
Meirols A–C: Bioactive Catecholic Compounds from the Marine-Derived Fungus Meira sp. 1210CH-42
 ,
,
Abstract
1. Introduction
2. Results and Discussion
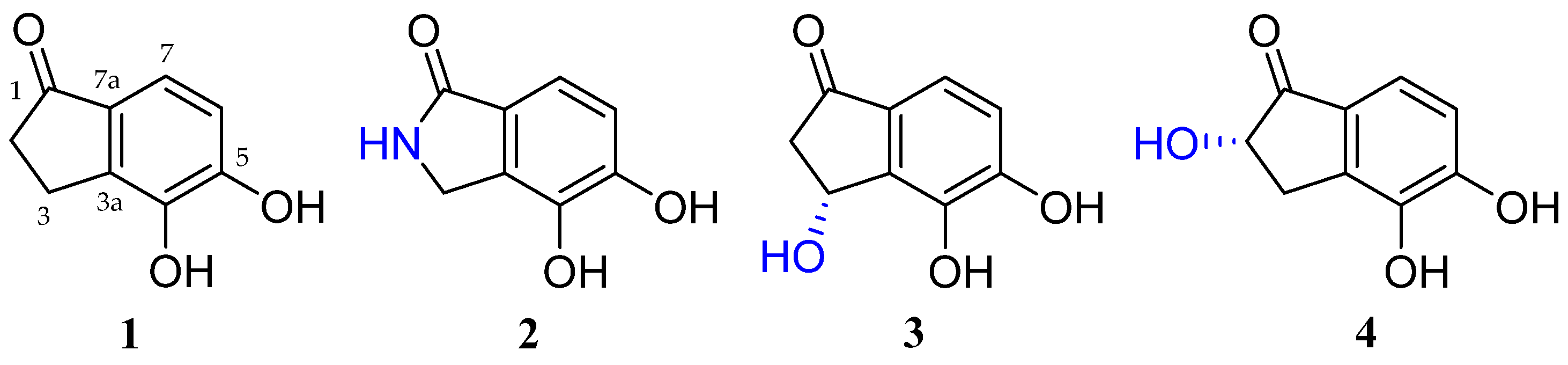
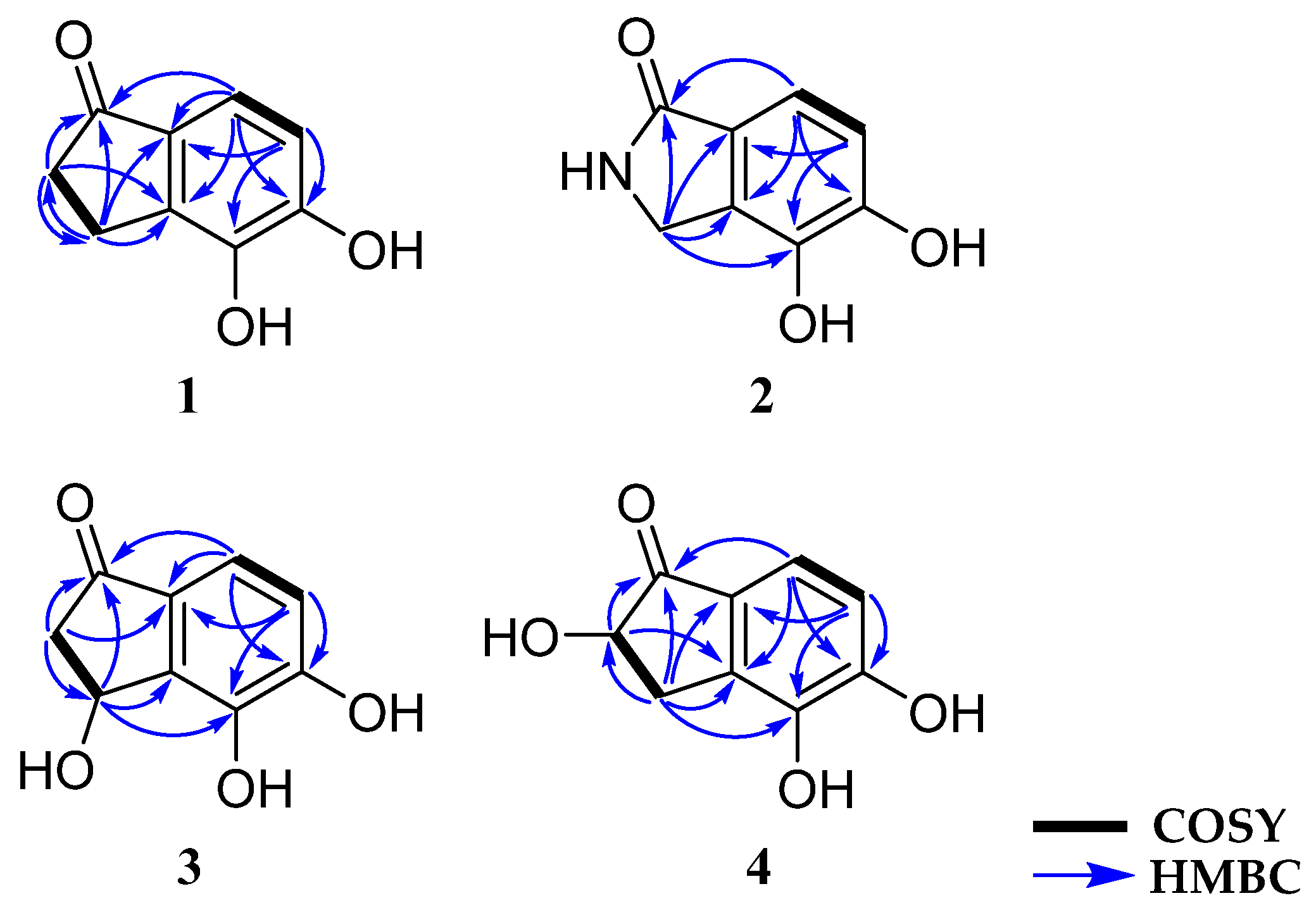
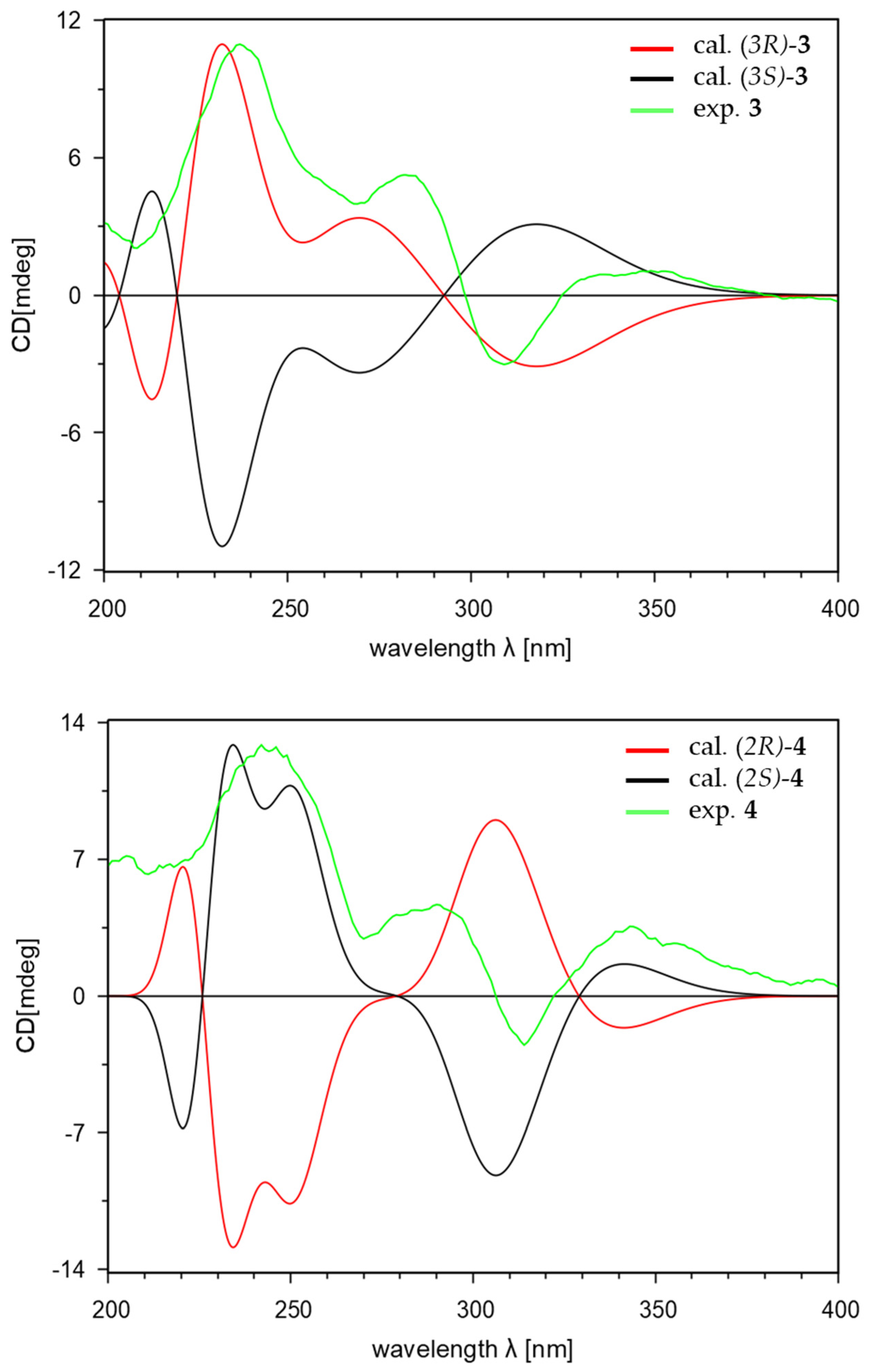
2.1. Structure Elucidation of New Compounds
2.2. Bioactivity Evaluation of Compounds
3. Materials and Methods
3.1. General Experimental Procedures and Reagents
3.2. Fungal Strain and Fermentation
3.3. Extraction and Isolation of Compounds 1–4
3.4. Computational Analysis
3.5. Antioxidant Activity Assay
3.6. α-Glucosidase Inhibitory Activity Assay
3.7. Cytotoxicity Assay
3.8. Tyrosinase Inhibitory Activity Assay
3.9. Antibacterial Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, S.; Lim, S.W.; Choi, J. Drug discovery inspired by bioactive small molecules from nature. Anim. Cells Syst. 2022, 26, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Bradford, P.A. Beta-Lactams and beta-Lactamase Inhibitors: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.J.; Zhao, Y.; Tao, X.; Li, J.; Chen, Y.; Holland, D.C.; Jin, T.Y.; Wang, A.Y.; Xiang, L. Catecholamine Derivatives: Natural Occurrence, Structural Diversity, and Biological Activity. J. Nat. Prod. 2023, 86, 2592–2619. [Google Scholar] [CrossRef] [PubMed]
- Sedo, J.; Saiz-Poseu, J.; Busque, F.; Ruiz-Molina, D. Catechol-based biomimetic functional materials. Adv. Mater. 2013, 25, 653–701. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Lee, K. Crosslinking Mechanisms of Phenol, Catechol, and Gallol for Synthetic Polyphenols: A Comparative Review. Appl. Sci. 2022, 12, 11626. [Google Scholar] [CrossRef]
- Boekhout, T.; Theelen, B.; Houbraken, J.; Robert, V.; Scorzetti, G.; Gafni, A.; Gerson, U.; Sztejnberg, A. Novel anamorphic mite-associated fungi belonging to the Ustilaginomycetes: Meira geulakonigii gen. nov., sp nov., Meira argovae sp. Nov. and Acaromyces ingoldii gen. nov., sp. nov. Int. J. Syst. Evol. Microbiol. 2003, 53, 1655–1664. [Google Scholar] [CrossRef]
- Rush, T.A.; Aime, M.C. The genus Meira: Phylogenetic placement and description of a new species. Anton. Leeuw. Int. J. G. 2013, 103, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Limtong, S.; Polburee, P.; Chamnanpa, T.; Khunnamwong, P.; Limtong, P. Meira siamensis sp. nov., a novel anamorphic ustilaginomycetous yeast species isolated from the vetiver grass phylloplane. Int. J. Syst. Evol. Microbiol. 2017, 67, 2418–2422. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Li, P.-D.; Zhao, J.; Wang, H.-K.; Jeewon, R.; Bhoyroo, V.; Aruna, B.; Lin, F.-C.; Wang, Q. Morph-molecular characterization of Meira nicotianae sp. nov., a novel basidiomycetous, anamorphic yeast-like fungus associated with growth improvement in tobacco plant. Phytotaxa 2018, 365, 169–181. [Google Scholar] [CrossRef]
- Kushnir, L.; Paz, Z.; Gerson, U.; Sztejnberg, A. The effect of three basidiomycetous fungal species on soil-borne, foliage and fruit-damaging phytopathogens in laboratory experiments. Biocontrol 2011, 56, 799–810. [Google Scholar] [CrossRef]
- Paz, Z.; Bilkis, I.; Gerson, U.; Kerem, Z.; Sztejnberg, A. Argovin, a novel natural product secreted by the fungus Meira argovae, is antagonistic to mites. Entomol. Exp. Appl. 2011, 140, 247–253. [Google Scholar] [CrossRef]
- Shin, H.J.; Lee, M.A.; Lee, H.S.; Heo, C.S. Thiolactones and Δ(8,9)-Pregnene Steroids from the Marine-Derived Fungus Meira sp. 1210CH-42 and Their alpha-Glucosidase Inhibitory Activity. Mar. Drugs 2023, 21, 246. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Yang, S.Q.; Li, X.M.; Hu, X.Y.; Li, X.; Wang, B.G. Aromatic Polyketides from the Deep-Sea Cold-Seep Mussel Associated Endozoic Fungus Talaromyces minioluteus CS-138. Mar. Drugs 2022, 20, 529. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Cho, H.; Lee, D.K.; Ha, J.; Choi, B.J.; Jeong, J.H.; Ryu, J.H.; Kang, J.S.; Jeon, R. Discovery of 5-Phenoxy-2-aminopyridine Derivatives as Potent and Selective Irreversible Inhibitors of Bruton’s Tyrosine Kinase. Int. J. Mol. Sci. 2020, 21, 8006. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- de Sa, J.D.M.; Pereira, J.A.; Dethoup, T.; Cidade, H.; Sousa, M.E.; Rodrigues, I.C.; Costa, P.M.; Mistry, S.; Silva, A.M.S.; Kijjoa, A. Anthraquinones, Diphenyl Ethers, and Their Derivatives from the Culture of the Marine Sponge-Associated Fungus Neosartorya spinosa KUFA 1047. Mar. Drugs 2021, 19, 457. [Google Scholar] [CrossRef] [PubMed]
- Heo, C.S.; Kang, J.S.; Kwon, J.H.; Van Anh, C.; Shin, H.J. Pyrrole-Containing Alkaloids from a Marine-Derived Actinobacterium Streptomyces zhaozhouensis and Their Antimicrobial and Cytotoxic Activities. Mar. Drugs 2023, 21, 167. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| δH, Mult (J in Hz) | δH, Mult (J in Hz) | δH, Mult (J in Hz) | δH, Mult (J in Hz) | |
| 2 | 2.63, t (5.6) | 2.47, d (18.6) 3.02, dd (18.6, 6.6) | 4.41, dd (7.7, 4.5) | |
| 3 | 3.01, t (5.6) | 4.34, s | 5.48, d (6.6) | 2.70, dd (16.7, 4.5) 3.51, dd (16.7, 7.7) |
| 6 | 6.84, d (8.0) | 6.90, d (8.0) | 6.91, d (8.0) | 6.87, d (8.1) |
| 7 | 7.15, d (8.0) | 7.16, d (8.0) | 7.14, d (8.0) | 7.17, d (8.1) |
| Position | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| δC, Type | δC, Type | δC, Type | δC, Type | |
| 1 | 209.1, C | 174.5, C | 205.6, C | 207.0, C |
| 2 | 37.6, CH2 | NH | 48.4, CH2 | 75.0, CH |
| 3 | 23.3, CH2 | 44.5, CH2 | 66.9, CH | 32.2, CH2 |
| 3a | 144.7, C | 132.4, C | 143.5, C | 139.8, C |
| 4 | 143.1, C | 141.6, C | 130.0, C | 128.1, C |
| 5 | 153.1, C | 150.4 C | 154.2, C | 153.8, C |
| 6 | 116.9, CH | 116.9, CH | 118.6, CH | 117.3, CH |
| 7 | 117.2, CH | 116.5, CH | 116.7, CH | 118.0, CH |
| 7a | 130.8, C | 125.2, C | 144.1, C | 143.0, C |
| Compound | DPPH Radical Scavenging Activity (EC50 ± SD, μM) a | α-Glucosidase Inhibitory Activity (IC50 ± SD, μM) b | Cytotoxic Activity (GI50 ± SD, μM) c | |
|---|---|---|---|---|
| RPMI-8402 d | NALM6 d | |||
| 1 | 7.47 ± 0.13 | 184.50 ± 2.93 | 21.00 ± 0.47 | 9.47 ± 0.41 |
| 2 | 6.82 ± 0.06 | 199.70 ± 1.87 | >30 | 29.55 ± 2.27 |
| 3 | 6.01 ± 0.07 | 367.43 ± 3.01 | >30 | >30 |
| 4 | 6.20 ± 0.12 | >555.00 | >30 | >30 |
| Ascorbic acid | 7.81 ± 0.25 | – | – | – |
| Acarbose | – | 301.93 ± 3.55 | – | – |
| Doxorubicin | – | – | 0.015 ± 0.13 × 10−2 | 0.003 ± 0.06 × 10−2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.A.; Kang, J.S.; Yang, J.-W.; Lee, H.-S.; Heo, C.-S.; Park, S.J.; Shin, H.J. Meirols A–C: Bioactive Catecholic Compounds from the Marine-Derived Fungus Meira sp. 1210CH-42. Mar. Drugs 2024, 22, 87. https://doi.org/10.3390/md22020087
Lee MA, Kang JS, Yang J-W, Lee H-S, Heo C-S, Park SJ, Shin HJ. Meirols A–C: Bioactive Catecholic Compounds from the Marine-Derived Fungus Meira sp. 1210CH-42. Marine Drugs. 2024; 22(2):87. https://doi.org/10.3390/md22020087
Chicago/Turabian StyleLee, Min Ah, Jong Soon Kang, Jeong-Wook Yang, Hwa-Sun Lee, Chang-Su Heo, Sun Joo Park, and Hee Jae Shin. 2024. "Meirols A–C: Bioactive Catecholic Compounds from the Marine-Derived Fungus Meira sp. 1210CH-42" Marine Drugs 22, no. 2: 87. https://doi.org/10.3390/md22020087
APA StyleLee, M. A., Kang, J. S., Yang, J.-W., Lee, H.-S., Heo, C.-S., Park, S. J., & Shin, H. J. (2024). Meirols A–C: Bioactive Catecholic Compounds from the Marine-Derived Fungus Meira sp. 1210CH-42. Marine Drugs, 22(2), 87. https://doi.org/10.3390/md22020087