Marine Toxins Targeting Ion Channels

Abstract
:1. Introduction
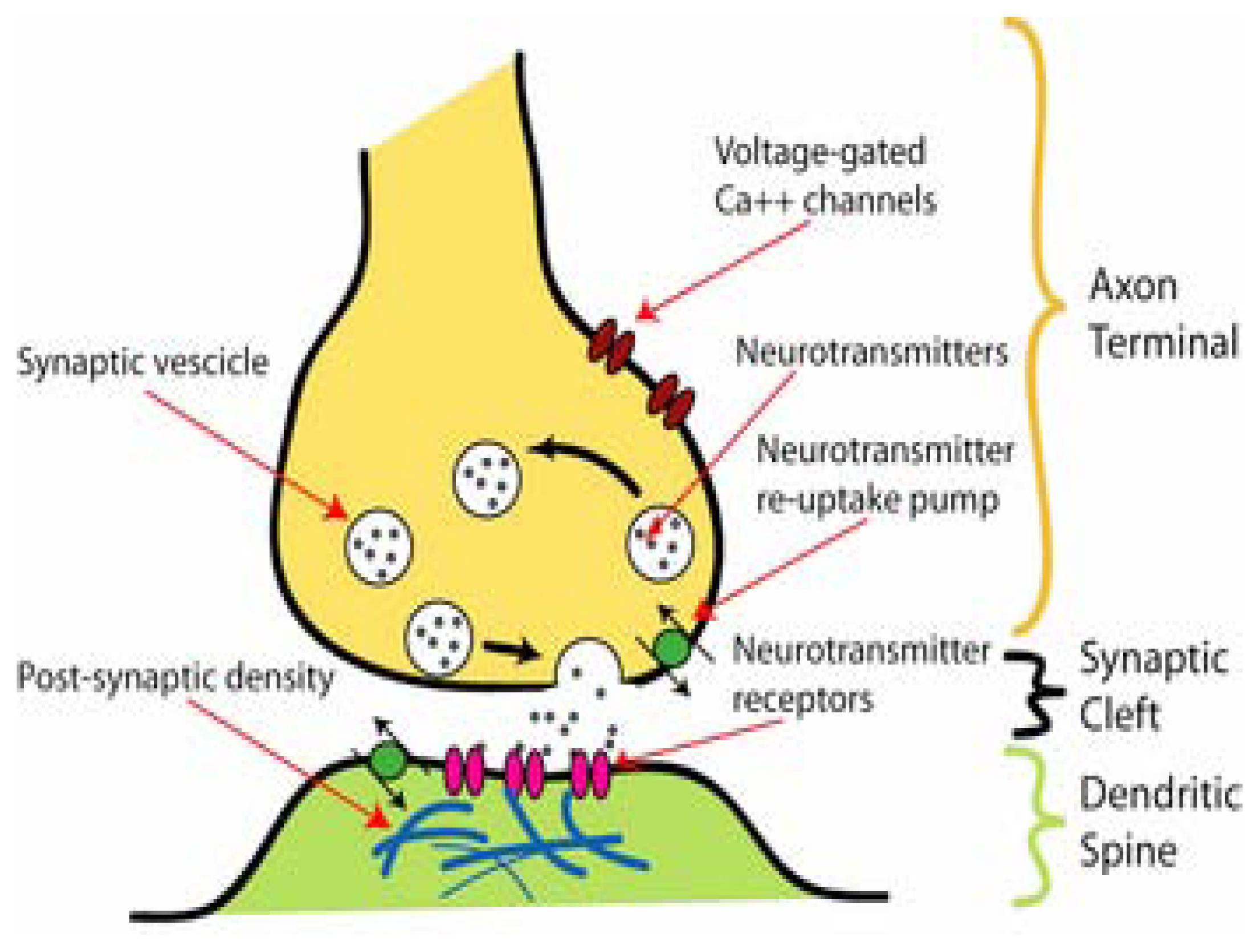
2. Ion channels
3. Voltage-Gated Ion Channels
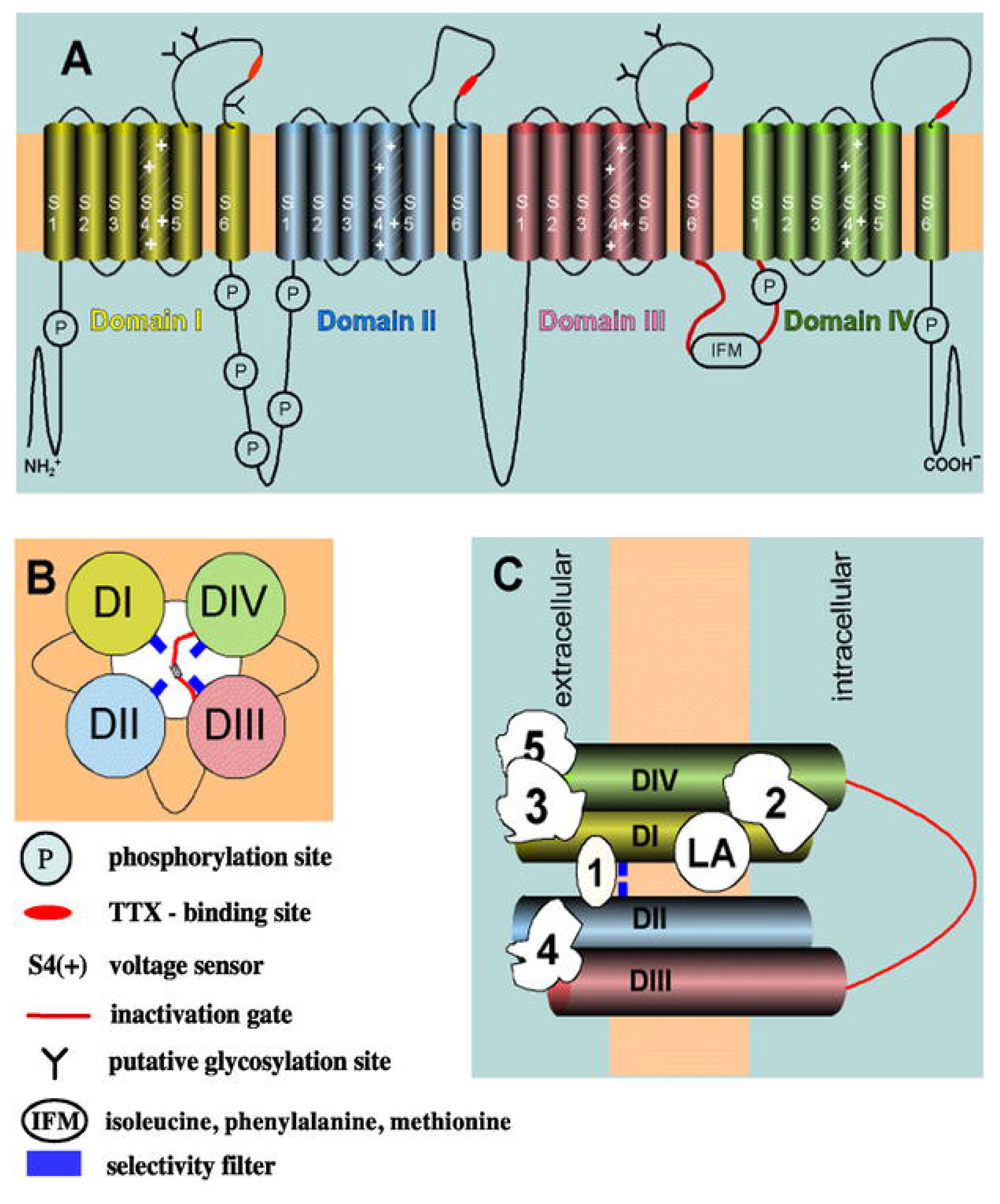
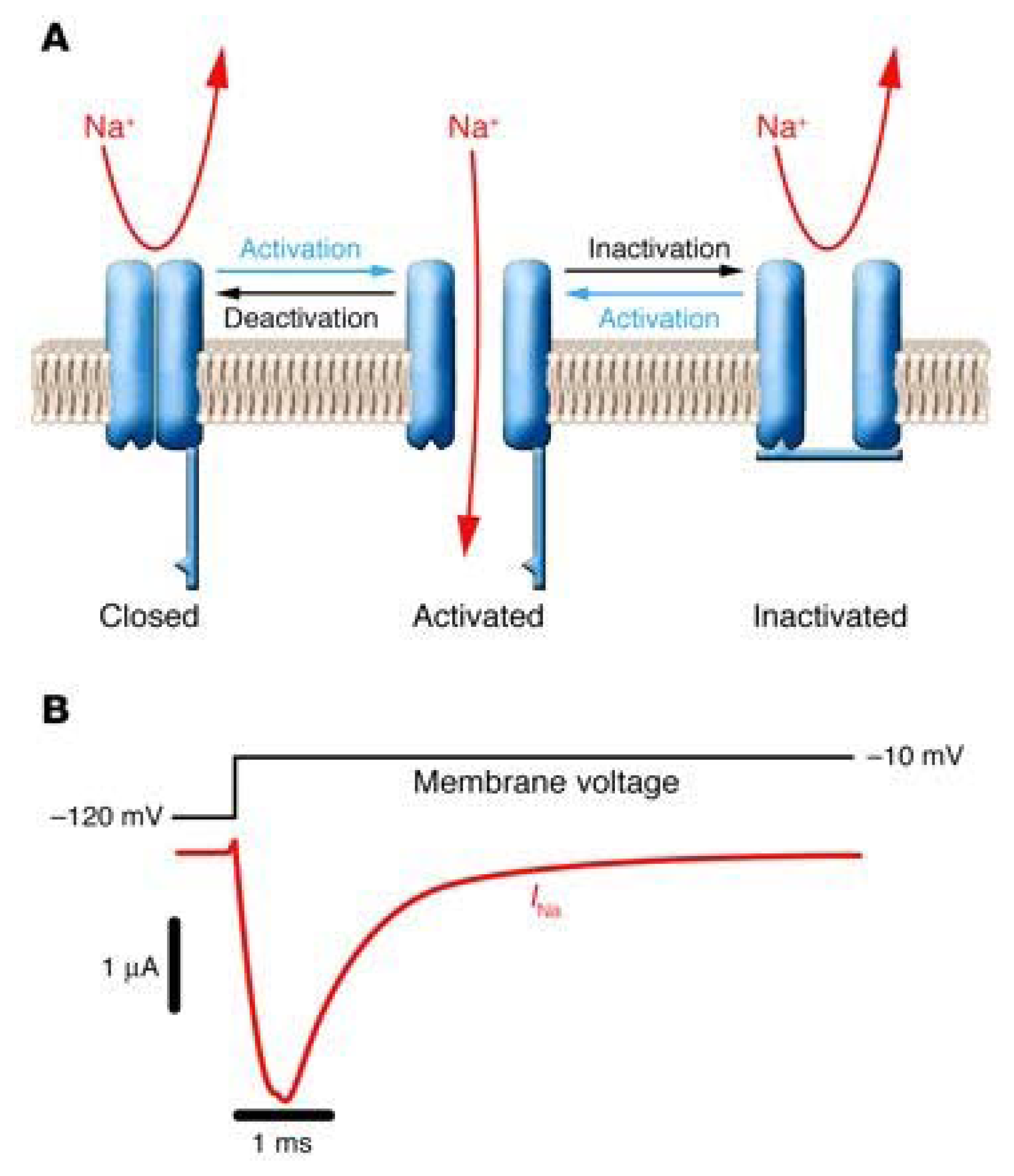
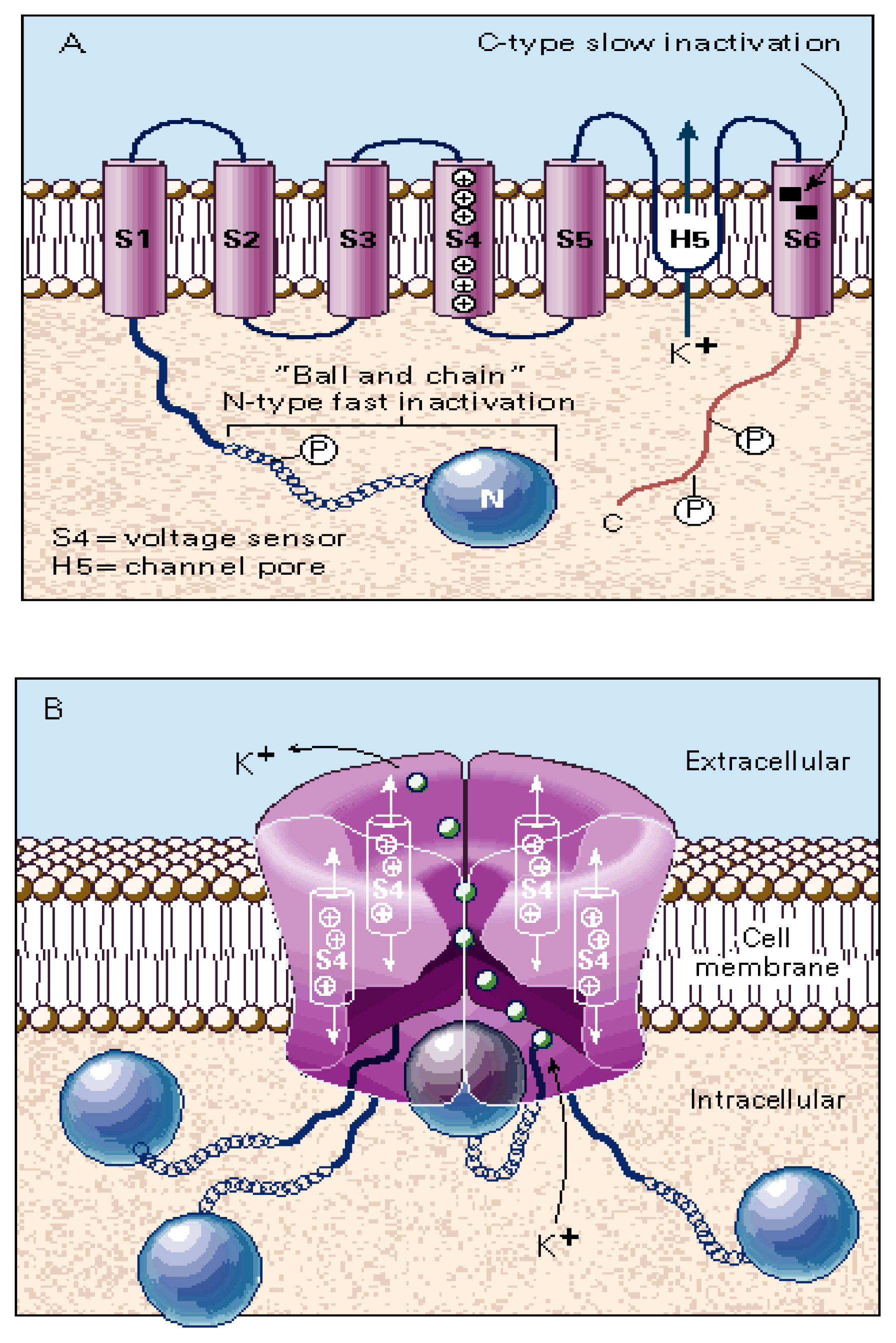
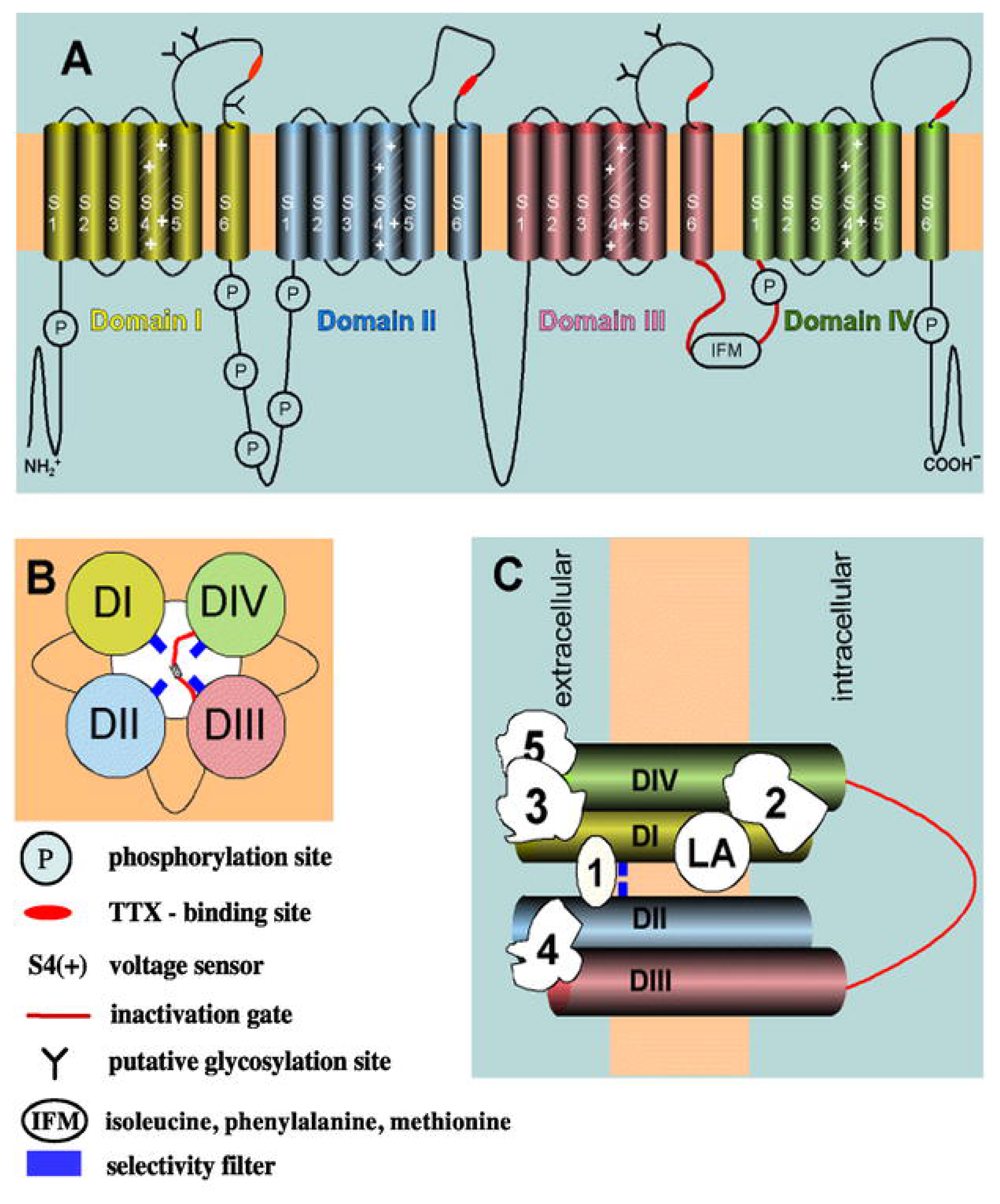
3.1. The Voltage-Gated Na+ Channel Superfamily
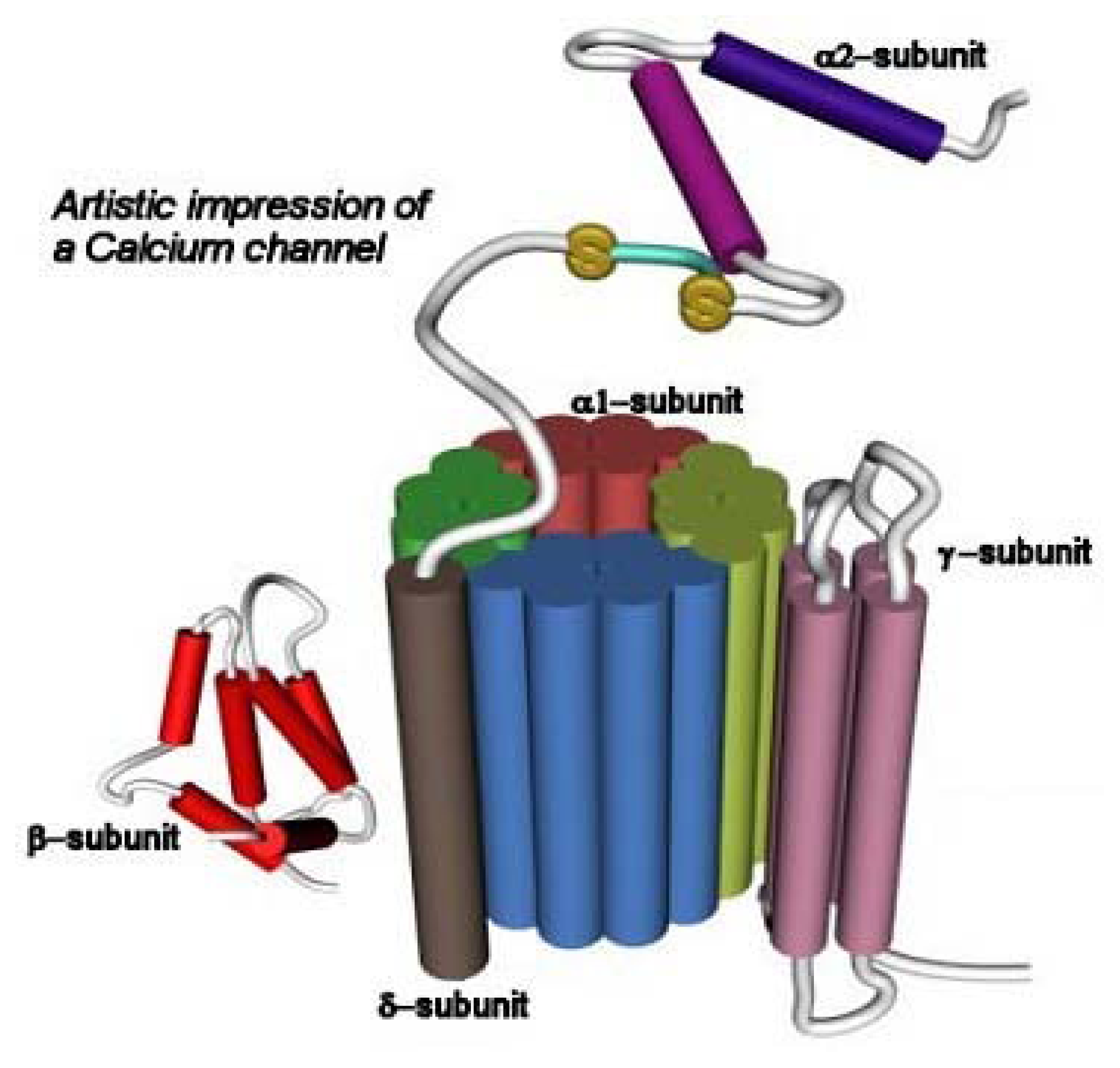
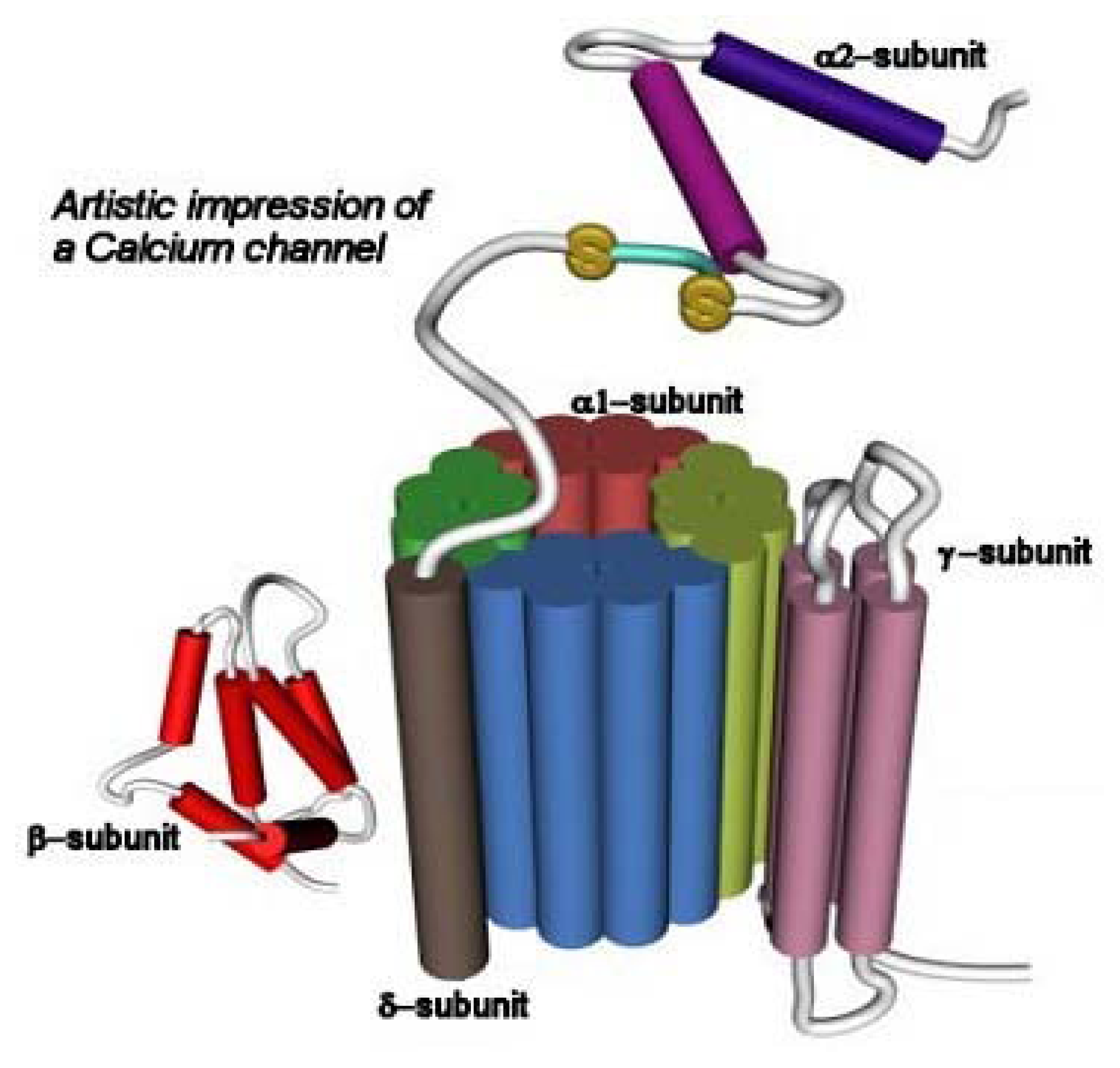
3.2. The Voltage-Gated Ca2+ Channel Superfamily
3.3. The Voltage-Gated K+ Channel Superfamily
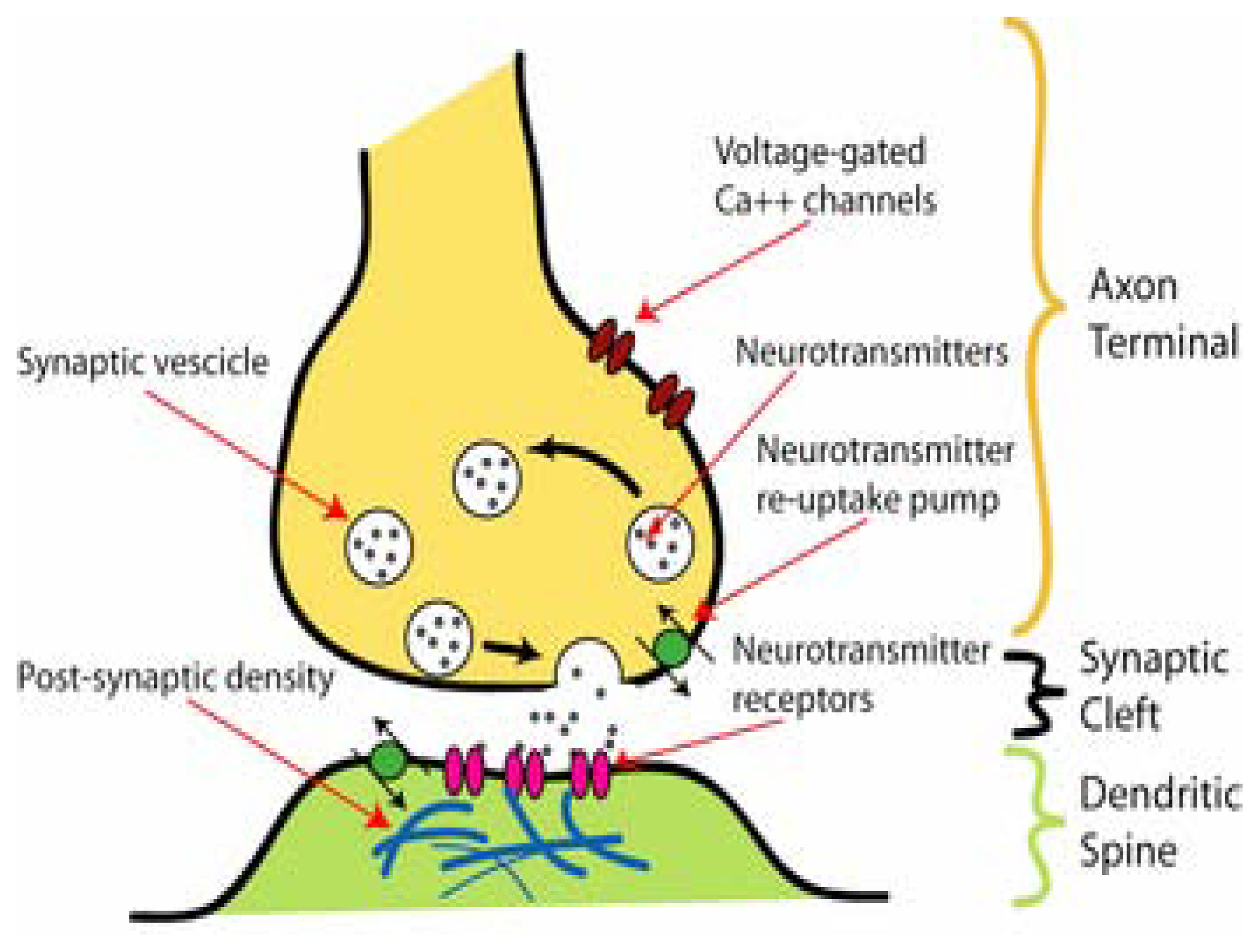
4. Ligand-Gated Ion Channels
4.1. Structural and Functional Similitude Among LGIC Superfamilies
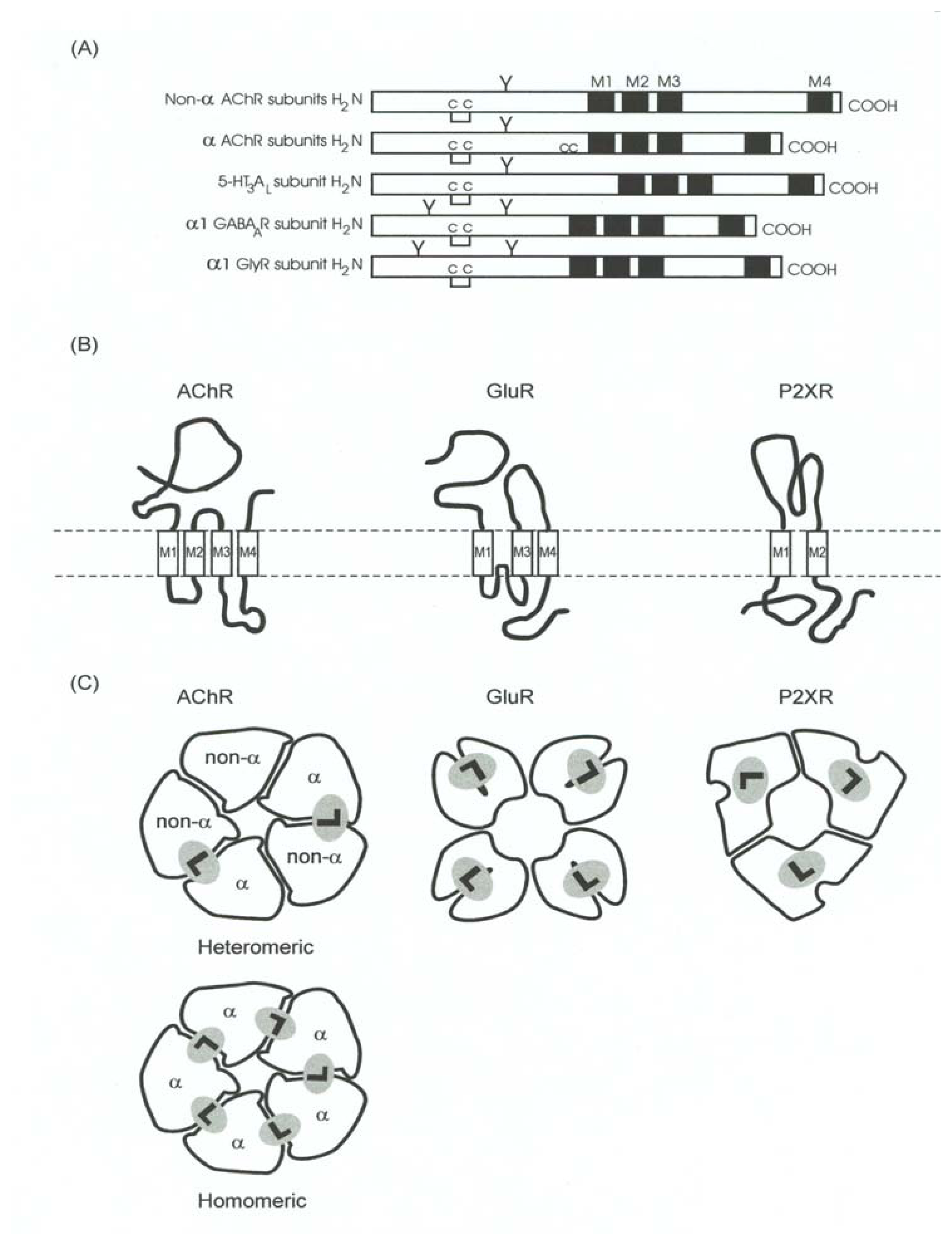
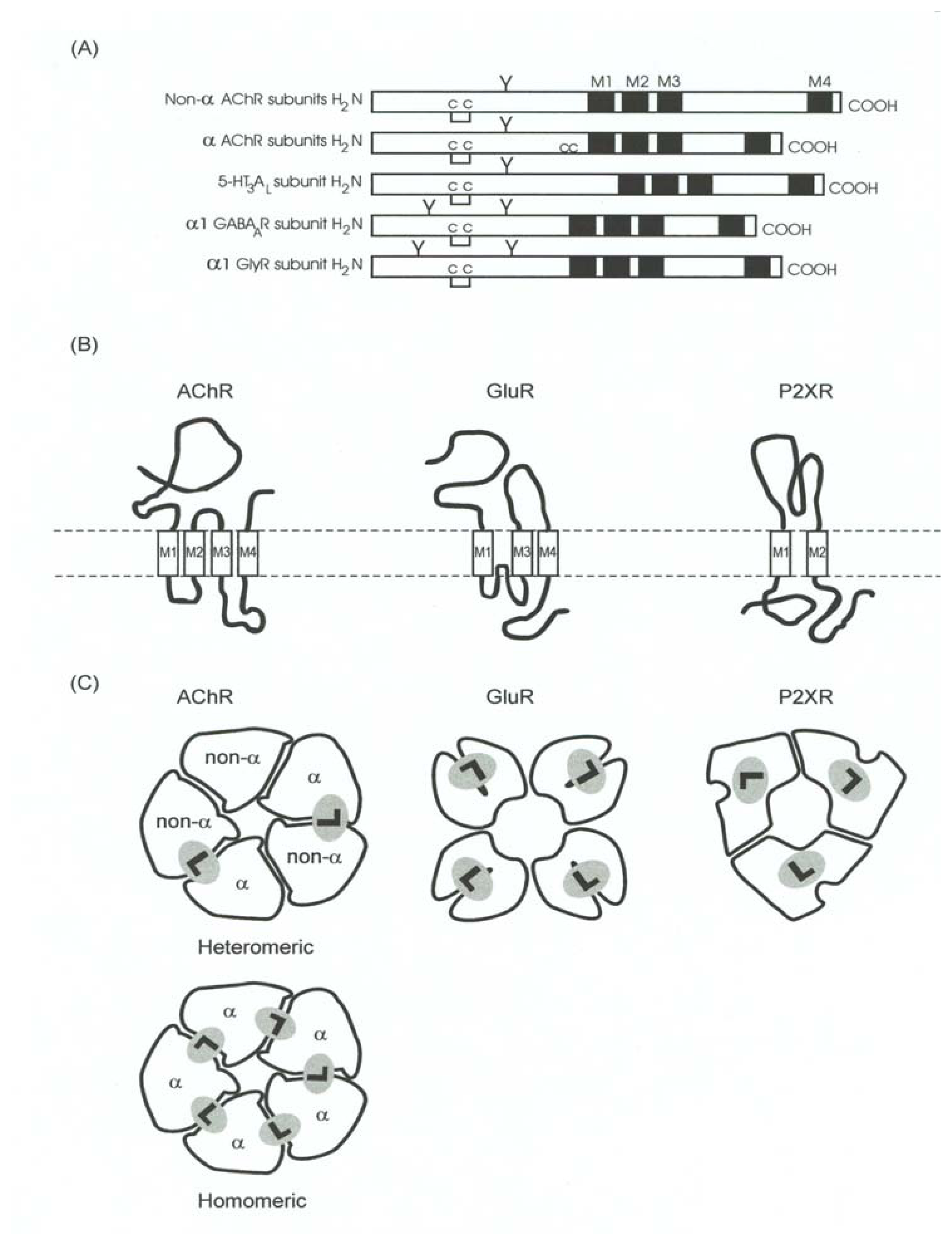
5. The Cys-Loop or Nicotinicoid Receptor Superfamily
- (1)
- High homology in amino acid [63,66] and pair base [78] sequence. Homology of ~30% is observed among different subunits, whereas ~70% homology is obtained considering just one subunit from different species. The genetic evidence also suggests that all members have evolved from the same ancestor protein [63,66,67,78].
- (2)
- A pentameric quaternary structure. This structural characteristic has been confirmed by electron microscopy of tubular crystals of native membranes containing high density of AChRs prepared from Torpedo electric organs. The enduring effort from Unwin’s laboratory has recently rendered a receptor three-dimensional image with a resolution of 4 Å [99].
- (3)
- A large N-terminal domain formed by ~220 residues located extracellularly, bearing the neurotransmitter binding sites. This funnel-shaped domain has an outer diameter of 70–80 Å and protrudes ~60 Å above the lipid membrane surface. This domain had been previously modeled mainly as antiparallel β-sheet secondary structures arranged perpendicular to the membrane surface [66]. This model was recently confirmed by determining the crystallographic structure of the AChBP from L. stagnalis[20], which resembles the extracellular portion of the AChR, in particular the α7 homomeric receptor, by 24%.
- (4)
- An interfacial location for the neurotransmitter binding sites. The comparison with the different AChBP structures, which also bind AChR agonists (e.g., ACh, CCh, nicotine, etc.) and competitive antagonists [e.g., α-bungarotoxin (α-BTx), d-tubocurarine, etc.], have confirmed that ligand sites are located between two subunits [18,20,24,25,27,48,87,106] (reviewed in [89]). Additional comparisons with other members of the superfamily including GABAARs [28,33,52,96], GlyRs [62], and 5-HT3Rs [83,84] also support such interfacial location.
- (5)
- An agonist binding site comprised by several loops (A-F) (reviewed in [7]), which have been re-named as “binding segments A-F” because some segments are actually in β-sheet rather than a loop conformation (reviewed in [69]). Binding segments A-C form the principal component of the agonist binding domain (which is located in the α subunit from AChRs and homologous subunits from other receptors), whereas binding segments D-F form the secondary component (which is located in the non-α subunit from AChRs and homologous subunits from other receptors). The evidence that supports this structural feature was provided by photoaffinity labeling and sitedirected mutagenesis experiments (reviewed in [7]), and more lately by cysteine- [92] and lysinescanning mutagenesis [88], as well as by comparative studies using the recently found AChBP structures as templates [see paragraph (4)].
- (6)
- Four transmembrane segments (M1–M4) per subunit (twenty transmembrane segments in total), where the M2 transmembrane segment from each subunit (five M2 segments in total) forms the wall of the ion channel. The ion channel is the main domain for the action of noncompetitive antagonists from endogenous as well as exogenous origin (reviewed in [5,6,9,10,12–14]). The transmembrane domain (including the phospholipid headgroup region) measure about 40 Å. Earlier hydropathy analysis suggested that the AChR have four transmembrane segments and that each segment is mainly in an α-helix secondary structure. This result has been confirmed by electron microscopy aided by computational methods [99,100], photoaffinity labeling (reviewed in [13,14]), substituted cysteine accessibility method (reviewed in [21,51], and references therein), tryptophan-scanning mutagenesis, as well as by circular dichroism and nuclear magnetic resonance spectroscopy (reviewed in [8,21]). However, these ion channels contain significant amounts of non-helical structures as well. For instance, β-sheet secondary structures have been observed in the external portion of M1 and other segments of M3.
- (7)
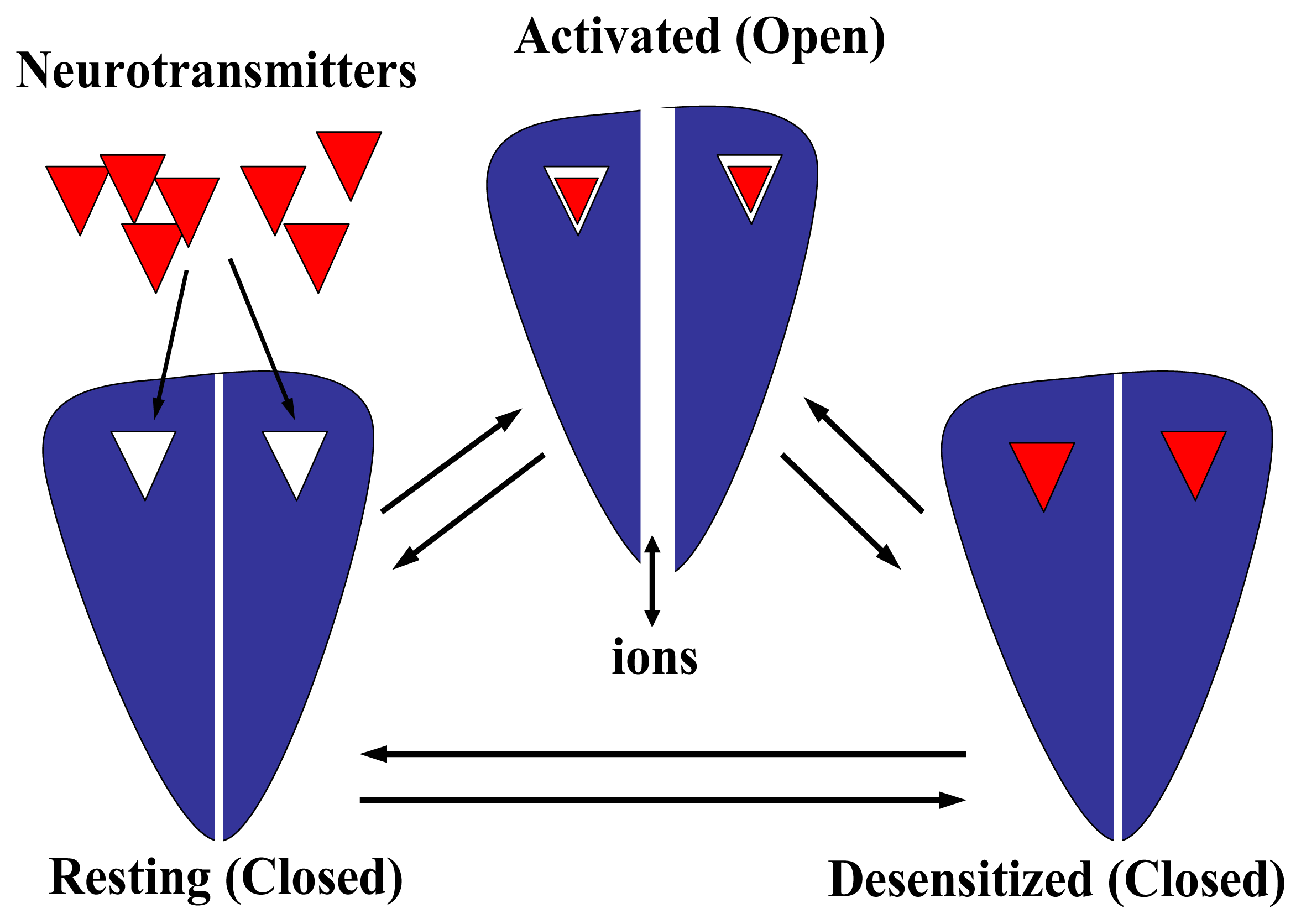
- A “hydrophobic girdle”, the so-called “kink”, in the middle of the M2 segment that is formed by residues that are conserved through all superfamily member subunits. This hydrophobic girdle constitutes the “gate” that is occluding ion flux in the resting (closed) state. In the activated (open) state, the rotation of one or two of the M2 segments would disrupt the hydrophobic interactions with adjacent helices allowing the passage of ions through the channel. In this regard, anticlockwise rotations in the extracellular domain as well as in the M2 domain were observed in refined electron microscopy images when the AChR was activated [100].
- (8)
- A dynamic mechanism, called “ion channel gating”, where agonist binding provokes a conformational change in the protein that finally leads the opening of the intrinsically-associated ion channel that is located far apart from the proper ligand binding domain. This modular mechanism has been studied in detail in the AChR by Auerbach’s laboratory. The main conclusion from these studies is that the chemical signal triggered by ACh binding is locally transmitted to the extracellular loop formed between M2 and M3 segments, the M2–M3 linker (reviewed in [21,30,31]), as a “conformational wave” that finally reaches the ion channel [45] (reviewed in [44]).Based on the evidence provided by low resolution electron microscopy, a concerted anticlockwise rotation of all extracellular domains has been hypothetized as a plausible mechanism for gating [98]. Additional evidence has been shown in other receptor members as well. For instance, electrostatic interactions were found between a Lys located in the M2–M3 linker and acidic residues located in the Cys-loop and in the loop 2 from the GABAAR [52]. Homologous residues in the GlyR [1] and the 5-HT3R (reviewed in [72]) were also found to be critical in the gating process.
- (9)
- A desensitization process, where the prolonged exposure of the receptor to agonists as well as some competitive and noncompetitive antagonists produces a conformational change in the protein that finally leads to a refractory state where the agonist does not open the ion channel (reviewed in [82]). This conformational state is depicted in Fig. 6. Qualitatively, the desensitization process is similar among the members of the Cys-loop receptor superfamily. Desensitization might be very important in the prevention of citotoxicity, in the process of nicotine addiction, or in other pathological states (reviewed in [82]). For instance, a Ser248 to Phe mutation in the pore domain of the α4 subunit results in faster desensitization of α4β2 AChRs causing a type of human cortical epilepsy [107].
- (10)
- A small cytoplasmic domain formed by the loop between M3 and M4 of variable length (from 80 to 265 amino acids). This region contains the greatest sequence divergence among the members of this receptor superfamily. This region is very important for receptor modulation by phosphorylation-deposphorylation processes (reviewed in [21,49,93]). For instance, tyrosine kinases have been found to reduce nicotine-induced currents, whereas phosphotyrosine phosphatases have been found to enhance nicotine-induced currents in chromaffin cells [105]. Receptor phosphorylation by serine/threonine and/or tyrosine kinases also increases the desensitization rate in several members of this LGIC superfamily (reviewed in [49,93]).The anchorage of certain receptors to the cytoskeleton is also mediated by the interaction of non-receptor proteins with the cytoplasmic domain. Among them we can name rapsyn for AChRs (reviewed in [108]) and the tubulin-binding element gephyrin for both GABAARs and GlyRs (reviewed in [21,57,73]). Protein-protein contacts may affect receptor assembly, trafficking, clustering, targeting, modulation, and turnover, among other processes.Finally, the cytoplasmic domain is considered an additional determinant for ion channel conductance (reviewed in [80]). Using different 5-HT3R chimaeras, a novel determinant of single-channel conductance was located to a putative amphipathic helix (the ‘HA stretch’), apart from the M2 domain. Key residues may be components of narrow openings within the inner vestibule of the channel, which contribute to the permeation pathway.
- (11)
- And finally, an extracellular location for the very short C-terminal. This small area is involved, at least in certain neuronal-type AChRs (e.g., α4β2 and α4β4), in the binding of neurosteroids [34,79] (reviewed in [5,6,9,10,13,14]). Neurosteroids are molecules synthesized in the CNS that modulate some of the functions of this receptor superfamily. For instance, 17β-estradiol may enhance the activity of α4β2 and α4β4 neuronal AChRs [34,79] (reviewed in [5,6,9,10,13,14,49,60]). GABAAR function is also enhanced by different neurosteroids (reviewed in [14,16]), which presents therapeutical opportunities for the treatment of several mood disorders. Opposite to that, neurosteroids other than 17β-estradiol (e.g., progesterone, hydrocortisone, etc.) inhibit noncompetitively several AChRs by interacting with the lipid-protein interface (reviewed in [5,6,9,10,13,14,49]).
5.1. The Nicotinic Acetylcholine Receptor Family
6. Conclusions
Acknowledgements
- Samples Availability: Available from the authors.
References
- Absalom, N. L.; Lewis, T. M.; Kaplan, W.; Pierce, K. D.; Schofield, P. R. Role of charged residues in coupling ligand binding and channel activation in the extracellular domain of the glycine receptor. J. Biol. Chem 2003, 278, 50151–50157. [Google Scholar]
- Ackerman, M. J.; Clapham, D. E. Ion channels − Basic science and clinical disease. N. Engl. J. Med 1997, 336, 1575–1586. [Google Scholar]
- Agnew, W. S.; Levinson, S. R.; Brabson, J. S.; Raftery, M. A. Purification of the tetrodotoxin-binding component associated with the voltage-sensitive sodium channel from Electrophorus electricus electroplax membranes. Proc. Natl. Acad. Sci. USA 1978, 75, 2602–2610. [Google Scholar]
- Alkondon, M.; Albuquerque, E. X. The nicotinic acetylcholine receptor subtypes and their function in the hippocampus and cerebral cortex. Prog. Brain Res 2004, 145, 109–120. [Google Scholar]
- Arias, H. R. Noncompetitive inhibition of nicotinic acetylcholine receptors by endogenous molecules. J. Neurosci. Res 1998, 52, 369–379. [Google Scholar]
- Arias, H. R. Binding sites for exogenous and endogenous non-competitive inhibitors of the nicotinic acetylcholine receptor. Biochim. Biophys. Acta Rev. Biomembr 1998, 1376, 173–220. [Google Scholar]
- Arias, H. R. Localization of agonist and competitive antagonist binding sites on nicotinic acetylcholine receptors. Neurochem. Int 2000, 36, 595–645. [Google Scholar]
- Arias, H. R. Biological and Biophysical Aspects of Ligand-Gated Ion Channel Receptor Superfamilies; Arias, H. R., Ed.; Research Signpost: India, 2006; Volume Chapter 1, pp. 1–25. [Google Scholar]
- Arias, H. R.; Bhumireddy, P. Anesthetics as chemical tools to study the structure and function of nicotinic acetylcholine receptors. Curr. Protein Pept. Sci 2005, 6, 451–472. [Google Scholar]
- Arias, H. R.; Bhumireddy, P.; Bouzat, C. Molecular mechanisms and binding site locations for noncompetitive antagonists of nicotinic acetylcholine receptors. Int. J. Biochem. Cell Biol. 2006, in press.. [Google Scholar]
- Arias, H. R.; Blanton, M. P. α-Conotoxins. Int. J. Biochem. Cell Biol 2000, 32, 1017–1028. [Google Scholar]
- Arias, H. R.; Blanton, M. P. Molecular and physicochemical aspects of local anesthetics acting on nicotinic acetylcholine receptor-containing membranes. Mini Rev. Med. Chem 2002, 2, 385–410. [Google Scholar]
- Arias, H. R.; Kem, W. R.; Trudell, J. R.; Blanton, M. P. Unique general anesthetic binding sites within distinct conformational states of the nicotinic acetylcholine receptor. Int. Rev. Neurobiol 2002, 54, 1–50. [Google Scholar]
- Arias, H. R.; Machu, T. K.; Trudell, J. R.; Blanton, M. P. Recent Research Developments in Biophysical Chemistry; Condat, C. A., Baruzzi, A., Eds.; Research Signpost: India, 2002; pp. 123–154. [Google Scholar]
- Becher, A.; Drenckhahn, A.; Pahner, I.; Margittai, M.; Jahn, R.; Ahnert-Hilger, G. The synaptophysin-synaptobrevin complex is developmentally upregulated in cultivated neurons but is absent in neuroendocrine cells. J. Neurosci 1999, 19, 1922–1931. [Google Scholar]
- Belelli, D.; Lambert, J. J. Neurosteroids: endogenous regulators of the GABAA receptor. Nat. Rev. Neurosci 2005, 6, 565–575. [Google Scholar]
- Bezanilla, F. Voltage-gated ion channels. IEEE Trans. Nanobiosci 2005, 4, 34–48. [Google Scholar]
- Bourne, Y.; Talley, T. T.; Hansen, S. B.; Taylor, P.; Marchot, P. Crystal structure of a Cbtx- AChBP complex reveals essential interactions between snake α-neurotoxins and nicotinic receptors. EMBO J 2005, 24, 1512–1522. [Google Scholar]
- Brandon, N.; Jovanovic, J.; Moss, S. Multiple roles of protein kinases in the modulation of γ-aminobutyric acidA receptor function and cell surface expression. Pharmacol. Ther 2002, 94, 113–122. [Google Scholar]
- Brejc, K.; van Dijk, W. J.; Klassen, R.V.; Schuumans, M.; van Der Oost, J.; Smit, A. B.; Sixma, T. K. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001, 403, 269–276. [Google Scholar]
- Cascio, M. Structure and function of the glycine receptor and related nicotinicoid receptors. J. Biol. Chem 2004, 279, 19383–19386. [Google Scholar]
- Catterall, W. A.; Goldin, A. L.; Waxman, S. G. International Union of Pharmacology. International Union of Pharmacology. XXXIX. Compendium of voltage-gated ion channels: sodium channels. Pharmacol. Rev 2003, 55, 575–578. [Google Scholar]
- Catterall, W. A.; Striessnig, J.; Snutch, T. P.; Perez-Reyes, E. International Union of Pharmacology. International Union of Pharmacology. XL. Compendium of voltage-gated ion channels: calcium channels. Pharmacol. Rev 2003, 55, 579–581. [Google Scholar]
- Celie, P. H. N.; Klaassen, R.V.; van Rossum-Fikkert, S. E.; van Elk, R.; van Nierop, P.; Smit, A. B.; Sixma, T. K. Crystal structure of acetylcholine-binding protein from Bulinus truncatus reveals the conserved structural scaffold and sites of variation in nicotinic acetylcholine receptors. Biol. Chem 2005, 280, 26457–26466. [Google Scholar]
- Celie, P. H. N.; van Rossum-Fikkert, S. E.; van Dijk, W. J.; Brejc, K.; Smit, A. B.; Sixma, T. K. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 2004, 41, 907. [Google Scholar]
- Changeux, J-P.; Edelstein, S. J. Allosteric receptors after 30 years. Neuron 1998, 21, 959–980. [Google Scholar]
- Chou, K-C. Insights from modelling the 3D structure of the extracellular domain of α7 nicotinic acetylcholine receptor. Biochem. Biophys. Res. Commun 2004, 319, 433–438. [Google Scholar]
- Chou, K-C. Modelling extracellular domains of GABA-A receptors: subtypes 1, 2, 3, and 5. Biochem. Biophys. Res. Commun 2004, 316, 636–642. [Google Scholar]
- Clement, J. G. Toxicology and pharmacology of bispyridium oximes-insight into the mechanism of action vs Soman poisoning in vivo. Fundam. Appl. Toxicol 1981, 1, 193–202. [Google Scholar]
- Colquhoun, D.; Sivilotti, L. G. Function and structure in glycine receptors and some of their relatives. Trends Neurosci 2003, 27, 337–344. [Google Scholar]
- Condat, C. A.; Lamberti, P. W.; Arias, H. R. Biological and Biophysical Aspects of Ligand- Gated Ion Channel Receptor Superfamilies; Arias, H. R., Ed.; Research Signpost: India, 2006; Volume Chapter 4. [Google Scholar]
- Connolly, C. N.; Wafford, K. A. The Cys-loop superfamily of ligand-gated ion channels: the impact of receptor structure on function. Biochem. Soc. Trans 2004, 32, 529–534. [Google Scholar]
- Cromer, B. A.; Morton, C. J.; Parker, M. W. Anxiety over GABAA receptor structure relieved by AChBP. Trends Biochem. Sci 2002, 27, 280–287. [Google Scholar]
- Curtis, L.; Buisson, B.; Bertrand, S.; Bertrand, D. Potentiation of human α4β2 neuronal nicotinic acetylcholine receptor by estradiol. Mol. Pharmacol 2002, 61, 127–135. [Google Scholar]
- Dahan, M.; Levi, S.; Luccardini, C.; Rostaing, P.; Riveau, B.; Triller, A. Diffusion dynamics of glycine receptors revealed by single-quantum dot tracking. Science 2003, 302, 442–445. [Google Scholar]
- Denac, H.; Mevissen, M.; Scholtysik, G. Structure, function and pharmacology of voltage-gated sodium channels. Naunyn-Schmiedeberg’s Arch. Pharmacol 2000, 362, 453–479. [Google Scholar]
- Escoubas, P.; Diochot, S.; Corzo, G. Structure and pharmacology of spider venos neurotoxins. Biochimie 2000, 82, 893–907. [Google Scholar]
- Farber, L.; Haus, U.; Spath, M.; Drechsler, S. Physiology and pathophysiology of the 5-HT3 receptor. Scand. J. Rheumatol. Suppl 2004, 119, 2–8. [Google Scholar]
- Farrant, M.; Nusser, Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat. Rev. Neurosci 2005, 6, 215–229. [Google Scholar]
- Fatt, P.; Katz, B. The electrical properties of crustacean muscle fibers. J. Physiol 1953, 120, 171–204. [Google Scholar]
- George, A. L., Jr. Inherited disorders of voltage-gated sodium channels. J. Clin. Invest 2005, 115, 1990–1999. [Google Scholar]
- Ghijsen, W. E.; Leenders, A. G. M. Differential signaling in presynaptic neurotransmitter release. Cell Mol. Life Sci 2005, 62, 937–954. [Google Scholar]
- Gotti, C.; Clementi, F. Neuronal nicotinic receptors: from structure to pathology. Progr. Neurobiol 2004, 74, 363–396. [Google Scholar]
- Grosman, C. Linear free-energy relationships and the dynamics of gating in the acetylcholine receptor channel. A Φ-value analysis of an allosteric transition at the single-molecule level. J. Biol. Phys 2002, 28, 267–277. [Google Scholar]
- Grosman, C.; Zhou, M.; Auerbach, A. Mapping the conformational wave of acetylcholine receptor channel gating. Nature 2000, 403, 773–776. [Google Scholar]
- Gutman, G. A.; Chandy, K. G.; Adelman, J. P.; Aiyar, J.; Bayliss, D. A.; Clapham, D. E.; Covarriubias, M.; Desir, G. V.; Furuichi, K.; Ganetzky, B.; Garcia, M. L.; Grissmer, S.; Jan, L. Y.; Karschin, A.; Kim, D.; Kuperschmidt, S.; Kurachi, Y.; Lazdunski, M.; Lesage, F.; Lester, H. A.; McKinnon, D.; Nichols, C. G.; O’Kelly, I.; Robbins, J.; Robertson, G. A.; Rudy, B.; Sanguinetti, M.; Seino, S.; Stuehmer, W.; Tamkun, M. M.; Vandenberg, C. A.; Wei, A.; Wulff, H.; Wymore, R. S. International Union of Pharmacology. International Union of Pharmacology. XLI. Compendium of voltage-gated ion channels: potassium channels. Pharmacol. Rev 2003, 55, 583–586. [Google Scholar]
- Hansen, S. B.; Talley, T. T.; Radić, Z.; Taylor, P. Structural and ligand recognition characteristics of an acetylcholine-binding protein from Aplysia californica. J. Biol. Chem 2004, 279, 24197–24202. [Google Scholar]
- Harel, M.; Kasher, R.; Nicolas, A.; Guss, J. M.; Balass, M.; Fridkin, M.; Smit, A.B.; Brejc, K.; Sixma, T. K.; Katchalski-Katzir, E.; Sussman, J. L.; Fuchs, S. The binding site of acetylcholine receptor as visualized in the X-Ray structure of a complex between α-bungarotoxin and a mimotope peptide. Neuron 2001, 32, 265. [Google Scholar]
- Hogg, R. C.; Raggenbass, M.; Bertrand, D. Nicotinic acetylcholine receptors: from structure to brain function. Rev. Physiol. Biochem. Pharmacol 2003, 147, 1–46. [Google Scholar]
- Huh, K. H.; Fuhrer, C. Clustering of nicotinic acetylcholine receptors: from the neuromuscular junction to interneuronal synapses. Mol. Neurobiol 2002, 25, 79–112. [Google Scholar]
- Karlin, A. Emerging structure of the nicotinic acetylcholine receptors. Nat. Rev. Neurosci 2002, 3, 102–114. [Google Scholar]
- Kash, T. L.; Trudell, J. R.; Harrison, N. L. Structural elements involved in activation of the gamma-aminobutyric acid type A (GABAA) receptor. Biochem. Soc. Trans 2004, 32, 540–546. [Google Scholar]
- Kauferstein, S.; Huys, I.; Lamthanh, H.; Stöcklin, R.; Sotto, F.; Menez, A.; Tytgat, J.; Mebs, D. A novel conotoxin inhibiting vertebrate voltage-sensitive potassium channels. Toxicon 2003, 42, 43–52. [Google Scholar]
- Kažić, T.; Gojković-Bukarica, L. Ion channels and drug development. Focus on potassium channels and their modulators. Facta Universitatis 1999, 6, 23–30. [Google Scholar]
- Keramidas, A.; Moorhouse, A. J.; Schofield, P. R.; Barry, P. H. Ligand-gated ion channels: mechanisms underlying ion selectivity. Progr. Biophys. Mol. Biol 2004, 86, 161–204. [Google Scholar]
- Kittler, J. T.; Moss, S. J. Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr. Opin. Neurobiol 2003, 13, 341–347. [Google Scholar]
- Kneussel, M.; Betz, H. Receptors, gephyrin and gephyrin-associated proteins: novel insights into the assembly of inhibitory postsynaptic membrane specializations. J. Physiol 2000, 525, 1–9. [Google Scholar]
- Koles, L.; Wirkner, K.; Illes, P. Modulation of ionotropic glutamate receptor channels. Neurochem. Res 2001, 26, 925–932. [Google Scholar]
- Kullmann, D. M.; Ruiz, A.; Rusakov, D. M.; Scott, R.; Semyanov, A.; Walker, M. C. Presynaptic, extrasynaptic and axonal GABAA receptors in the CNS: where and why? Prog. Biophys. Mol. Biol 2005, 87, 33–46. [Google Scholar]
- Lambert, J. J.; Belelli, D.; Harney, S. C.; Peters, J. A.; Frenguelli, B. G. Modulation of native and recombinant GABAA receptors by endogenous and synthetic neuroactive steroids. Brain Res. Brain Res. Rev 2001, 37, 68–80. [Google Scholar]
- Langley, J. N. On the reaction of cells and of nerve endings to certain poisons, chiefly as regards the reaction of striated muscle to nicotine and to curare. J. Physiol (London) 1905, 33, 374–413. [Google Scholar]
- Laube, B.; Maksay, G.; Schemm, R.; Betz, H. Modulation of glycine receptor function: a novel approach for therapeutic intervention at inhibitory synapses? Trends Pharmacol. Sci 2002, 23, 519–527. [Google Scholar]
- Le Novére, N.; Changeux, J-P. Molecular evolution of the nicotinic acetylcholine receptor: an example of multigene family in excitable cells. J. Mol. Evol 1995, 40, 155–172. [Google Scholar]
- Le Novére, N.; Changeux, J-P. LGICdb: the ligand-gated ion channel database. Nucleic Acids Res 2001, 29, 294–295. [Google Scholar]
- Le Novére, N.; Changeux, J-P. The ligand gated ion channel database: an example of a sequence database in neuroscience. Philos. Trans. R. Soc. Lond. B Biol. Sci 2001, 356, 1121–1130. [Google Scholar]
- Le Novére, N.; Corringer, P. J.; Changeux, J-P. Improved secondary structure predictions for a nicotinic receptor subunit: incorporation of solvent accessibility and experimental data into a twodimensional representation. Biophys. J 1999, 76, 2329–2345. [Google Scholar]
- Le Novére, N.; Corringer, P. J.; Changeux, J-P. The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J. Neurobiol 2002, 53, 447–456. [Google Scholar]
- Leonard, S.; Bertrand, D. Neuronal nicotinic receptors: from structure to function. Nicotine Tobacco Res 2001, 3, 203–223. [Google Scholar]
- Lester, H. A.; Dibas, M. I.; Dahan, D. S.; Leite, J. F.; Dougherty, D. A. Cys-loop receptors: new twists and turns. Trends Neurosci 2004, 27, 329–336. [Google Scholar]
- Lloyd, G. K.; Williams, M. Neuronal nicotinic acetylcholine receptors as novel drug targets. J. Pharmacol. Exp. Ther 2000, 292, 461–467. [Google Scholar]
- Long, S. B.; Campbell, E. B.; Mackinnon, R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005, 309, 897–903. [Google Scholar]
- Lummis, S. C. R. The transmembrane domain of the 5-HT3 receptor: its role in selectivity and gating. Biochem. Soc. Trans 2004, 32, 535–539. [Google Scholar]
- Lynch, J. W. Molecular structure and function of the glycine receptor chloride channel. Physiol. Rev 2004, 84, 1051–1095. [Google Scholar]
- MacDermott, A. B.; Role, L. W.; Siegelbaum, S. A. Presynaptic ionotropic receptors and the control of transmitter release. Annu. Rev. Neurosci 1999, 22, 443–485. [Google Scholar]
- Millar, N. S. Assembly and subunit diversity of nicotinic acetylcholine receptors. Biochem. Soc. Trans 2003, 31, 869–874. [Google Scholar]
- Miller, C. An overview of the potassium channel family. Genome Biol. 2000, 4, 0004:1–0004:5. [Google Scholar]
- Miller, R. J. Presynaptic receptors. Annu. Rev. Pharmacol. Toxicol 1998, 38, 201–227. [Google Scholar]
- Ortells, M. O.; Lunt, G. G. Evolutionary history of the ligand-gated ion-channel superfamily of receptors. Trends Neurosci 1995, 18, 121–127. [Google Scholar]
- Paradiso, K.; Zhang, J.; Steinbach, J. H. The C terminus of the human nicotinic α4β2 receptor forms a binding site required for potentiation by an estrogenic steroid. J. Neurosci 2001, 21, 6561–6568. [Google Scholar]
- Peters, J. A.; Kelley, S. P.; Dunlop, J. I.; Kirkness, E. F.; Hales, T. G.; Lambert, J. J. The 5- hydroxytryptamine type 3 (5-HT3) receptor reveals a novel determinant of single-channel conductance. Biochem. Soc. Trans 2004, 32, 547–552. [Google Scholar]
- Pichon, Y.; Prime, L.; Benquet, P.; Tiaho, F. Some aspects of the physiological role of ion channels in the nervous system. Eur. Biophys. J 2004, 33, 211–226. [Google Scholar]
- Quick, M. W.; Lester, R. A. J. Desensitization of neuronal nicotinic receptors. J. Neurobiol 2002, 53, 457–478. [Google Scholar]
- Reeves, D. C.; Lummis, S. C. R. The molecular basis of the structure and function of the 5-HT3 receptor: a model ligand-gated ion channel. Mol. Membr. Biol 2002, 19, 11–26. [Google Scholar]
- Reeves, D. C.; Sayed, M. F. R.; Chau, P. L.; Price, K. L.; Lummis, S. C. R. Prediction of 5-HT3 receptor agonist-binding residues using homology modeling. Biophys. J 2003, 84, 2338–2344. [Google Scholar]
- Reid, C. A.; Bekkers, J. M.; Clements, J. D. Presynaptic Ca2+ channels: a functional patchwork. Trends Neurosci 2003, 26, 683–687. [Google Scholar]
- Roux, B. Ion conduction and selectivity in K+ channels. Annu. Rev. Biophys. Biomol. Struct 2005, 34, 153–171. [Google Scholar]
- Schapira, M.; Abagyan, R.; Totrov, M. Structural model of nicotinic acetylcholine receptor isotypes bound to acetylcholine and nicotine. BMC Struct. Biol 2002, 2, 1–8. [Google Scholar]
- Sine, S. M.; Wang, H. L.; Bren, N. Lysine scanning mutagenesis delineates structural model of the nicotinic receptor ligand binding domain. J Biol Chem. 2002, 277, 29210–29223. [Google Scholar]
- Sixma, T. K.; Smit, A.B. Acetylcholine binding protein (AChBP): a secreted glial protein that provides a high-resolution model for the extracellular domain of pentameric ligand-gated ion channels. Annu. Rev. Biophys. Biomol. Struct 2003, 32, 311–334. [Google Scholar]
- Smit, A. B.; Syed, N. I.; Schaap, D.; van Minnen, J.; Klumperman, J.; Kits, K. S.; Lodder, H.; van der Schors, R. C.; van Elk, R.; Sorgedrager, B.; Brejc, K.; Sixma, T. K.; Geraerts, W. P. A. gliaderived acetylcholine-binding protein that modulates synaptic transmission. Nature 2001, 411, 261–268. [Google Scholar]
- Stevens, C. F. Presynaptic function. Curr. Opin. Neurobiol 2004, 14, 341–345. [Google Scholar]
- Sullivan, D.; Chiara, D. C.; Cohen, J. B. Mapping the agonist binding site of the nicotinic acetylcholine receptor by cysteine scanning mutagenesis: antagonist footprint and secondary structure prediction. Mol. Pharmacol 2002, 61, 463–472. [Google Scholar]
- Swope, S. L.; Moss, S. J.; Raymond, L. A.; Huganir, R. L. Regulation of ligand-gated ion channels by protein phosphorylation. Adv. Second Messenger Phosphoprot. Res 1999, 33, 49–78. [Google Scholar]
- Tempel, T. M.; Papazian, D. M.; Schwarz, T. L.; Jan, Y. N.; Jan, L.Y. Sequence of a probable potassium channel component encoded at Shaker locus in Drosophila. Science 1987, 237, 770–775. [Google Scholar]
- Trimmer, J. S.; Rhodes, K. J. Localization of voltage-gated ion channels in mammalian brain. Annu. Rev. Physiol 2004, 66, 477–519. [Google Scholar]
- Trudell, J. Unique assignment of inter-subunit association in GABAA α1β3γ2 receptors determined by molecular modeling. Biochim. Biophys. Acta 2002, 1565, 91–96. [Google Scholar]
- Tuppo, E. E.; Arias, H. R. The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol 2005, 37, 289–305. [Google Scholar]
- Unwin, N. The Croonian Lecture 2000. Nicotinic acetylcholine receptor and the structural basis of fast synaptic transmission. Philos. Trans. R. Soc. London Ser B 2000, 355, 1813–1829. [Google Scholar]
- Unwin, N. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J. Mol. Biol 2005, 346, 967–989. [Google Scholar]
- Unwin, N.; Miyazawa, A.; Li, J.; Fujiyoshi, Y. Activation of the nicotinic acetylcholine receptor involves a switch in conformation of the α subunits. J. Mol. Biol 2002, 319, 1165–1176. [Google Scholar]
- Van der Kloot, W.; Molgó, J. Quantal acetylcholine release at the vertebrate neuromuscular junction. Physiol. Rev 1994, 74, 899–991. [Google Scholar]
- Vazquez, G.; Wedel, B. J.; Aziz, O.; Trebak, M.; Putney, J.W., Jr. The mammalian TRPC cation channels. Biochim. Biohphys. Acta 2004, 1742, 21–36. [Google Scholar]
- Vial, C.; Roberts, J.A.; Evans, R.J. Molecular properties of ATP-gated P2X receptor ion channels. Trends Pharmacol. Sci 2004, 25, 487–493. [Google Scholar]
- Waites, C. L.; Craig, A. M.; Garner, C. C. Mechanisms of vertebrate synaptogenesis. Annu. Rev. Neurosci 2005, 28, 251–274. [Google Scholar]
- Wang, K.; Hackett, J. T.; Cox, M. E.; van Hoek, M.; Lindstrom, J. M.; Parsons, S. J. Regulation of the neuronal nicotinic acetylcholine receptor by SRC family tyrosine kinases. J. Biol. Chem 2004, 279, 8779–8786. [Google Scholar]
- Wei, D.; Sirois, S.; Du, Q-S.; Arias, H.R.; Chou, K-C. Theoretical studies of Alzheimer’s disease drug candidate 3-[(2,4-dimethoxy)benzylidene]-anabaseine (GTS-21) and its derivatives. Biochem. Biophys. Res. Commun. 2005, 338, 1059–1064. [Google Scholar]
- Weiland, S.; Witzemann, V.; Villarroel, A.; Propping, P.; Steinlein, O. An amino acid exchange in the second transmembrane segment of a neuronal nicotinic receptor causes partial epilepsy by altering its desensitization kinetics. FEBS Lett 1996, 398, 91–96. [Google Scholar]
- Willmann, R.; Fuhrer, C. Neuromuscular synaptogenesis: clustering of acetylcholine receptors revisited. Cell Mol. Life Sci 2002, 59, 1296–1316. [Google Scholar]
- Yamakage, M.; Namiki, A. Calcium channels - basic aspects of their structure, function and gene encoding; anesthetic action on the channels - a review. Can. J. Anesth 2002, 49, 151–164. [Google Scholar]
- Yu, F. H.; Catterall, W. A. Overview of the voltage-gated sodium channel family. Genome Biol 2003, 4, 207. [Google Scholar]
- Ziv, N. E.; Garner, C. C. Cellular and molecular mechanisms of presynaptic assembly. Nat. Rev. Neurosci 2004, 5, 385–399. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bindig Site | Toxin/Drug (origin) | Pharmacological Effect |
|---|---|---|
| 1 | Tetrodotoxin (marine) Saxitoxin (marine) μ-Conotoxins GIIIA and GIIIB (marine) | Ion channel block |
| 2 | Veratridine (plant) Batrachotoxin (animal) Aconitine (plant) Grayanotoxin (plant) | Persistent activation |
| 3 | Scorpion α-toxins (animal) Sea anemone toxins (marine) δ-Atracotoxins (animal) δ–Conotoxins PVIA, TxIA, TxIB, and TxIIA (marine) | Slow inactivation Enhancement of persistent activation |
| 4 | Scorpion β-toxins (animal) | Shift voltage-dependent activation to more negative potentials |
| 5 | Brevetoxins (marine) Ciguatoxins (marine) | Shift voltage-dependent activation to more negative potentials |
| 6 | Pyrethroids (plant) DDT (synthetic) | Slow activation, inactivation and deactivation processes |
| Local anesthetic | Local anesthetics [synthetics in general; plant product (e.g., cocaine)] | Ion channel block (use-dependent) |
| Additional binding sites | DPI 201-106 (synthetic) BDF9148 (synthetic) | Prolongation of action potential |
| Additional binding sites | Pronase (synthetic) N-bromoacetamide (synthetic) Chloramine-T (synthetic) | Disruption of fast inactivation |
| Channel Type | Current | Tissue Localization | Drugs (origin) | Pharmacological effect |
|---|---|---|---|---|
| CaV1 Channels | ||||
| CaV1.1 | L | Skeletal muscle Transverse tubules | Dihydropiridines (synthetic) Phenylalkylamines (synthetic) Benzothiazepines (synthetic) | Contraction |
| CaV1.2 | L | Cardiac myocytes Endocrine cells Neuronal cell bodies and proximal dendrites | Dihydropiridines (synthetic) Phenylalkylamines (synthetic) Benzothiazepines (synthetic) | Contraction Hormone release Synaptic integration |
| CaV1.3 | L | Endocrine cells Neuronal cell bodies and proximal dendrites | Dihydropiridines (synthetic) Phenylalkylamines (synthetic) Benzothiazepines (synthetic) | Hormone release Synaptic integration |
| CaV1.4 | L | Retina | Not established | Neurotransmitter release |
| CaV2 Channels | ||||
| CaV2.1 | P/Q | Nerve terminals and dendrites | ω-Agatoxins IVA and IVB (animal) | Inhibits neurotransmitter release |
| CaV2.2 | N | Nerve terminals and dendrites | ω-Conotoxins GVIA, MVIIC, and MVIIA (marine) | Inhibits neurotransmitter release |
| CaV2.3 | R | Neuronal cell bodies and dendrites | SNX-482 (animal) | Inhibits repetitive firing |
| CaV3 Channels | ||||
| CaV3.1 | T | Neuronal cell bodies and dendrites Cardiac myocytes | Kurtoxin (animal) Mibefradil (synthetic) Pimozide (synthetic) | Inhibits repetitive firing Inhibits pacemaker |
| CaV3.2 | T | Neuronal cell bodies and dendrites Cardiac myocytes | Kurtoxin (animal) Mibefradil (synthetic) Pimozide (synthetic) | Inhibits repetitive firing Inhibits pacemaker |
| CaV3.3 | T | Neuronal cell bodies and dendrites | Kurtoxin (animal) Mibefradil (synthetic) Pimozide (synthetic) | Inhibits repetitive firing |
| Superfamily of Cys-Loop or Nicotinicoid Receptors | |||
| Family of AChRs | Family of 5-HT3Rs | Family of GABA receptors | Family of GlyRs |
| Subfamily of epithelial receptors | Subfamily of GABAARs | Subfamily of GlyRs | |
| Subfamily of neuronal α-bungarotoxin-sensitive receptors | Subfamily of GABACRs | ||
| Subfamily of muscle receptor | Subfamily of GABA-induced cation channel receptors (invertebrates) | ||
| Subfamily of heteromeric neuronal receptors | |||
| Subfamily of heteromeric protostomian receptors | |||
| Subfamily of ACh-induced anion channel receptors (invertebrates) | |||
| Superfamily of Ionotropic Glutamate Receptors | |||
| Family of excitatory GluRs | Family of inhibitory GluRs | ||
| Subfamily of NMDAR subunits | Subfamily of Glu-induced anion channel receptors (invertebrates) | ||
| Subfamily of AMPAR subunits | |||
| Subfamily of KAR subunits | |||
| Subfamily of kainate binding proteins | |||
| Subfamily of delta subunits | |||
| Superfamily of Inotropic ATP Receptors | |||
| Family of P2XnRs | |||
© 2006 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Arias, H.R. Marine Toxins Targeting Ion Channels. Mar. Drugs 2006, 4, 37-69. https://doi.org/10.3390/md403037
Arias HR. Marine Toxins Targeting Ion Channels. Marine Drugs. 2006; 4(3):37-69. https://doi.org/10.3390/md403037
Chicago/Turabian StyleArias, Hugo R. 2006. "Marine Toxins Targeting Ion Channels" Marine Drugs 4, no. 3: 37-69. https://doi.org/10.3390/md403037
APA StyleArias, H. R. (2006). Marine Toxins Targeting Ion Channels. Marine Drugs, 4(3), 37-69. https://doi.org/10.3390/md403037