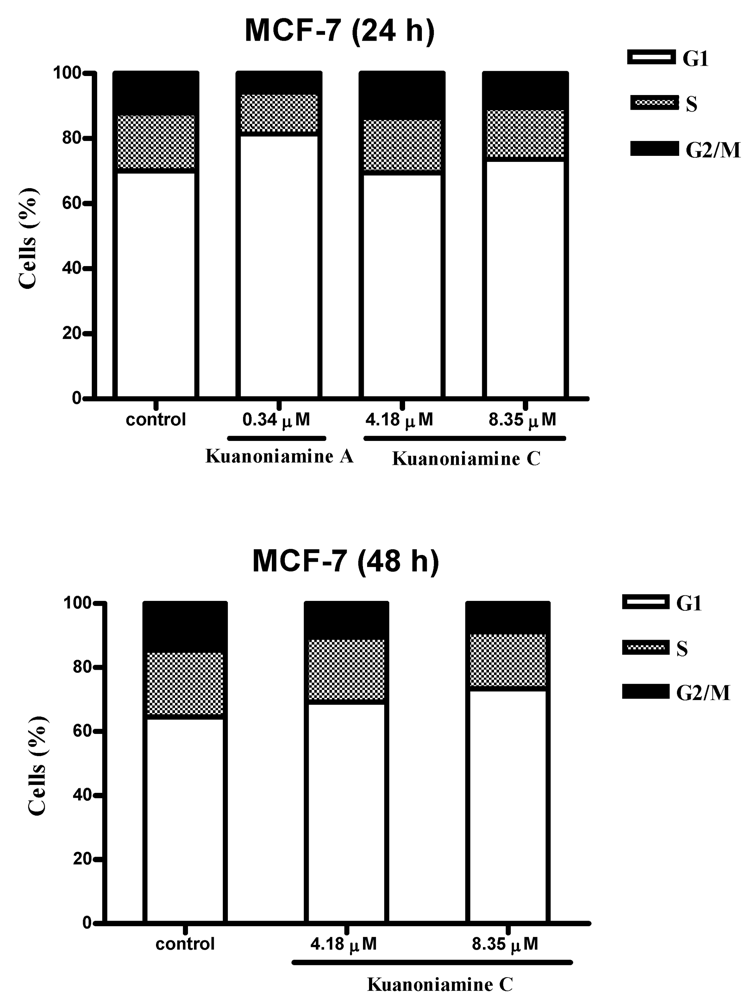
To further investigate the effect of these two compounds on MCF-7 cell proliferation, the cells were exposed for 24 and 48 h to 0.34 μM of kuanoniamine A (6) and to 4.18 and 8.35 μM of kuanoniamine C (7) and the distribution of cells in each phase of the cell cycle was determined by flow cytometric analysis of DNA content and the results are shown in Figure 5.
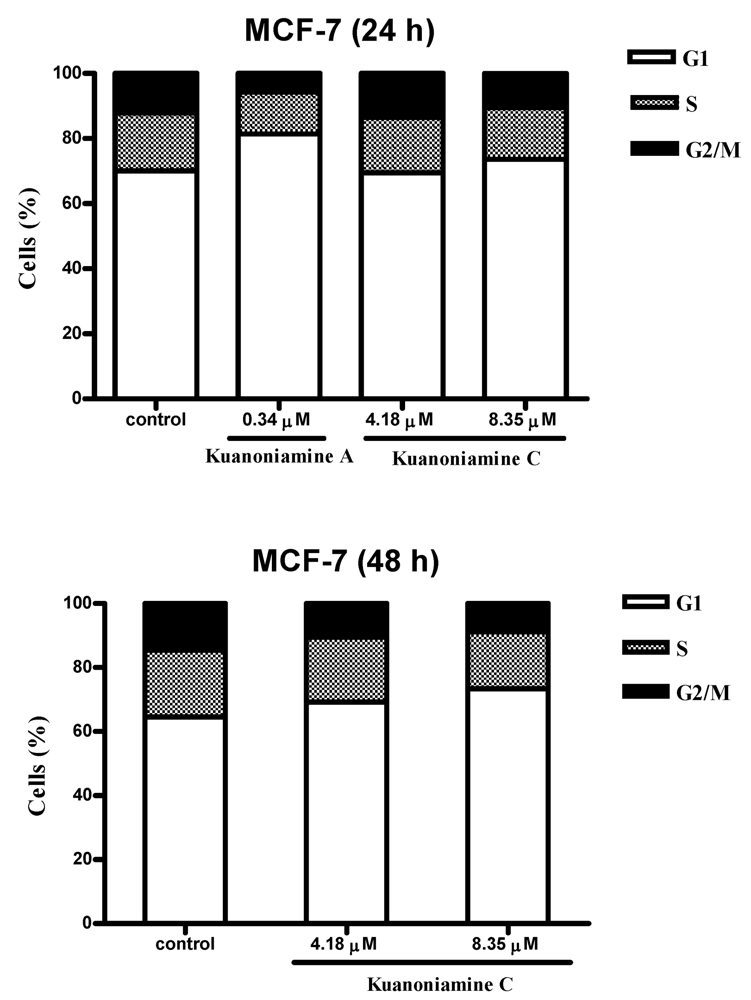
Figure 5.
Effect of kuanoniamine A (6) and kuanoniamine C (7) on the cell cycle of MCF-7 cells. Untreated control cells and cells treated with 0.34 mM of kuanoniamine A (6) for 24 h and treated with 4.18 and 8.35 mM of kuanoniamine C (7) for 24 and 48 h, stained with propidium iodide (PI) and analyzed by flow cytometry for DNA content.
From this figure, it can be concluded that an extensive reduction of cells in G2/M phase (5.71%) with a consequent accumulation of cells in G1 phase (81.41%) and a reduction on the cellular fraction in S phase (12.88%) was observed in cells treated with 0.34 μM of kuanoniamine A (6) for 24 h when compared with these phases of untreated control cells (12.17%, 70.04% and 17.79 %, respectively). In contrast to what has been observed for kuanoniamine A (6), no considerable effect was detected in cells treated with 4.18 and 8.35 μM of kuanoniamine C (7) for 24 h. However, the increase in the exposure time to 48 h has also led to an increase in the number of cells in G1 (73.34%) and a decrease of G2/M-phase (8.89%) and S-phase (17.77%) population when compared to control cells (64.01%, 13.28% and 22.71%, respectively). It is not surprising that both compounds have caused the reduction of the S phase cellular fraction as the concentrations used in these experiments were found to inhibit severely DNA synthesis.
During the experiments we have observed that the proliferative effect of kuanoniamine C (7), associated with treatments with low concentrations and short exposure times, as previously detected by thymidine incorporation, was not accompanied by an enhancement of MTT reducing activity or by an increase of cellular protein content. This observation can be explained by a low sensitivity of the MTT and SRB assay for the analysis of cell proliferation when compared with the 3H-thymidine uptake method incorporation.
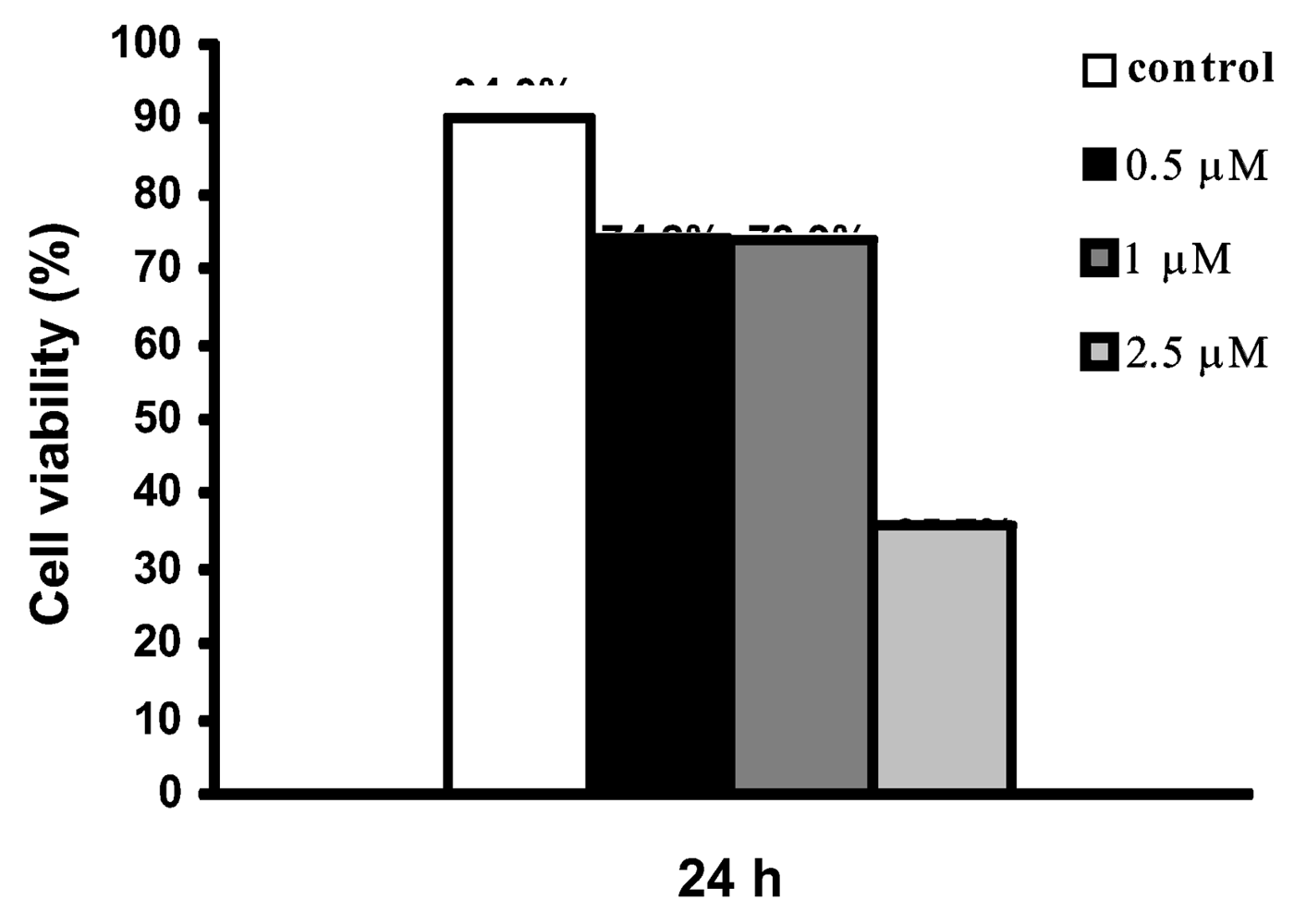
In order to investigate if the growth inhibitory effect of the kuanoniamine A (6) and kuanoniamine C (7) on MCF-7 was exerted through the involvement of apoptosis, the cells were treated for 24 h with inhibitory concentrations of kuanoniamine A (0.5, 1.0 and 2.5 μM) and kuanoniamine C (1.0 and 2.5 μM) and then evaluated for DNA fragmentation by the TUNEL assay. With this method, a significant increase in the number of apoptotic cells was detected after treatment with kuanoniamine A (6.19 % and 7.2 % for 0.5 and 1.0 μM, respectively) as well as with kuanoniamine C (5.21 % and 9.25 % for 1.0 and 2.5 μM, respectively) when compared with untreated control cells (1.41 %). This observation led to a conclusion that the antiproliferative effects of these two compounds were in part associated with apoptosis. Although treatments with 0.5 and 1.0 μM of kuanoniamine A (6) for 24 h were associated with an inhibition of DNA synthesis, cell viability (as measured by the trypan blue assay, Figure 6) remained moderately high ( 70%), which allowed apoptosis estimation. On the contrary, the concentration of 2.5 μM showed to be very toxic to cells causing the drastic decrease of viability for the values of 35.7% impairing apoptosis evaluation.
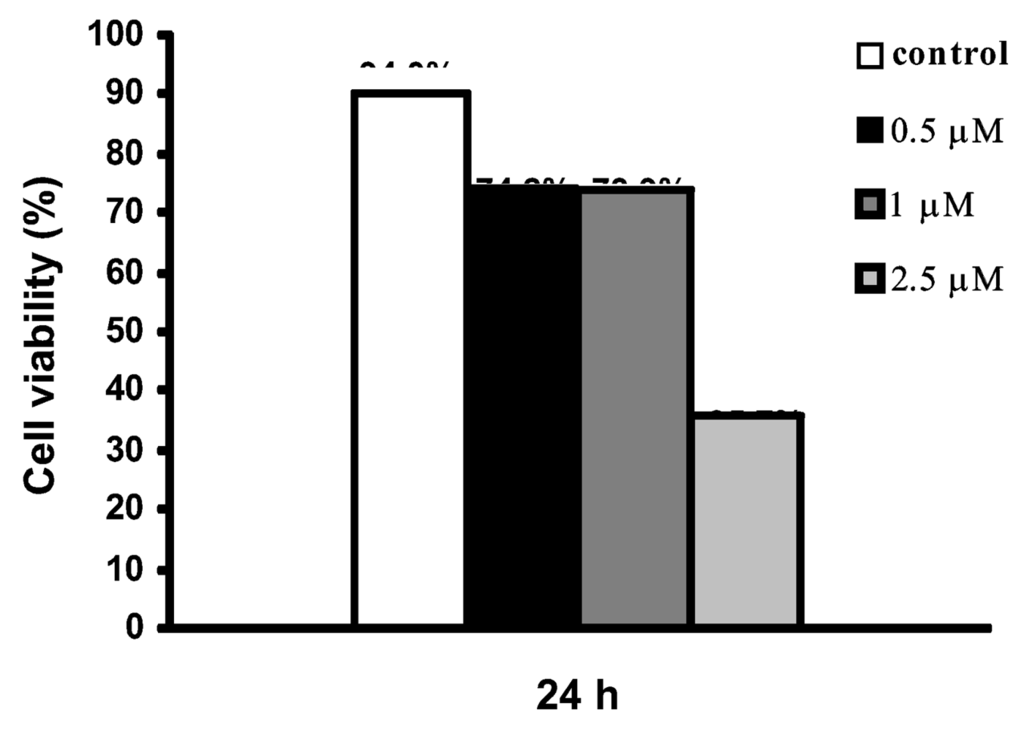
Figure 6.
Effect of kuanoniamine A (6) on the viability of MCF-7 cells determined by trypan blue exclusion assay. Cells were exposed to 0.5, 1 and 2.5 μM kuanoniamine A (6) for 24 h.
Finally, kuanoniamine A (6) and kuanoniamine C (7) were evaluated for their effect on the mitogenic response of peripheral human lymphocytes to PHA. Both compounds were shown to inhibit the proliferation of human lymphocytes (Table 2). Kuanoniamine A (6) was found to be a very potent inhibitor, exhibiting a IC50 value in the same order of cyclosporin A (1.50 μM versus 1.1 μM, respectively).
Table 2.
Effect of kuanoniamine A (6) and kuanoniamine C (7) on the proliferation of human lymphocytes induced by PHA.*
3. Conclusion
The pyridoacridine alkaloids kuanoniamines A and C, isolated from the Thai collection of the marine sponge Oceanapia sagittaria (Sollas), have been evaluated for their cytotoxic effect against five human tumour cell lines and one human non-tumour cell lines by the SRB method. Kuanoniamine A (6) has shown to be a potent growth inhibitor of all the human tumor cell lines as well as the non-tumour cell line. Though kuanoniamine C (7) was found to be much less potent than kuanoniamine A, it was found to possess a high selectivity toward the estrogen dependent (ER+) breast cancer cell line. As the inhibitory effect of kuanoniamine C on the estrogen independent (ER−) breast cancer cell line was less intense than that on the estrogen dependent (ER+) counterpart, it was suggested that the estrogen receptors could be involved in this antiproliferative effect. Using [3H] thymidine incorporation assay, it was found that the DNA synthesis of the MCF-7 cells was dramatically affected by kuanoniamine A and it was dependent on the concentration of kuanoniamine A rather and the exposure time. On the other hand, kuanoniamine C showed a stimulatory effect at low concentration but an inhibitory effect at high concentrations. The biphasic effect of kuanoniamine C on DNA synthesis of the MCF-cells resembled with that caused by phytoestrogens on the estrogen dependent breast cancer cells. Using the MTT-reducing capacity assay, it was found that the effect of kuanoniamine A was much more potent than kuanoniamine C on the cell viability of the MCF-7 cells and this loss of cell viability caused by kuanoniamine A was in accordance with its drastic effect on DNA synthesis observed after these treatments. Flow cytometric analysis of the DNA content has showed that kuanoniamine A has caused an extensive reduction of the MCF-7 cells in G2/M phase with a consequent accumulation of the cells in G1 phase and a reduction of the cellular fraction in S phase while kuanoniamine C showed no considerable effect on these cells. Using the TUNEL assay to measure a prelytic DNA fragmentation, it was found that both kuanoniamines A and C have caused an increase of the apoptotic cells, indicating that their antiproliferative effects on the MCF-7 cancer cell lines could in part be related to the phenomenon of apoptosis. However, more studies are needed to confirm and characterize the capacity of these compounds in inducing apoptosis since DNA cleavage may also accompany necrosis which is another mechanism of cell death. Contrary to apoptosis, necrotic cell death is often associated with an in vivo extensive tissue damage which, in turn, results in an intense inflammatory response. It is advocated that the inflammatory component of the necrotic cell death could have the potential advantage of stimulating an immune response to the tumor and a balance between apoptotic and necrotic cell death has been referred as an advantage on cancer therapy [22].
4. Experimental
4.1. General
Melting points were recorded on a Bock monoscope and are uncorrected. Rotations were determined on a Polax-2L instrument. Si gel for chromatography was silica gel 60 (0.2–0.5 mm Merck) for analytical work and for preparative work TLC silica gel 60 GF 254 Merck.
4.2. Animal material
Oceanapia sagittaria (Sollas, 1902), order Haplosclerida, family Oceanapiidae, was collected by Scuba diving in the Gulf of Thailand near Koh Samed (3 meter depth) in April 2005 and frozen immediately at −20ºC prior to extraction. The sponge material was identified by Dr. Sumaitt Putchakarn and the voucher registered as BIMS-I 2004 was deposited at Bangsaen Institute of Marine Science, Burapha University, Thailand.
4.3. Extraction, isolation and characterization of the constituents
The sample (3.5 kg net weight) was thawed, homogenized with EtOH (7L), allowed to stand for 24 h in the dark chamber and filtered. The residue on the filter paper was again extracted with EtOH (2x7L), the aqueous alcoholic extracts were combined, evaporated at reduced pressure to give ca. 800 ml and extracted with EtOAc (3 x 1L). The EtOAc extracts were combined and concentrated at reduced pressure to give a crude EtOAc extract (13.1g).
The crude EtOAc extract (13.1 g) was applied over a flash chromatography column of Si gel 60 (150g) and eluted with a solvent gradient system of petrol-CHCl3, CHCl3, CHCl3,-MeOH, EtOAc and MeOH, with 200 ml fractions being collected as follows: Frs. 1–2 (petrol-CHCl3,1:4), 3–7 (CHCl3), 8–17 (CHCl3,-MeOH, 49:1), 18–22 CHCl3,-MeOH, 19:1), 23–24 (CHCl3,-MeOH, 9:1), 25–26 (EtOAc), 27–30 (MeOH). Frs. 3–4 were combined (2.6 g) and recrystallized from a mixture of petrol and CHCl3 to give white crystal of 24 α-methylcholestanol (1, 436 mg). Frs. 6–7 were combined (870 mg) and chromatographed over Si Gel 60 (20 g) and eluted with petrol-CHCl3, CHCl3-Me2CO, and CHCl3-MeOH, 200 ml subfractions were collected as follows: Sfrs. 1–7 (petrol-CHCl3, 2:3), 8–15 (petrol-CHCl3, 1:4), 16–24 (CHCl3), 25–30 (CHCl3-Me2CO, 19:1), 31–34 (CHCl3-Me2CO, 9:1), 35–38 (CHCl3-Me2CO, 4:1), 39–43 (CHCl3-Me2CO, 3:2), 44–45 (MeOH). Sfrs. 3–6 were combined (156 mg) and purified by TLC (Si gel, CHCl3) to give 33.2 mg of a mixture of p-hydroxybenzoic acid (3) and phenylacetic acid (4). Subfrs. 7–15 were combined and purified by TLC (Si gel, CHCl3-Me2CO-HCO2H, 18:1:0.1) to give kuanoniamine A (6, 20 mg). Frs. 8–10 were combined (390 mg) and purified by TLC (Si gel, CHCl3, Me2CO, HCO2H, 95:5:0.1) to give p-hydroxybenzaldehyde (2, 15.7 mg) and 3-formylindole (5, 18.6 mg). Frs. 11–13 were combined (346 mg) and purified by TLC (Si gel, CHCl3, MeOH, HCO2H, 98:2:0.1) to give kuanoniamine A (6, 32 mg). Frs. 20–22 were combined (355 mg) and applied over a flash chromatography column of ODS gel (10g) and eluted with a gradient system of MeOH-H2O and MeOH-CHCl3-H2O, 100 ml fractions being eluted as follows: sfrs 1–2 (MeOH-H2O, 7:3), 3–4 (MeOH-H2O, 4:1), 5–6 (MeOH-H2O, 9:1), 7–10 (MeOH), 11–14 MeOH-CHCl3-H2O, 7:3:0.5). Sfrs. 5–6 were combined (81 mg) and recrystallized from a mixture of CHCl3 and Me2CO to give yellow crystals of kuanonamine C (7, 44 mg).
4.4. Biological activity
4.4.1. Material and Methods
4.4.1.1. Reagents
Fetal bovine serum (FBS), L-glutamine, phosphate buffered saline (PBS) and trypsin were from Gibco Invitrogen Co. (Scotland, UK). RPMI-1640 medium were from Cambrex (New Jersey, USA). Acetic acid, cyclosporin A, dimethyl sulfoxide (DMSO), doxorubicin, ethylenediaminetetraacetic acid (EDTA), Histopaque®-1077, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), penicillin, phytohemagglutinin (PHA), propidium iodide (PI), RNase, streptomycin, sulforhodamine B (SRB), Triton X-100, trypan blue were from SigmaChemical Co (Saint Louis, USA). Magnesium chloride, paraformaldehyde, tricloroacetic acid (TCA), Tris, sodium citrate and sodium dodecyl sulphate (SDS) were sourced from Merck (Darmstadt, Germany). Dimethylformamide (DMF) from Romil Chemicals (Cambridge, England). Heparin was sourced from Sanofi Winthrop Inc. (Paris, France). Vectashield, Vectashield containing DAPI (0.5 μg/ml) were from Vector Laboratories (UK). FACSCalibur flow was from Becton Dickinson (Mountain View, CA), [3H]-thymidine from Amersham (Illinois, USA) and scintillation liquid from PerkinElmer (Boston, USA).
4.4.1.2. Samples
Stocks solutions of kuanoniamine A (6) and kuanoniamine C (7) were prepared in DMSO and kept at − 20°C. Appropriate dilutions of the compounds were freshly prepared just prior the assays.
4.4.1.3. Cell cultures
Five human tumour cell lines, MCF-7 (breast adenocarcinoma, estrogen-dependent ER (+)), MDA-MB-231 (breast adenocarcinoma, estrogen-independent ER (−)), NCI-H460 (non-small cell lung cancer), SF-268 (glioma) and UACC-62 (melanoma) and one non-tumour cell line, MRC-5 (diploid embryonic lung fibroblast) were used. MCF-7, MDA-MB-231 and MRC-5 were obtained from the European Collection of Cell Cultures (ECACC, Salisbury, UK) while NCI-H460, SF-268 and UACC-62 cell lines were provided by the National Cancer Institute (NCI, Bethesda, U.S.A.). Cells, growing as monolayer, were routinely maintained in RPMI-1640 medium supplemented with 5% heat-inactivated FBS, 2 mM glutamine and antibiotics (penicillin 100 U/ml, streptomycin 100 μg/ml), at 37°C in a humidified atmosphere containing 5% CO2. Exponentially growing cells were obtained by plating 1.5×105cells/ml for MCF-7, MDA-MB-231, SF-268 and MRC-5, 1.0 × 105cells/ml for UACC-62 and 0.75 × 104cells/ml for NCI-H460, followed by 24 h incubation. The effect of the vehicle solvent (DMSO) on the growth of these cell lines was evaluated in all the experiments by exposing untreated control cells to the maximum concentration (0.5%) of DMSO used in each assay.
4.4.1.4. Cells growth assay
The effects of the kuanoniamine A (6) and kuanoniamine C (7) on the growth of human tumour and non-tumour cell lines were evaluated according to the procedure adopted by the National Cancer Institute (NCI, USA) in the “In vitro Anticancer Drug Discovery Screen” that uses the protein-binding dye sulforhodamine B to assess cell growth [18, 19]. Exponentially growing cells were then exposed for 48 h to five serial concentrations of kuanoniamine A (0.14; 0.69; 3.46; 17.30 and 86.51 μM) and kuanoniamine C (0.11; 0.53; 2.67; 13.37 and 66.84 μM) that were starting from a maximum of 25 μg/ml (86.51 μM and 66.84 μM, respectively).
Following this exposure period adherent cells were fixed in situ with 50% TCA, washed with distillated water and stained with 0.4% SRB solubilized in 1% acetic acid. The bound stain was solubilized in 10 mM Tris and the absorbance was measured at 492 nm in a microplate reader (Biotek Instruments Inc., Powerwave XS, Wincoski, USA). For each cell line a dose-response curve was obtained and the growth inhibition of 50% (GI50), corresponding to the concentration of kuanoniamine A and kuanoniamine C that inhibited 50% of the net cell growth was calculated as described elsewhere [19]. Doxorubicin was used as a positive control and tested in the same manner.
4.4.1.5. Lymphocytes proliferation assay
The effect of kuanoniamine A and kuanoniamine C on the mitogenic response of human lymphocytes to PHA (10 μg/ml) was evaluated using a modified version of the colorimetric MTT-assay [20]. This has been intensively described elsewhere by our group [21]. Briefly, mononuclear cells, isolated from heparinized peripheral blood of healthy volunteers by Histopaque®-1077 density centrifugation, were adjusted to 2–3 x 106 cells/ml in RPMI-1640 supplemented with 10% FBS, 2 mM glutamine and antibiotics (penicillin 100 U/ml, streptomycin 100 μg/ml). They were then harvested in 96-well plates and exposed for 4 days to seven serial concentrations of kuanoniamine A (0.12; 0.36; 1.07; 3.20; 9.61; 28.84 and 86.51 μM) and of kuanoniamine C (0.09; 0.28; 0.83; 2.48; 7.43; 22.28 and 66.84 μM) at 37 ºC. Following this incubation period, the MTT solution (1 mg/ml) was added and plates were incubated for more 4 h. The water insoluble formazan dye formed was solubilized with SDS/DMF solution (20% SDS in a 50% solution of DMF, pH 4.7) overnight at 37 °C. The absorbance of the colored solution was then measured in a microplate reader (Biotek Instruments Inc., Powerwave XS, Wincoski, USA) at 550 nm. The concentration of compounds that inhibited 50% of lymphocyte proliferation (IC50) was calculated by the curve obtained (% inhibition versus concentration). Cyclosporin A was used as a positive control. Seven serial concentrations (0.0014; 0.01; 0.04; 0.18; 0.89; 4.44 and 22.20 μM) were studied in the same manner.
4.4.1.6. Evaluation of MCF-7 cells growth by [3H] thymidine incorporation, SRB and MTT assays
MCF-7 cells growing exponentially in 96-well plates were treated with a range of concentrations of kuanoniamine A (0.04; 0.08; 0.17; 0.34; 0.68; 1.35; 2.70; 5.41; 10.81; 21.63; 43.26 and 86.51 μM) and of kuanoniamine C (0.03; 0.07; 0.13; 0.26; 0.52; 1.04; 2.09; 4.18; 8.36; 16.71; 33.42 and 66.84 μM) for 6, 12, 24 and 48 h. Following this incubation period cell growth was assessed by [3H]-thymidine incorporation, MTT and SRB assays. In the [3H]-thymidine incorporation assay cells were further incubated with 1 μCi of [3H] thymidine for 4 h. Pulsed cells were then harvested on a glass filter 102 x 256 mm (Skatron, Norway) using a semiautomatic cell harvester (Skatron Instruments, Norway) and allowed to dry. Incorporation of radioactive thymidine was determined in a scintillation counter (LS 6500, Beckman Instruments, CA and USA) and defined by comparing the arithmetic mean of counts per minute (cpm) of treated cells with that of untreated control cells.
In the MTT assay, MTT was added (5 mg/ml) and cells were incubated for a further 4 h. Formazan product were solubilized with SDS/DMF solution overnight at 37ºC. Absorbance was measured at 550 nm in the microplate reader. The percentage of MTT reduction was calculated comparing the absorbance of treated cells to that of untreated control cells.
In the SRB assay adherent cells were fixed in situ with 50% TCA, washed with distillated water and stained with the protein binding dye (0.4% SRB) solubilized in 1% acetic acid, and then the bound stain was solubilized in 10 mM Tris, as described [18]. The bound stain was solubilized and the absorbance was measure at 492 nm in the microplate reader.
4.4.1.7. Cell cycle analysis
MCF-7 cells growing exponentially in 25 cm2 flasks were treated with 0.08, 0.17, 0.34 and 0.68 μM of kuanoniamine A for 24 h and 4.18 and 8.35 μM of kuanoniamine C for 24 and 48 h. After treatment, attached cells were released by trypsinization and mixed with non-adherent cells. Cells were centrifuged, washed twice with PBS and fixed with 70% ice-cold ethanol. Fixed cells (1 x 106 cells/ml) were resuspended in the DNA staining solution containing 50 μg/ml propidium iodide, 0.5 mg/ml RNase in 10 mM Tris and 5 mM MgCl2. DNA cellular content was analyzed with a FACSCalibur flow cytometer (Becton Dickinson, Mountain View, CA) with excitation at 488 nm. Data was acquired in a listmode data file, gated to 30.000 events in cell cycle, using the CellQuest Pro software, version 4.0.2 (Becton Dickinson Mountain View, CA) included in the system. Cell cycle was analysed using the ModFit LT 3.0 software program (Verity Software Topsham) with an activated gate to eliminate aggregates (FL2-A/FL2-W).
4.4.1.8. Apoptosis assay
Fragmentation of the cellular DNA was evaluated using the In Situ Cell Death Detection Kit (TUNEL) Fluorescein (Boehringer Mannheim, Germany). Exponential MCF-7 cells growing on coverglasses in 6-well plates were exposed to 0.5 and 1.0 μM of kuanoniamine A and 1.0 and 2.5 μM of kuanoniamine C for 24 h. After treatment, plates were centrifuged at 2000 rpm for 5 min at room temperature in order to sediment non-adherent cells and fixed with 4% paraformaldehyde (in PBS) for 1 hour at room temperature. Fixed cells were washed 4 times for 5 minutes and permeabilized (0.1% Triton X-100, 0.1% sodium citrate in distillated water) for 2 min on ice (4°C) and washed with PBS 4 times for 5 minutes. Coverglasses were mounted in Vectashield containing 50 μg/ml PI and 0.5 μg/ml RNase. All preparations were observed under the fluorescence microscope (Eclipse E400, Nikon, Japan). The images were acquired from the photograph system ACT.2U from Nikon. Apoptotic cells were quantified by counting a minimum of 400 cells from at least five different random areas of the slide.
4.4.1.9. Trypan blue exclusion assay
Exponential MCF-7 cells growing in 6-well plates were treated with 0.5, 1.0 and 2.5 μM of kuanoniamine A for 24h. After treatment, attached cells were released by trypsinization and mixed with non-adherent cells. Cells were stained with 0.2% trypan blue and viable and non-viable cells were counted in a hemocytometer. Results were expressed in terms of percentage of viable cells in the total cell number.
Acknowledgments
This work was supported by FTC- Fundação para a Ciência e Tecnologia (Project POCI/MAR/58114/2004). Work in Thailand was supported by the Thailand Research Fund (No. TRG 4780003). We thank Dr. Sumaitt Puchakarn, BIMS, Burapha University, Thailand, for collection and identification of the sponge material and Dr. Graham Eaton, Department of Chemistry, University of Leicester, UK for HRMS.
- Samples Availability: Available from the authors.
References
- Hooper, JNA; Kelly-Borges, M; Riddle, M. Oceanapia sagittaria from the Gulf of Thailand. Mem Qd Mus 1993, 33, 61–72. [Google Scholar]
- Nicholas, GM; Hong, TW; Molinski, TF; Lerch, ML; Cancilla, M; Lebrilla, CB. Oceanapiside, an antifungal bis- α,ϖ-aminoacid alcohol glycoside from the marine sponge Oceanapia phillipensis. J Nat Prod 1999, 62, 1678–1681. [Google Scholar]
- Makarieva, TN; Denisenko, VA; Dmitrenok, PS; Guzii, AG; Santalova, EA; Stonik, VA; MacMillan, JB; Molinski, TF. Oceanalin A, a hybrid α,ϖ-bifunctionalized sphingoid tetrahydroisoquinoline β-glycoside from the marine sponge Oceanapia sp. Org Lett 2005, 7, 2897–2900. [Google Scholar]
- Capon, RJ; Skene, C; Liu, EHT; Lacey, E; Gill, JH; Heiland, K; Friedel, T. The isolation and synthesis of novel nematocidal dithiocyanates from an Australian marine sponge, Oceanapia sp. J Org Chem 2001, 66, 7765–7769. [Google Scholar]
- Capon, RJ; Skene, C; Liu, EHT; Lacey, E; Gill, JH; Heiland, K; Friedel, T. Nematocidal thiocyanatins from a Southern Australian marine sponge Oceanapia sp. J Nat Prod 2004, 67, 1277–1282. [Google Scholar]
- Matsugana, S; Okada, Y; Fusetani, N; van Soest, RWM. An antimicrobial C14 acetylenic acid from a marine sponge Oceanapia species. J Nat Prod 2000, 63, 690–691. [Google Scholar]
- Boyd, KG; Harper, MK; Faulker, DJ. Oceanapamine, a sesquiterpene alkaloid from the Phillipine sponge Oceanapia sp. J Nat Prod 1995, 58, 302–305. [Google Scholar]
- Mancini, I; Guella, G; Sauvain, M; Debitus, C; Duigou, AG; Ausseil, F; Menou, JL; Pietra, F. New 1,2,3,4-tetrahydropyrrolo[1,2-a]pyrimidinium alkaloids (phloeodictynes) from the New Caledonian shallow-water haplosclerid sponge Oceanapia fistulosa. Structure elucidation from mainly LC-tandem-MS-soft-ionization techniques and discovery of antiplasmodial activity. Org Biomol Chem 2004, 2, 783–787. [Google Scholar]
- Solomon, CE; Faulkner, DJ. Sagitol, a pyridoacridine alkaloid from the sponge Oceanapia sagittaria. Tetrahedron Lett 1996, 37, 9147–9148. [Google Scholar]
- Eder, C; Schupp, P; Proksch, P; Wray, V; Steube, K; Müller, CE; Frobenius, W; Herderich, M; van Soest, RWM. Bioactive pyridoacridine alkaloids from the Micronesian sponge Oceanapia sp. J Nat Prod 1998, 68, 301–305. [Google Scholar]
- Solomon, CE; Deerinck, T; Ellisman, MH; Faulker, DJ. The cellular localization of dercitamide in the Palauan sponge Oceanapia sagitaria. Mar Biol 2001, 139, 313–319. [Google Scholar]
- Schupp, P; Eder, C; Paul, V; Proksch, P. Distribution of secondary metabolites in the sponge Oceanapia sp. and its ecological implications. Mar Biol 1999, 135, 573–580. [Google Scholar]
- Carroll, AR; Ngo, A; Quinn, RJ; Redburn, J; Hooper, JNA. Petrosamine B, an inhibitor of the Helicobacter pylori enzyme aspartyl semialdehyde dehydrogenase from the Australian sponge Oceanapia sp. J Nat Prod 2005, 68, 804–806. [Google Scholar]
- Carroll, AR; Sheuer, PJ. Kuanoniamines A, B, C, and D: Pentacyclic alkaloids from a Tunicate and its mollusc predator Chelynotus semperi. J Org Chem 1990, 55, 4426–4431. [Google Scholar]
- Kitahara, Y; Ankara, S; Yonezawa, T; Nagatsu, M; Shibano, Y; Kubo, A. Synthetic studies on pentacyclic aromatic alkaloids, kuanoniamine A, 11-hydroxyascididemin, and neocalliactine acetate. Tetrahedron 1997, 63, 8024–8026. [Google Scholar]
- Wang, C; Kurzer, MS. Phytoestrogen concentration determines effects on DNA synthesis in human breast cancer cells. Nutr Cancer 1997, 28, 236–247. [Google Scholar]
- Wang, C; Kurzer, MS. Effects of Phytoestrogens on DNA synthesis in MCF-7 cells in the presence of estradiol or growth factors. Nutr Cancer 1998, 31, 90–100. [Google Scholar]
- Skehan, P; Storeng, R; Scudiero, D; Monks, A; McMahon, J. Vistica D, Warren JT, Bokesch H, Kenny S, Boyd MR. New colorimetric cytotoxic assay for anticancer drug screening. J Natl Cancer Inst 1990, 82, 1107–1112. [Google Scholar]
- Monks, A; Scudiero, D; Skehan, P; Shoemaker, R; Paul, K; Vistica, D; Hose, C; Langley, J; Cronise, P; Vaigro-Wolff, A; Gray-Goodrich, M; Campbell, H; Mayo, J; Boyd, M. Feasibility of a high-flux anticancer drugs screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst 1991, 83, 757–776. [Google Scholar]
- Mosman, T. apid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxic assays. J Immunol Meth 1983, 65, 55–63. [Google Scholar]
- Gonzalez, MJ; Nascimento, MSJ; Cidade, MM; Pinto, MM; Kijjoa, A; Anantachoke, C; Silva, AMS; Herz, W. Immunomodulatory activity of xanthones from Callophyllum teysmannii var. inophylloide. Planta Med 1999, 65, 368–371. [Google Scholar]
- Edinger, AL; Thompson, CB. Death by design: apoptosis, necrosis and autophagy. Current Opinion in Cell Biology 2004, 16, 663–669. [Google Scholar]