Monoindole Alkaloids from a Marine Sponge Spongosorites sp.

Abstract
:Introduction
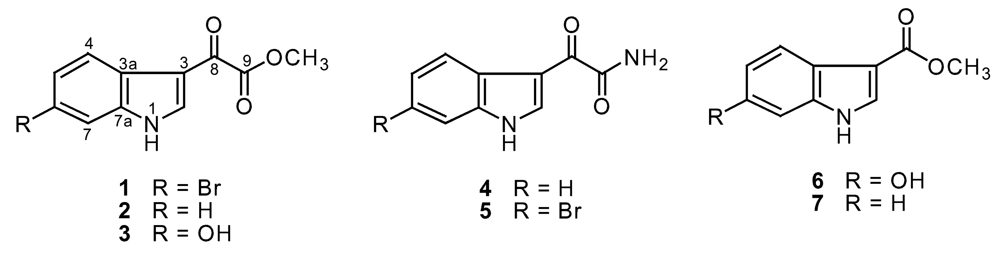
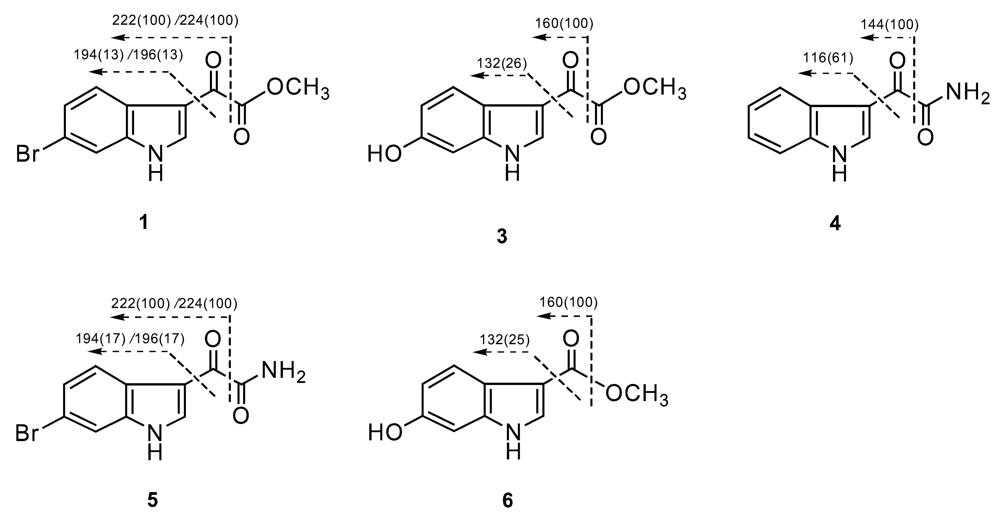
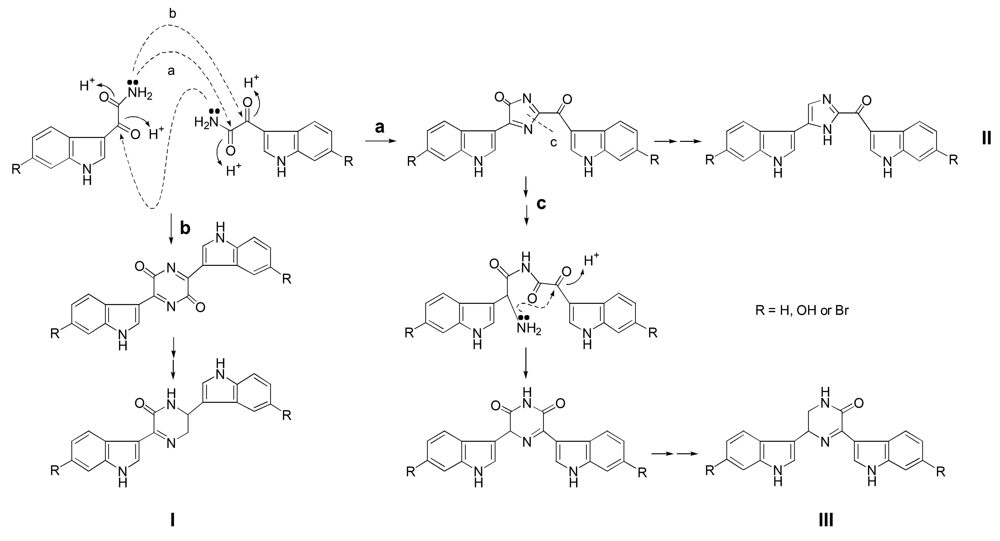
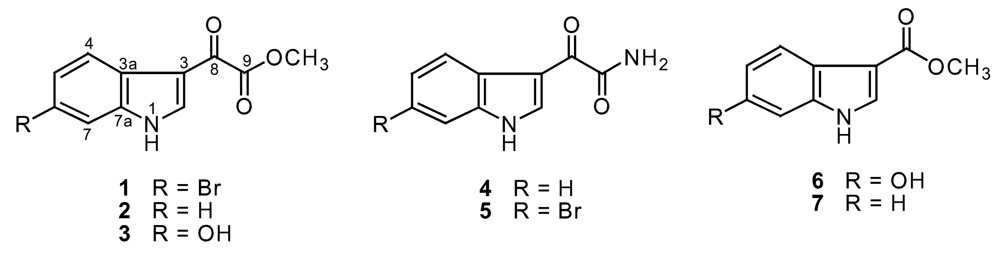
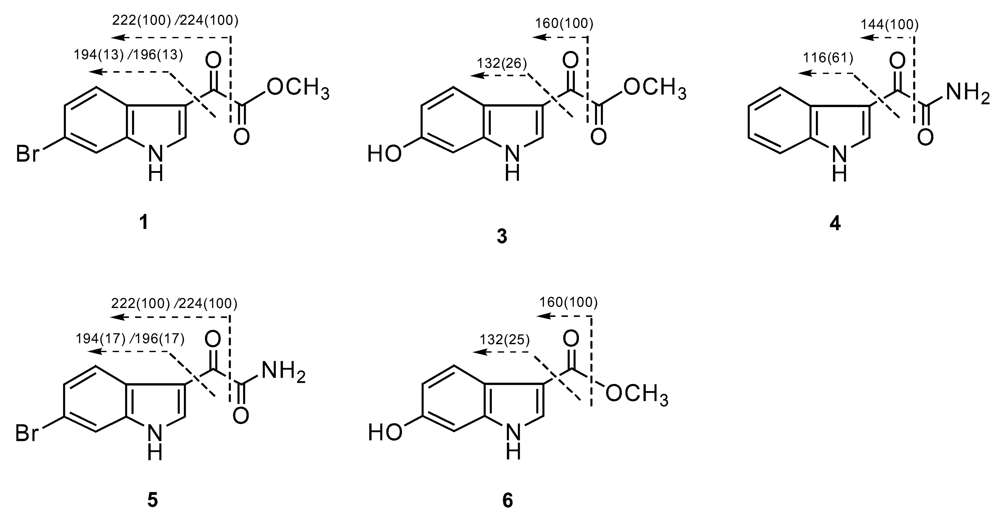
Result and discussion
Experimental
General Experimental Procedures
Animal Material
Extraction and Isolation
Evaluation of Cytotoxicity
Acknowledgments
- Samples Availability: Not available.
References
- Kobayashi, J; Murayama, T; Ishibashi, M; Kosuge, S; Takamatsu, M; Ohizumi, Y; Kobayashi, H; Ohta, T; Nozoe, S; Sasaki, T. Hyrtiosins A and B, new indole alkaloids from the Okinawan marine sponge Kyrtios erecta. Tetrahedron 1990, 46, 7699–7702. [Google Scholar]
- Segraves, NL; Crews, P. Investigation of brominated tryptophan alkaloids from two Thorectidae sponges: Thorectandra and Smenospongia. J Nat Prod 2005, 68, 1484–1488. [Google Scholar]
- Li, H; Matsunaga, S; Fusetani, N. Bioactive marine meabolites. Part 52. Simple antifungal metabolites from a marine sponge, Halichondria sp. Comp Biochem Phys B: Biochem Mol Bio 1994, 2, 261–264. [Google Scholar]
- Rasmussen, T; Christophersen, C; Nielsen, PH; Rajagopal, R. Auxin activity of brominated indoles from the marine sponge Pseudosuberites hyalinus. J Mar Biotechnol 1995, 2, 167–169. [Google Scholar]
- Kobayashi, J; Cheng, J; Yamamura, S; Sasaki, T; Ohizumi, Y. Penaresin, a new sarcoplasmic reticulum Ca-inducer from the Okinawan marine sponge Penares sp Heterocycles 1990, 31, 2205–2208.
- Lindquist, N; Fenical, W. Polyandrocarpamides A–D, novel metabolites from the marine ascidian Polyandrocarpa sp. Tetrahedron Lett 1990, 31, 2521–2524. [Google Scholar]
- Peters, L; König, GM; Terlau, H; Wright, AD. Four new bromotryptamine derivatives from the marine bryozoan Flustra foliacea. J Nat Prod 2002, 65, 1633–1637. [Google Scholar]
- Zheng, L; Yan, X; Xu, J; Chen, H; Lin, W. Hymeniacidon perleve associated bioactive bacterium Psedomonas sp. NJ6-3-1. Appl Biochem Micro 2005, 41, 29–33. [Google Scholar]
- Li, Y; Li, X; Kim, D; Choi, H; Son, B. Indolyl alkaloid derivatives, Nb-acetyltrptamine and oxaline from a marine-derived fungus. Arch Pharm Res 2003, 26, 21–23. [Google Scholar]
- Van Lear, GE; Morton, GO; Fulmor, W. New antibacterial bromoindole metabolites from the marine sponge Polyfibrospongia maynardii. Tetrahedron Lett 1973, 4, 299–300. [Google Scholar]
- Bao, B; Sun, Q; Yao, X; Hong, J; Lee, CO; Sim, CJ; Im, KS; Jung, JH. Cytotoxic bisindole alkaloids from a marine sponge Spongosorites sp. J Nat Prod 2005, 68, 711–715. [Google Scholar]
- Bao, B; Sun, Q; Yao, X; Hong, J; Lee, CO; Cho, HY; Im, KS; Jung, JH. Bisindole alkaloids of the topsentin and hamacanthin classes from a marine sponge Spongosorites sp. J Nat Prod 2007, 70, 2–8. [Google Scholar]
- Hughes, TV; Cava, MP. Total synthesis of didemnimide A and B. Tetrahedron Lett 1998, 39, 9629–9630. [Google Scholar]
- Jimènez, C; Quiñoà, E; Adamczeski, M; Hunter, LM; Crews, P. Novel sponge-derived amino acids. 12. Tryptophan-derived pigments and accompanying sesterterpenes from Fascaplysinopis reticulata. J Org Chem 1991, 56, 3403–3410. [Google Scholar]
- Dumdei, E; Andersen, RJ. Igzamide, a metabolite of the marine sponge Plocamissma Igzo. J Nat Prod 1993, 56, 792–794. [Google Scholar]
- Bokesch, HR; Pannell, LK; McKee, TC; Boyd, MR. Coscinamides A, B and C, three new bis indole alkaloids from the marine sponge Coscinoderma sp. Tetrahedron Lett 2000, 41, 6305–6308. [Google Scholar]
- Faul, MM; Winneroski, LL; Krumrich, CA. Synthesis of rebeccamycin and 11-dechlororebeccamycin. J Org Chem 1999, 64, 2465–2470. [Google Scholar]
- Santos, LS; Pilli, RA; Rawal, VH. Enantioselective total syntheses of (+)-arborescidine A, (–)-arborescidine B, and (–)-arborescidine C. J Org Chem 2004, 69, 1283–1289. [Google Scholar]
- Miyake, FY; Yakushijin, K; Horne, DA. Synthesis of marine sponge bisindole alkaloids dihydrohamacanthins. Org Lett 2002, 4, 941–943. [Google Scholar]
- Fedouloff, M; Hossner, F; Voyle, M; Ranson, J; Powles, J; Riley, G; Sanger, G. Synthesis and pharmacological activity of metabolites of the 5-HT4 receptor antagonist SB-207266. Bioorg Med Chem 2001, 9, 2119–2128. [Google Scholar]
- Hu, S; Tan, R; Hong, K; Yu, Z; Zhu, H. Methyl indole-3-carboxylate. Acta Cryst 2005, E61, 1654–1656. [Google Scholar]
- Bano, S; Ahmad, VU; Perveen, S; Bano, N; Shafiuddin; Shameel, M. Marine natural products; II. Chemical constituents of red alga Botryocladia Leptopoda. Planta Med 1987, 53, 117–118. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 12.19 | 11.52 | ||||
| (br s) | (br s) | |||||
| 2 | 8.45 | 8.44 | 8.22 | 8.69 | 8.68 | 7.86 |
| (d, J=2.0 Hz) | (s) | (s) | (s) | (s) | (s) | |
| 4 | 8.07 | 8.16 | 7.82 | 8.22 | 8.12 | 7.74 |
| (d, J=8.0 Hz) | (d, J=7.0 Hz) | (d, J=8.0 Hz) | (d, J=6.0 Hz) | (d, J=8.5 Hz) | (d, J=8.5 Hz) | |
| 5 | 7.40 | 7.27 | 6.74 | 7.25 | 7.36 | 6.68 |
| (dd, J=8.0, 2.0 Hz) | (t, J=7.0 Hz) | (dd, J=8.0, 2.0 Hz) | (t, J=6.0 Hz) | (dd, J=8.5, 2.0 Hz) | (dd, J=8.5, 2.0 Hz) | |
| 6 | 7.30 | 7.25 | ||||
| (t, J=7.0 Hz) | (t, J=6.0 Hz) | |||||
| 7 | 7.73 | 7.55 | 6.87 | 7.52 | 7.70 | 6.81 |
| (d, J=2.0 Hz) | (d, J=7.0 Hz) | (d, J=2.0 Hz) | (d, J=6.0 Hz) | (d, J=2.0 Hz) | (d, J=2.0 Hz) | |
| -OCH3 | 3.89 | 3.90 (s) | 3.87 | 3.76 | ||
| (s) | (s) | (s) | ||||
| -NH2 | 8.06 | 8.05 | ||||
| (br s) | (br s) | |||||
| 7.69 | 7.67 | |||||
| (br s) | (br s) | |||||
| -OH | 9.17 | |||||
| (br s) |
| position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 2 | 139.5 | 136.8 | 134.5 | 138.1 | 140.2 | 130.6 |
| 3 | 112.5 | 112.7 | 112.5 | 112.0 | 112.0 | 106.3 |
| 3a | 124.8 | 125.5 | 118.5 | 126.1 | 125.6 | 118.7 |
| 4 | 122.5 | 121.1 | 121.4 | 121.2 | 122.8 | 120.8 |
| 5 | 125.3 | 122.8 | 112.2 | 122.4 | 125.0 | 111.6 |
| 6 | 116.2 | 123.8 | 154.4 | 123.3 | 115.6 | 153.7 |
| 7 | 115.5 | 112.4 | 97.7 | 112.4 | 115.6 | 97.2 |
| 7a | 138.6 | 138.4 | 138.5 | 136.2 | 140.0 | 137.4 |
| 8 | 178.2 | 178.6 | a | 182.9 | 180.0 | 164.8 |
| 9 | 164.0 | 164.9 | 164.4 | 165.9 | 165.9 | |
| -OCH3 | 52.4 | 52.5 | 51.9 | 50.4 |
Share and Cite
Bao, B.; Zhang, P.; Lee, Y.; Hong, J.; Lee, C.-O.; Jung, J.H. Monoindole Alkaloids from a Marine Sponge Spongosorites sp. Mar. Drugs 2007, 5, 31-39. https://doi.org/10.3390/md502031
Bao B, Zhang P, Lee Y, Hong J, Lee C-O, Jung JH. Monoindole Alkaloids from a Marine Sponge Spongosorites sp. Marine Drugs. 2007; 5(2):31-39. https://doi.org/10.3390/md502031
Chicago/Turabian StyleBao, Baoquan, Ping Zhang, Yoonmi Lee, Jongki Hong, Chong-O. Lee, and Jee H. Jung. 2007. "Monoindole Alkaloids from a Marine Sponge Spongosorites sp." Marine Drugs 5, no. 2: 31-39. https://doi.org/10.3390/md502031
APA StyleBao, B., Zhang, P., Lee, Y., Hong, J., Lee, C.-O., & Jung, J. H. (2007). Monoindole Alkaloids from a Marine Sponge Spongosorites sp. Marine Drugs, 5(2), 31-39. https://doi.org/10.3390/md502031