Abstract
In this study, a detailed chemical investigation of a streptomycin-resistant strain of the deep-sea marine, actinomycete Amycolatopsis sp. WP1, yielded six novel amycolachromones A–F (1–6), together with five known analogues (7–11). Amycolachromones A–B (1–2) possessed unique dimer skeletons. The structures and relative configurations of compounds 1–11 were elucidated by extensive spectroscopic data analyses combined with X-ray crystal diffraction analysis. Plausible biogenetic pathways of amycolachromones A–F were also proposed.
1. Introduction
Marine microbial natural products, especially those derived from marine actinomycetes, have become an important source of novel bioactive compounds [1,2,3]. However, traditional screening strategies generally do not provide access to the full array of secondary metabolites encoded within actinomycete genomes [4]. For example, Streptomyces coelicolor initially produces four classes of metabolites using laboratory fermentation, despite genome sequencing revealing the capacity to produce >30 families of metabolites [5,6]. To solve this problem, various strategies have been proposed to activate the expression of otherwise silent biosynthetic gene clusters, including the ‘one strain many compounds’ (OSMAC) approach [7], co-cultivation with other microorganisms [8] and chemical epigenetics [9]. Recently, a ribosome engineering approach that targets ribosomal proteins or RNA polymerase (RNAP) has shown promise for expression of cryptic gene clusters. This method selects for mutants that are resistant to antibiotics that target the bacterial ribosome, presumably activating the expression of bacterial cryptic genes by resistant mutants [10,11]. Shima and co-workers demonstrated this method in actinomycetes by activating the biosynthetic pathway for actinorhodin in mutant Streptomyces that developed resistance to streptomycin [12]. Recent adoptions of this approach demonstrated the ability of streptomycin-resistant mutants to enhance production of actinolactomycin [13], fredericamycin A and chlorinated alkaloids, inducamides A–C [14,15].
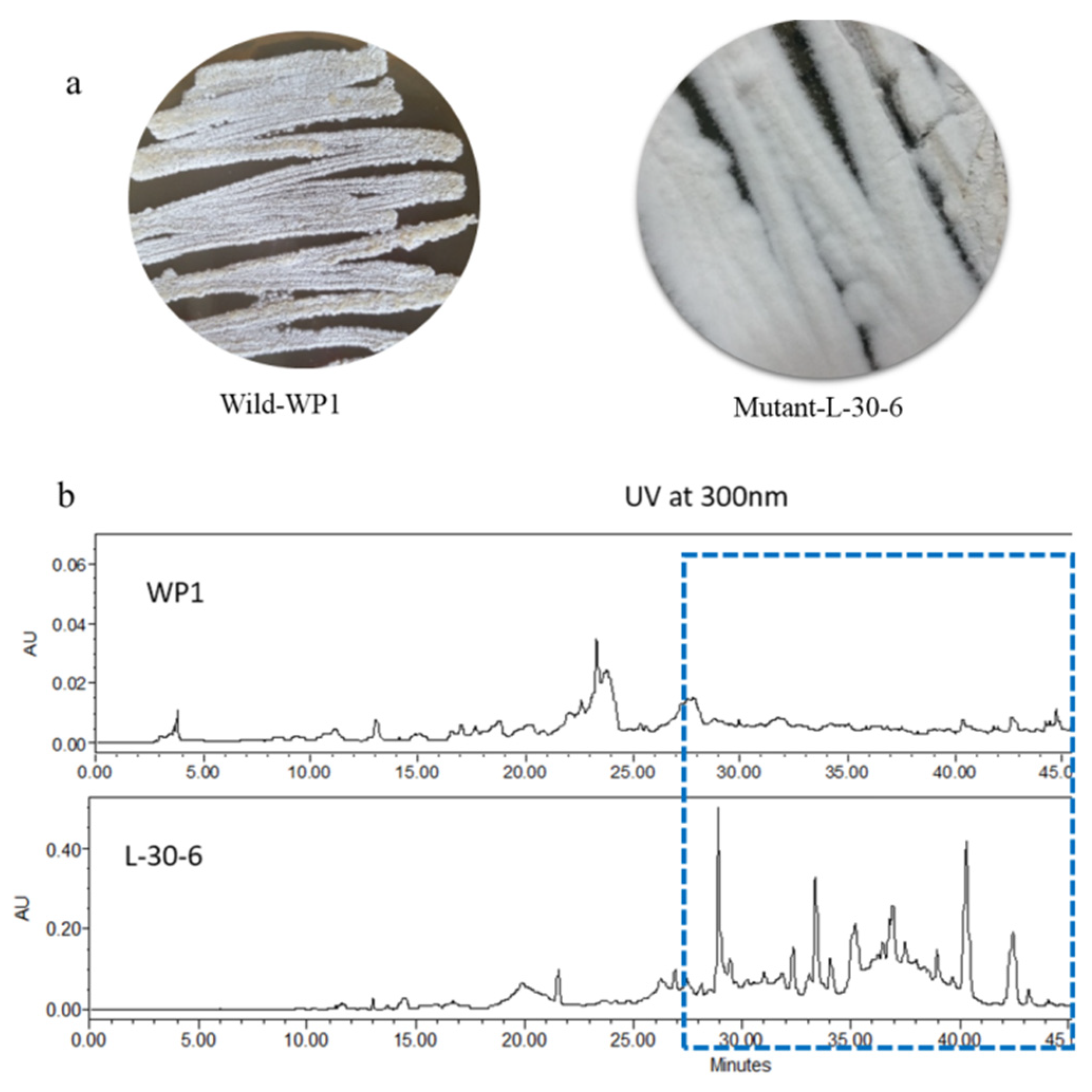
Chromones are oxygen-containing heterocyclic compounds with a chromone benzoannelated γ-pyrone ring (4H-chromen-4-one, 4H-1-benzopyran-4-one) that are widely distributed in bacteria, fungi and plant [16]. Chromones and analogues can be considered privileged structures in drug discovery due to their numerous biological activities, such as anti-inflammatory, antiplatelet, anticancer, antimicrobial, anti-neurodegenerative and anti-obesity effects [17]. In this paper, we undertook a ribosome engineering approach for activating biosynthetic pathways in Amycolatopsis sp. WP1, a deep sea actinomycete isolated from sediments collected at −2945 m in the Indian Ocean. A streptomycin-resistant strain, designated as L-30-6 (Figure 1), was observed to produce six new chromone derivatives, designated as the amycolachromones A–F (1–6), and five known chromone derivatives (7–11) (Figure 2).
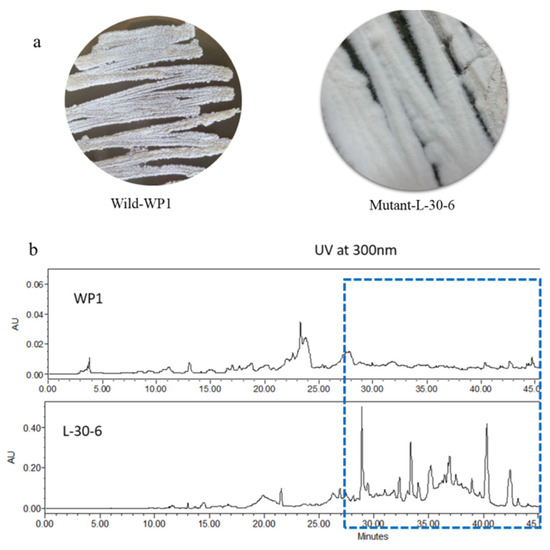
Figure 1.
(a) Wild-type strain WP1 and streptomycin-resistant strain l-30-6 grown under identical conditions on ISP2 media. (b) HPLC traces of wt-WP1 and mutant l-30-6 showing the production of new compounds (UV detection at 300 nm).
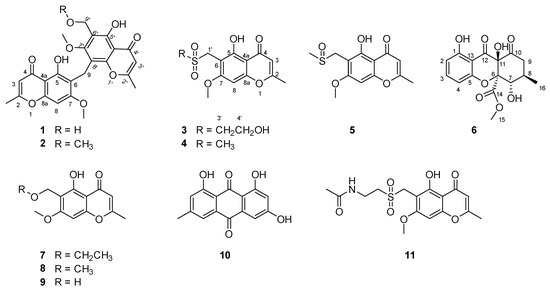
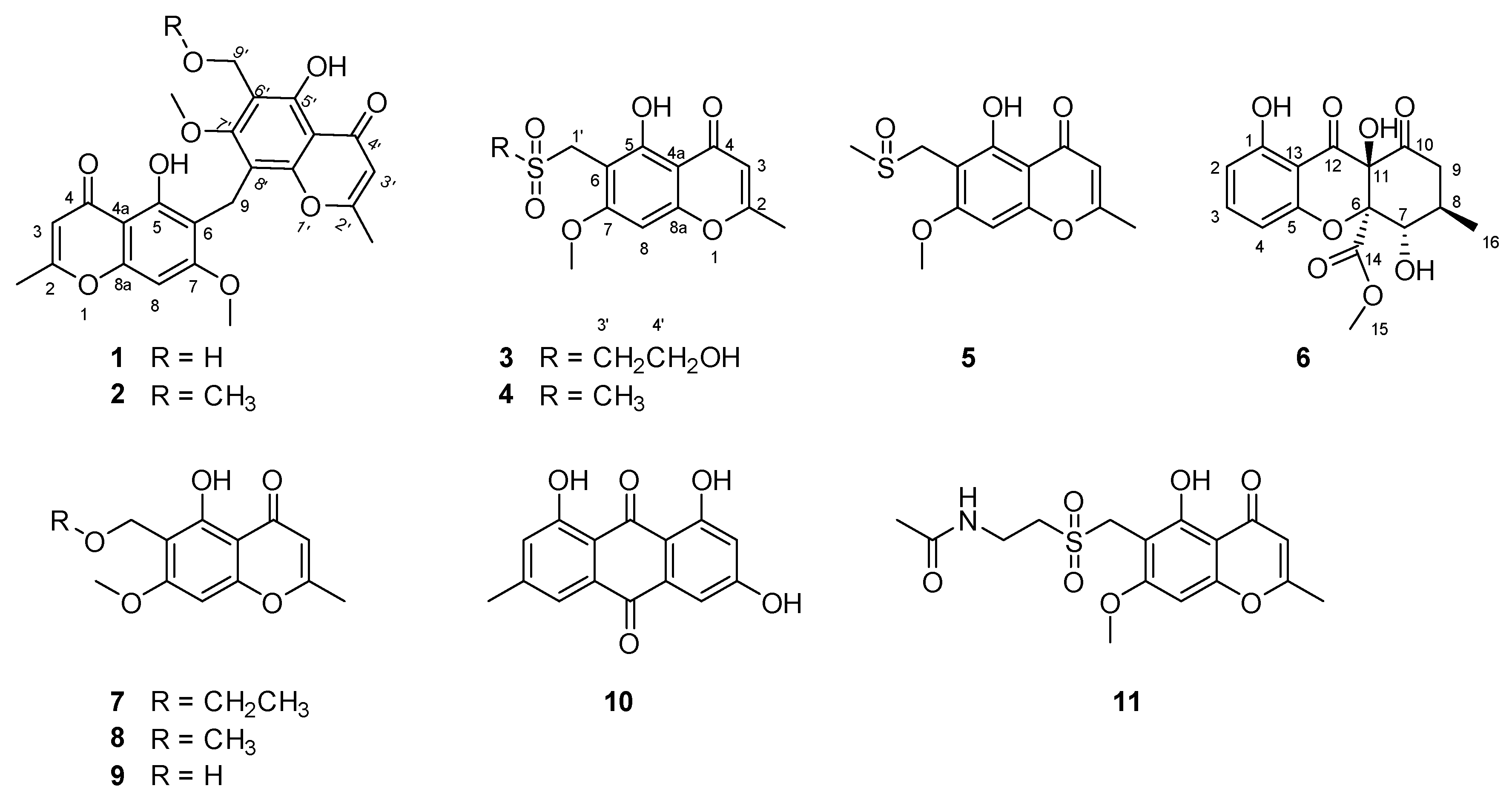
Figure 2.
Chemical structures of compounds 1–11.
2. Results and Discussion
Amycolachromone A (1) displayed HRESIMS peak at m/z 477.1172 [M + Na]+ (calcd 477.1162) corresponding to the molecular formula C24H22O9, indicating fourteen degrees of unsaturation. Analysis of the NMR data of 1 (Table 1, see Supplementary Materials) revealed three aromatic protons at δH 6.64 (1H, s, H-8), 6.22 (1H, s, H-3′), 6.20 (1H, s, H-3), two methoxy groups at δH 3.83 (3H, s, CH3O-7) and 3.75 (3H, s, CH3O-7′), two methyl groups at δH 2.35 (3H, s, CH3-2) and 3.21 (3H, s, CH3-2′), two methylenes at δH 4.45 (2H, d, J = 4.8 Hz, H-9′) and 3.98 (2H, s, H-9), two phenolic hydroxyl groups at δH 13.13 (1H s, OH-5) and 13.10 (1H s, OH-5′), a hydroxyl group at δH 4.78 (1H, t, J = 5.2 Hz, OH-9′). The 13C NMR (Table 1) revealed 24 carbon signals: the two carbonyls C-4 (δC 183.2) and C-4′ (δC 182.5), the three aromatic carbons C-3 (δC 108.7), C-3′ (δC 108.4) and C-8 (δC 90.6), five non-oxygenated quaternary aromatic carbons at C-4a (δC 110.8), C-6 (δC 104.4), C-4a’ (δC 106.7), C-6′ (δC 117.6), and C-8′ (δC 112.5), eight oxygenated quaternary aromatic carbons at C-2 (δC 168.5), C-2′ (δC 168.3), C-7 (δC 163.8),C-7′ (δC 163.5), C-5 (δC 158.5), C-5′ (δC 158.3), C-8a (δC 156.7), and C-8a’ (δC 154.8), two methoxy groups CH3O-7 (δC 63.2) and CH3O-7′ (δC 56.7), two methyl groups CH3-2 (δC 20.4) and CH3-2′ (δC 20.0), and two methylenes C-9′ (δC 52.1) and C-9 (δC 16.9). Analysis of the 1H and 13C NMR data of 1 revealed the presence of the same 5-hydroxy-4H-chromen-4-one moiety as found in xanthones [18,19], and therefore suggested a compound comprising two xanthone building blocks.

Table 1.
1H (500 MHz) and 13C (125 MHz) NMR data of compounds 1 and 2 in DMSO-d6.
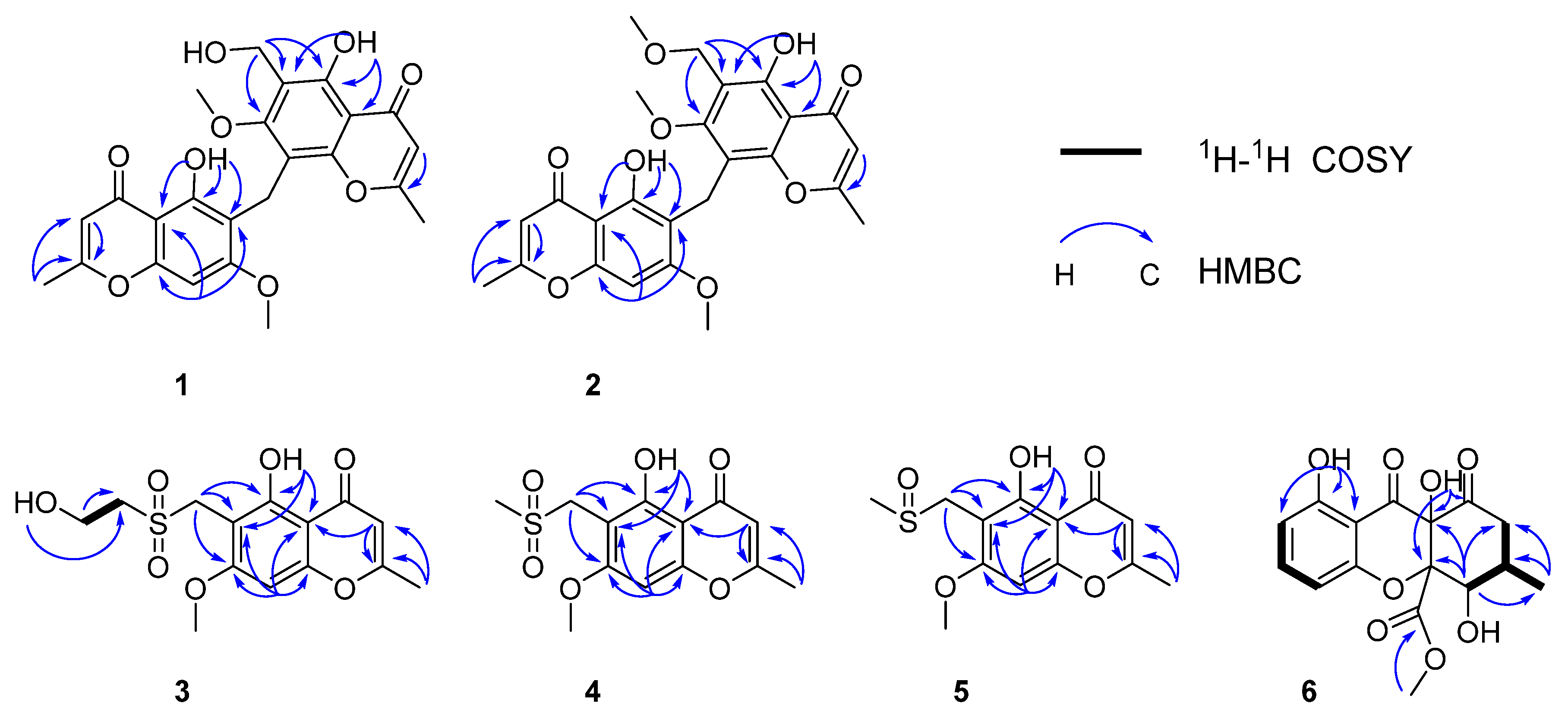
1H-1H COSY correlations were observed from H-9′ to OH-9′. Further confirmation was found for HMBC correlations of 5-OH to C-5, C-6, 4a; H-8 to C-6, C-4a, C-8a; H-3 to C-2; 2-CH3 to C-2, C-3, indicating the same Eugenin. HMBC correlations from 5′-OH to C-5′, C-6′, 4a′; H-3′ to C-2′; 2′-CH3 to C-2′, C-3′; H-9′ to C-5′, C-6′, C-7′, indicated the same 6-Hydroxymethyleugenin (10) [20]. HMBC correlations from H-9 to C-7, C-5, C-4a, C8′, C-7, C-8a′, indicated that Eugenin and 6-hydroxymethyleugenin are linked with C-9. Selected key correlations in the observed NMR spectrum are shown in Figure 3. On the basis of these results, the structure of compound 1 was established as shown.
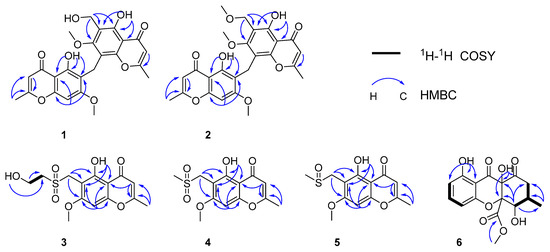
Figure 3.
Key HMBC and COSY correlations of compounds 1–6.
Amycolachromone B (2) displayed HRESIMS peak at m/z 469.1502 [M + H]+ (calcd 469.1499), m/z 491.1333 [M + Na]+ (calcd 469.1318), corresponding to the molecular formula C25H24O9 (fourteen degrees of unsaturation). Analysis of the NMR data of 2 (Table 1) revealed for three aromatic protons at δH 6.64 (1H, s, H-8), 6.22 (1H, s, H-3′), 6.23 (1H, s, H-3), two methoxy groups at δH 3.82 (3H, s, CH3O-7) and 3.76 (3H, s, CH3O-7′), two methyl groups at δH 2.22 (3H, s, CH3-2) and 3.37 (3H, s, CH3-2′), two methylene at δH 4.35 (2H, s, H-9′) and 3.98 (2H, s, H-9), two phenolic hydroxyl groups at δH 13.21 (1H s, OH-5) and 13.11 (1H s, OH-5′). The 13C NMR (Table 1) revealed 25 carbon signals: the two carbonyl group C-4 (δC 183.2), C-4′ (δC 182.4), three aromatic carbon C-3 (δC 108.7), C-3′ (δC 108.5) and C-8 (δC 90.6), five nonoxygenated quaternary aromatic carbons at C-4a (δC 110.8), C-6 (δC 100.9), C-4a′ (δC 106.7), C-6′ (δC 114.1), and C-8′ (δC 112.5), eight oxygenated quaternary aromatic carbons at C-2 (δC 168.8), C-2′ (δC 168.2), C-7 (δC 164.2), C-7′ (δC 163.4), C-5 (δC 158.9), C-5′ (δC 158.6),C-8a (δC 156.7), and C-8a′ (δC 154.8), three methoxy groups CH3O-7(δC 62.5), CH3O-9′ (δC 57.7), and CH3O-7′ (δC 56.6), two methyl groups CH3-2 (δC 20.4) and CH3-2′ (δC 20.0), two methylene C-9′ (δC 63.2) and C-9 (δC 16.9). Analysis of the 1H and 13C NMR data of 2 revealed the presence of the same 5-hydroxy-4H-chromen-4-one moiety as found in xanthones [18,19], and comprised two xanthones. In contrast, the NMR data of 2 showed them to be nearly identical except for a methoxy group linked with C-9′. Further confirmation was found for HMBC correlations of 5-OH to C-5, C-6, 4a; H-8 to C-6, C-4a, C-8a, C-7; H-3 to C-2; 2-CH3 to C-2, C-3, indicated that same as Eugenin [20]. HMBC correlations from 5′-OH to C-5′, C-6′, 4a′; H-3′ to C-2′; 2′-CH3 to C-2′, C-3′; H-9′ to C-5′, C-6′, C-7′, indicated that same as 6-Methoxymethyleugenin (9) [21]. HMBC correlations from H-9 to C-7, C-5, C-4a, C8′, C-7, C-8a′, indicated that Eugenin and Methoxymethyleugenin are linked with C-9. Selected key correlations in the observed NMR spectrum are shown in Figure 3. On the basis of these results, the structure of compound 2 was established as shown.
Amycolachromone C (3) displayed HRESIMS ion at m/z 351.0515 [M + Na]+ (calcd 351.0514), corresponding to the molecular formula C14H16O7S, indicating nine degrees of unsaturation. Analysis of the NMR data of 3 (Table 2) revealed two aromatic protons at δH 6.78 (1H, s, H-8), 6.32 (1H, s, H-3), a methoxy group at δH 3.90 (3H, s, CH3O-7), a methyl group at δH 2.39 (3H, s, CH3-2), three methylenes at δH 4.39 (2H, s, H-1′), δH 3.80 (2H, q, J =6.1 Hz, H-4′), and 3.21 (2H, t, J = 6.3, H-3′), a phenolic hydroxyl group at δH 11.40 (1H s, OH-5), and a hydroxyl group at δH 5.03 (1H, t, J = 5.4 Hz, 1H, OH-4′). Examination of the 13C NMR spectrum (Table 2) revealed 14 carbon signals: a carbonyl group C-4 (δC 182.4), the two aromatic carbons C-3 (δC 108.9) and C-8 (δC 91.3), three non-oxygenated quaternary aromatic carbons at C-8a (δC 158.2), C-4a (δC 104.6), and C-6 (δC 101.0), and three oxygenated quaternary aromatic carbons at C-2 (δC 168.9), C-5 (δC 159.9), and C-7 (δC 163.9) and the methylene C-1′ (δC 49.3). Analysis of the 1H and 13C NMR data of 2 revealed the presence of the same 5-hydroxy-4H-chromen-4-one moiety as found in xanthones [18,19].
Table 2.
1H (500 MHz) and 13C (125 MHz) NMR data of compounds 3–5 in DMSO-d6.
In the 1H-1H COSY spectrum, there were correlations from H-4′ to OH-5′ and H-3′. According to the HMBC, there were correlations from H-1′ to C-6, C-5, and C-7, H-4′ to C-3′, OH-4′ to C-3′. The sulfur atom present in 3 was shown to be attached at C-1′and C-3′, indicated that C-1′was attached at C-6. Further confirmation was found for HMBC correlations of OH-5 to C-4a, C-6, C-5; CH3-2 to C-3, C-2; CH3O-7 to C7, H-3 to CH3-2, C-4a, C-2; H-8 to C-4a, C-6, C-8a, C-7, C-4, a hydroxyl group could be located at C-5, a methoxy groups could be located at C-7, a methyl group could be located at C-2. Selected key correlations in the observed NMR spectrum are shown in Figure 3. On the basis of these results, the structure of compound 3 was established as shown.
Amycolachromone D (4) displayed HRESIMS peak at m/z 321.0406 [M + Na]+ (calcd 321.0409), corresponding to the molecular formula C13H14O6S (nine degrees of unsaturation). Analysis of the NMR data of 4 (Table 2) revealed for two aromatic protons at δH 6.76 (1H, s, H-8), 6.30 (1H, s, H-3), a methoxy groups at δH 3.90 (3H, s, CH3O-7), two methyl groups at δH 2.91 (3H, s, H-3′) and 2.39 (3H, s, CH3-2), a methylene group at δH 4.33 (2H, s, H-1′). The 13C NMR (Table 2) revealed 13 carbon signals: a carbonyl group C-4 (δC 182.3), two aromatic carbon C-3 (δC 108.9) and C-8 (δC 91.1), three nonoxygenated quaternary aromatic carbons at C-8a (δC 158.2),C-4a (δC 104.7), and C-6 (δC 101.2), three oxygenated quaternary aromatic carbons at C-2 (δC 168.7), C-5 (δC 159.1), and C-7 (δC 163.7), a methoxy groups CH3O-7 (δC 57.2), two methyl groups C-3′ (δC 42.0) and CH3-2 (δC 20.4), a methylene group C-1′ (δC 49.4). Analysis of the 1H and 13C NMR data of 4 revealed the presence of the same 5-hydroxy-4H-chromen-4-one moiety as found in xanthones [18,19]. A side-by-side comparison of the NMR spectroscopic data with those of 3 showed them to be nearly identical except for the final hydroxymethyl unit on the side chain.
According to the HMBC correlations from H-1′ to C-6, C-5 and C-7, the sulfur atom present in 4 was shown to be attached at C-1′and C-3′, indicating that C-1′ was attached at C-6, Further confirmation was found for HMBC correlations of CH3O-7 to C7; H-3 to CH3-2, C-4a, C-2; H-8 to C-4a, C-6, C-8a, C-7, C-4, a methoxy group could be located at C-7 and a methyl group could be located at C-2. Selected key correlations in the observed NMR spectrum are shown in Figure 3. On the basis of these results, the structure of compound 4 was established as shown.
Amycolachromone E (5) displayed HRESIMS peak at m/z 305.0462 [M + Na]+ (calcd 305.0460), corresponding to the molecular formula C13H14O5S (eight degrees of unsaturation). Analysis of the NMR data of 5 (Table 2) revealed for two aromatic protons at δH 6.76 (1H, s, H-8), 6.29 (1H, s, H-3), a methoxy groups at δH 3.90 (3H, s, CH3O-7), two methyl groups at δH 2.54 (3H, s, H-3′) and 2.39 (3H, s, CH3-2) and a methylene group at δH 3.99 (2H, d, J = 6.7 Hz, H-1′). The 13C NMR (Table 2) revealed 13 carbon signals: a carbonyl group C-4 (δC 182.3), two aromatic carbon C-3 (δC 108.9) and C-8 (δC 91.2), three nonoxygenated quaternary aromatic carbons at C-8a (δC 157.8), C-4a (δC 104.7), and C-6 (δC 102.3), three oxygenated quaternary aromatic carbons at C-2 (δC 168.8), C-5 (δC 159.6), and C-7 (δC 163.6), a methoxy groups CH3O-7 (δC 57.2), two methyl groups C-3′ (δC 39.1) and CH3-2 (δC 20.4), a methylene group C-1′ (δC 48.4). Analysis of the 1H and 13C NMR data of 5 revealed the presence of the same 5-hydroxy-4H-chromen-4-one moiety as found in xanthones [18,19]. A side-by-side comparison of the NMR spectroscopic data with those of 3 showed them to be nearly identical except for the final sulfur monoxide unit on the side chain.
According to the HMBC correlations from H-1′ to C-6, C-5 and C-7, H-3′ to C-1′, the sulfur atom present in 5 was shown to be attached at C-1′and C-3′, indicating that C-1′ was attached at C-6, Further confirmation was found for HMBC correlations of CH3O-7 to C7; H-3 to CH3-2, C-4a, C-2; H-8 to C-4a, C-6, C-8a, C-7 and C-4, a methoxy group could be located at C-7, a methyl group could be located at C-2. Selected key correlations in the observed NMR spectrum are shown in Figure 3. On the basis of these results, the structure of compound 5 was established as shown.
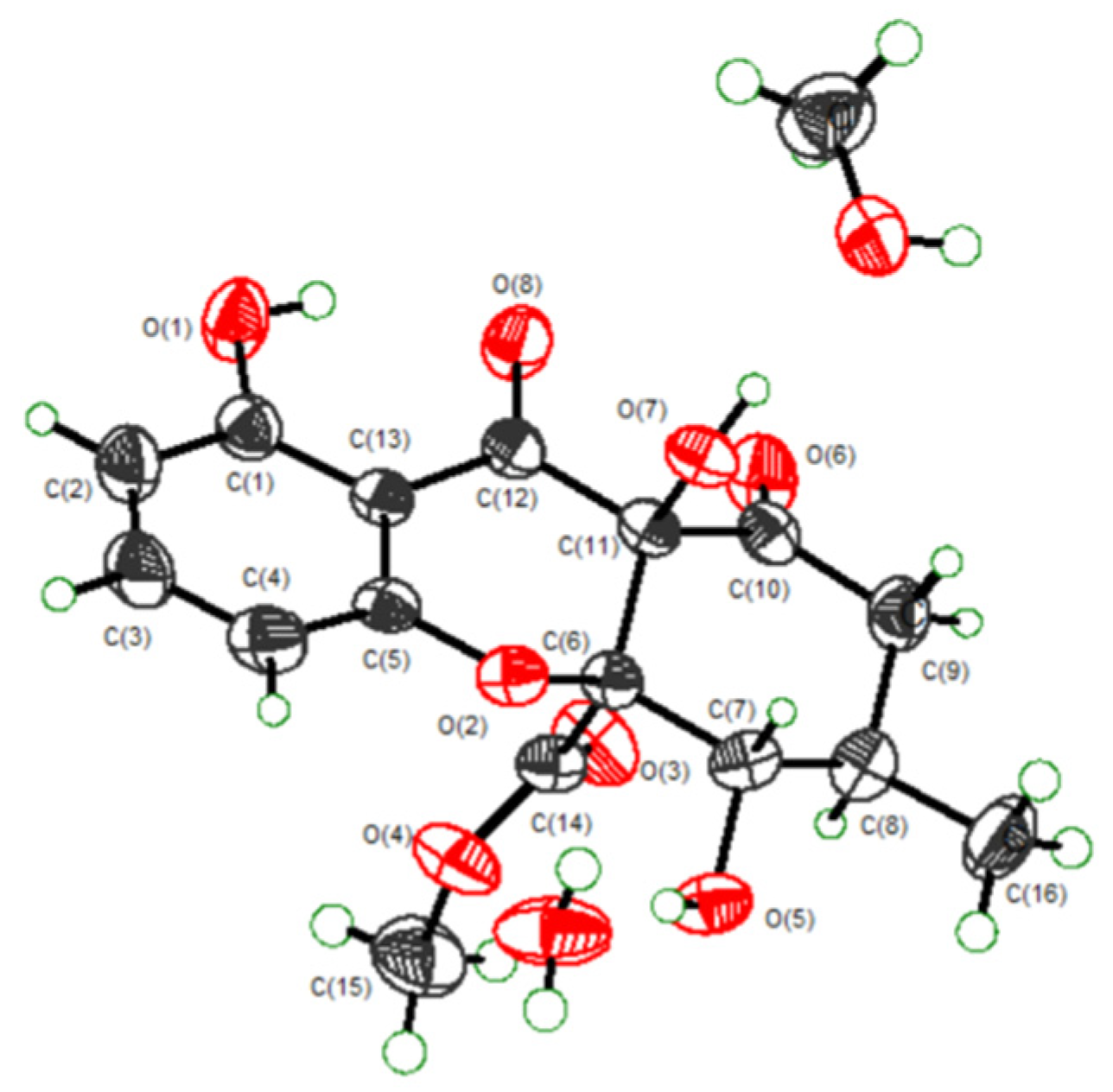
Amycolachromone F (6), −54 (c 0.1, MeOH), displayed HRESIMS peak at m/z 337.0915 [M + H]+ (calcd 337.0923), corresponding to the molecular formula C16H16O8 (nine degrees of unsaturation). Analysis of the 1H data of 6 (Table 3) revealed resonance for three aromatic protons at δH 7.52 (1H, t, J = 8.3 Hz, H-3), 6.60 (1H, d, J = 8.3 Hz, H-4), 6.53 (1H, d, J = 8.3 Hz, H-2), a methoxy group at δH 3.50 (3H, s, H-15), a methyl group at δH 1.06 (3H, d, J = 6.4 Hz, H-16), a methylene at δH 2.81 (1H, dd, J = 14.4, 12.9 Hz, H-9a) and 2.25 (1H, dd, J = 14.5, 5.3 Hz, H-9b), a oxygenated methine at δH 4.20 (1H, dd, J = 10.5, 6.0 Hz, H-7), a methine at δH 1.97–1.86 (1H, m, H-8), three hydroxyl groups at δH 11.35 (1H s, OH-1), 8.09 (1H, s, OH-11), and 5.91 (1H, d, J = 6.0 Hz, OH-7). The 13C NMR (Table 3) revealed sixteen carbon signals: three carbonyl group C-10 (δC 198.6), C-12 (δC 191.8) and C-14 (δC 168.5), three aromatic carbon C-3 (δC 138.7), C-2 (δC 109.5), and C-4 (δC 107.4), a nonoxygenated quaternary aromatic carbons at C-13 (δC 106.5), two oxygenated quaternary aromatic carbons at C-1 (δC 161.9) and C-5 (δC 158.9), two sp3-quaternary carbon C-11 (δC 90.0) and C-6 (δC 73.0), a methoxy group C-15 (δC 52.7), a methyl group C-16 (δC 18.6), an oxygenated methine C-7 (δC 71.8), a methine C-8 (δC 31.1) and a methylene C-9 group (δC 43.1). Analysis of the 1H and 13C NMR data of 6 revealed the presence of the same 5-hydroxy-4H-chromen-4-one moiety as found in xanthones [18,19]. In the 1H-1H COSY spectrum, the correlations from H-7 to H-8 and OH-7, from H-8 to H-9 and H-16. Further confirmation was found for HMBC correlations of H-7 to C-16, C-6, C-11 and C-9; H3-16 to C-7, C-8 and C-9, indicated that C-16 was attached to C-8, and OH-7 was located at C-7. HMBC correlations from the O-methyl proton signal H3-15 to the carboxylic carbon C-14 confirmed that the O-methyl group was located at C-14. HMBC correlations from OH-11 to C-11, C-6 and C-10, OH-1 to C-2, C-13 and C-1 indicated that OH-1 and OH-11 were attached to C-1 and C-11, respectively [22,23]. Selected key correlations in the observed NMR spectrum are shown in Figure 3. Thus, the planar structure of 6 was established. Moreover, the relative configuration of 6 was established to be 6R*, 7S*, 8R* and 11R* by X-ray crystallography using Mo Ka radiation (Figure 4).
Table 3.
1H (500 MHz) and 13C (125 MHz) NMR data of compound 6 in DMSO-d6.
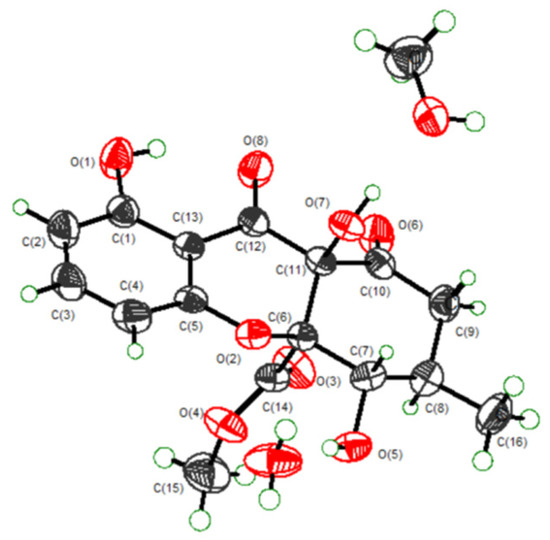
Figure 4.
ORTEP diagram for the single-crystal X-ray of Amycochromone F (6).
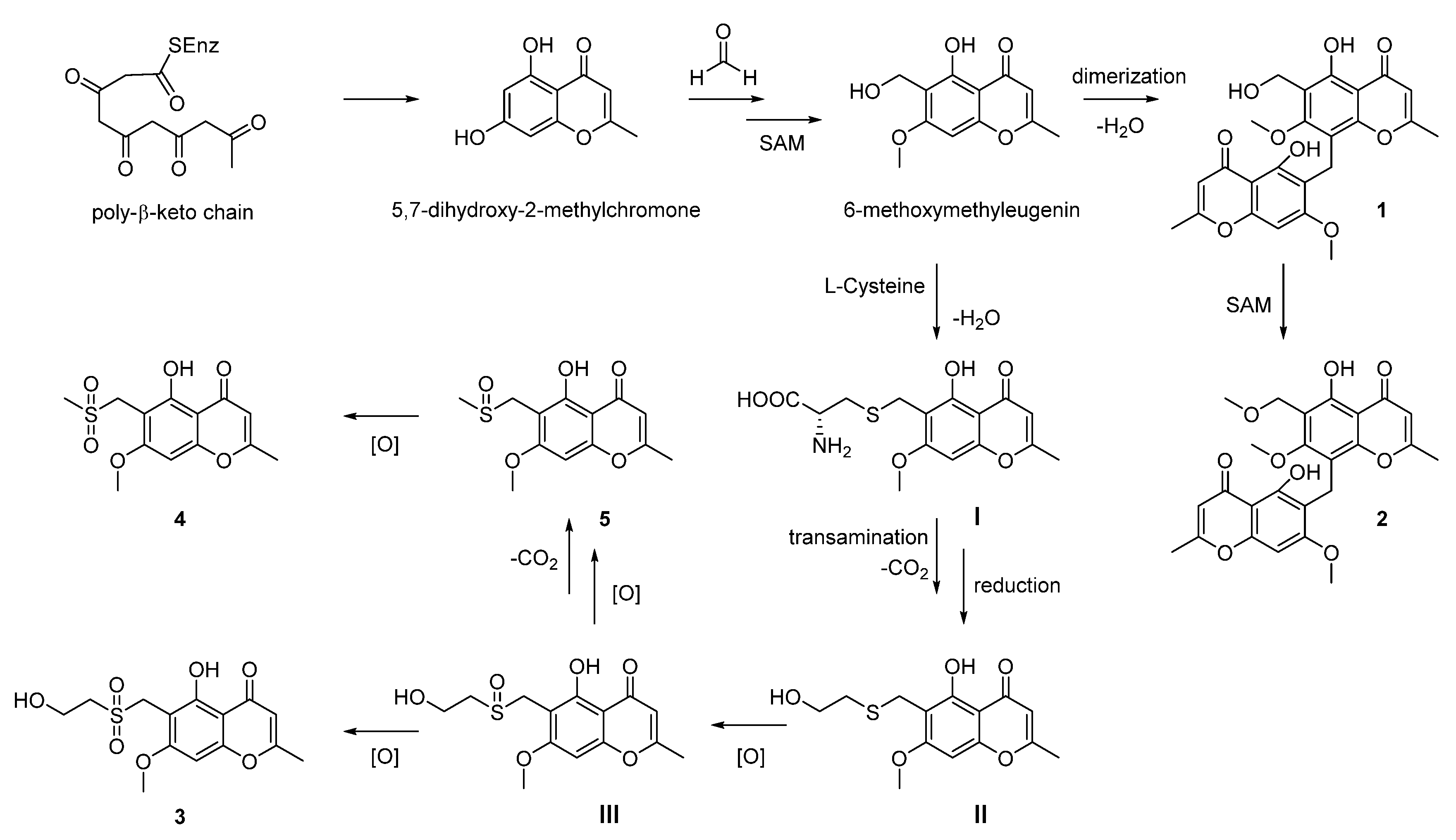
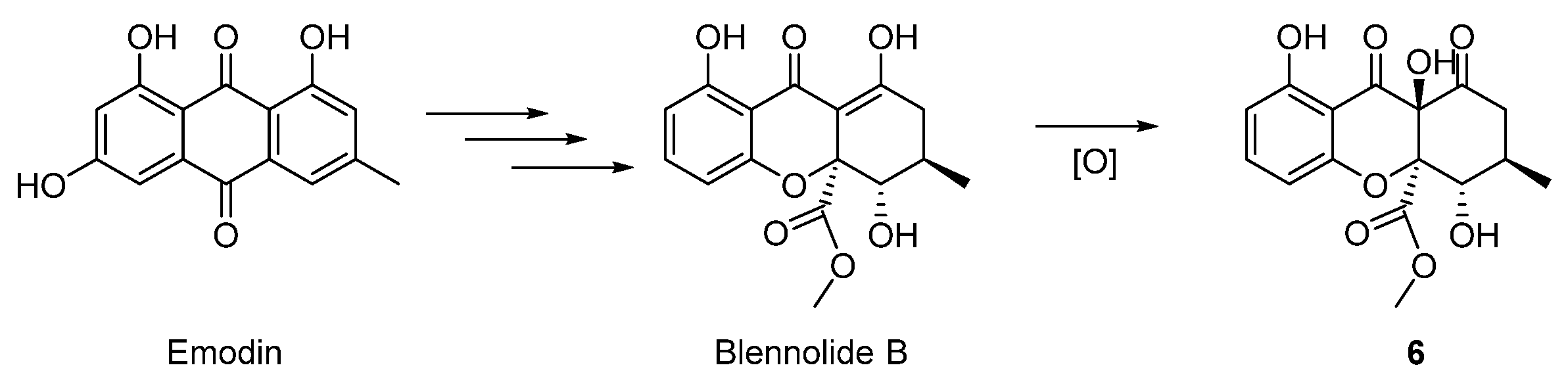
Further analysis of the structures allowed us to raise a plausible biosynthetic pathway of compounds 1–6. As outlined in the Scheme 1, compounds 1–5 were structurally related to the known metabolite 6-methoxymethyleugenin, which was derived from the widely existing 5,7-dihydroxy-2-methylchromone via the hydroxymethylation with formaldehyde and the methylation with SAM (S-adenosyl methionine). The compound 1 was the dimerization of 6-methoxymethyleugenin, and the sequential methylation with SAM could afford the related compound 2. For compound 3–5, we proposed that the sulfur in these structures was from l-cysteine. Thus, the Michael addition of l-cysteine to the ortho-quinone methide intermediate from 6-methoxymethyleugenin gave the compound I. Then, transamination, decarboxylation and reduction sequence of I furnished the 2-sulfo-ethanol II occurred. An oxidation of sulfur in II gave the compound III. Finally, compound 3 was obtained through the double oxidation of sulfur in II. The oxidation of the hydroxyl group in III to the corresponding carboxylic acid occurred and followed with a decarboxylation afforded for compound 5. Furthermore, compound 4 was the oxidation product of 5 [24,25]. In addition, compound 6 was the oxidation product of the known natural product blennolide B, which was proposed by Franck to be a derivative of emodin (Scheme 2) [26].
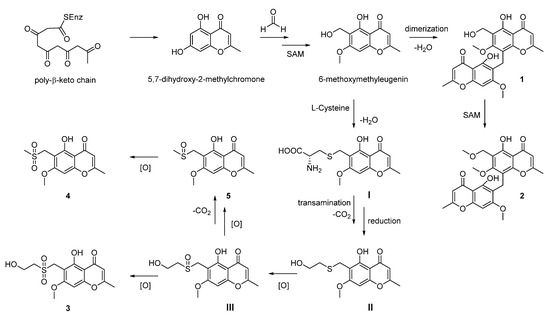
Scheme 1.
Proposed hypothetical biosynthesis pathway of 1–5.
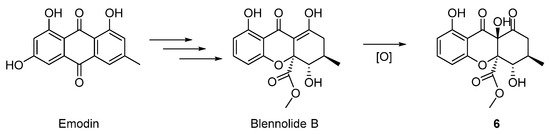
Scheme 2.
Proposed hypothetical biosynthesis pathway of 6.
The structures of five known compounds were identified as 6-ethoxymethyleugenin (7), 6-methoxymethyleugenin (8), 6-hydroxymethyleugenin (9), emodin (10) and the ascomycete metabolite chaetoquadrin D (11) by comparison of spectroscopic data with reported values and are described here for the first time as produced by Amycolatopsis sp.
The AlkB family of DNA repair enzymes utilize an α-ketoglutarate/Fe(II)-dependent mechanism to oxidize the aberrant alkyl groups, finally repairing alkyl DNA bases [27,28]. Compounds 1–11 were evaluated for their in vitro ABH2 inhibitory activities. Compounds 1–11 exhibited weak inhibitory activity against the ABH2 enzyme. However, in 2019, a paper was published that tested emodin (10). It exhibited strong inhibitory activity for the ALKH3 enzyme with IC50 of 8.8 μM [29]. This hinted that these compounds might inhibit other members of the AlkB family of enzymes.
In conclusion, the chemical investigation of a streptomycin-resistant strain of the deep-sea marine actinomycete, Amycolatopsis sp. WP1, led to the isolation and identification of six novel compounds, amycolachromones A–F (1–6) and five known analogues (7–11). Among them, amycolachromones A–B (1–2) represents an unusual fused skeleton between two 6-hydroxymethyleugenin, and the relative configuration of amycolachromones F (6) was determined by the signal-crystal X-ray diffraction. The discovery of amycolachromones A–F not only expanded the chemical diversity of natural products and inspire further synthetic studies, but also provided a template for the exploration of inhibitors of other members of the AlkB family of enzymes.
3. Materials and Methods
General experimental procedures. All chemical reagents and solvents were purchased from Sigma–Aldrich (Shanghai, China). UV spectra were acquired with a DU 800 UV/vis spectrophotometer (Beckman Coulter, Brea, CA, USA). IR spectra were acquired with a Nicolet 380 FT-IR (Thermo Electron Corporation, Beverly, MA, USA). NMR experiments were conducted using an Agilent NMR 500 MHz spectrometer (Santa Clara, CA, USA) and BRUKER NMR 600 MHz spectrometer (San Jose, CA, USA) with (CD3)2SO as the solvent (referenced to residual DMSO at δH 2.54 and δC 39.5) at 25 °C. Electrospray ionization mass spectra (ESIMS) were acquired using an AB Sciex TripleTOF 4600 spectrometer (Boston, MA, USA) in the positive and negative ion mode. HPLC experiments were performed on a Hitachi Elite LaChrom system (Tokyo, Japan) equipped with a diode array detector model L-2450, pump L-2130 and autosampler L-2200. Semipreparative HPLC experiments were completed with a Waters XBridge Prep C18 (Miflord, CO, USA) 5 μm, 10 mm × 250 mm column and Phenomenex Luna C18 5 μm, 250 mm × 21.2 mm column.
Bacterial Strain and Culture Conditions. The WP1 strain (CGMCC No. 10738) was isolated from deep-sea sediments of the Southwest Indian Ocean and identified as Amycolatopsis sp. by 16S rRNA sequence comparison. WP1 was grown in ISP2 medium consisting of 1.0% (w/v) malt extract, 0.4% (w/v) yeast extract, 0.4% (w/v) glucose and 3% (w/v) sea salt, the pH of medium was adjusted to 7.4 using 2 M HCl and 2 M NaOH.
Mutants of strain WP1. The WP1 strain suspensions were spread onto ISP2 plates containing different concentrations (0, 10, 20, 30, 40, 50 and 60 mg/mL) of streptomycin. The plates were incubated at 37 °C for 7 days. Mutant colonies producing the white pigment different than the WP1 strain were selected, generating mutant strain L-30-6, which was obtained on the IPS2 plate containing 30 mg/mL streptomycin.
Extraction and isolation. The mutant L-30-6 strain was inoculated into ISP2 broth with 3% sea salt in 250 mL Erlenmeyer flasks, at 30 °C on a rotary shaker at 180 rpm for 2 days as seed culture. Each of the seed cultures (32 mL) was transferred into 1 L Erlenmeyer flasks containing 400 mL of ISP2 supplemented with 3% sea salt. These flasks were incubated at 30 °C on a rotary shaker at 180 rpm for 6 days. The resulting cultures (60 L) were centrifuged to yield the supernatant and a mycelial pellet. The supernatant was adsorbed onto macroporous resin XAD16N (DOW, St. Louis, Missouri, CA, USA) and eluted with linear gradient of 0–100% EtOH in H2O to afford six fractions (A–F).
Fraction C (3.8 g) was subjected to semipreparative HPLC (Phenomenex Luna C18, 250 mm × 21.2 mm, 5 µm, 10 mL/min) using a gradient solvent from 40–90% MeOH in H2O over 30 min to give five fractions (C1–C5). Fraction C2 was further purified by semipreparative HPLC (Waters XBridge Prep C18 5 μm, 10 mm× 250 mm, 4 mL/min) using an isocratic solvent system of CH3CN:H2O (15:85) over 30 min to afford compound 6 (10.2 mg) and C2A. Subfraction C2A was further purified by preparative HPLC with MeOH-H2O (45:55) to provide compound 7 (2.6 mg). Fraction C3 was further purified by semipreparative HPLC with MeOH-H2O (45:55) to yield compound 12 (6.5 mg), 3 (3.1 mg) and 4 (2.2 mg). Fraction C4 was further purified by semipreparative HPLC with MeOH:H2O (45:55) to afford compound 5 (2.2 mg).
Fraction D (2.1 g) was subjected to semipreparative HPLC (Phenomenex Luna C18, 250 mm × 21.2 mm, 5 µm,10 mL/min) using a gradient solvent from 50–80% MeOH in H2O over 30 min to generate five fractions (D1–D5). Fraction D3 was further purified by semipreparative HPLC using an isocratic solvent system of MeCN:H2O (50:50) to afford compound 1 (1.7 mg) and compound 2 (1.8 mg). D4 was subjected to preparative HPLC with MeCN:H2O (30:70) to provide compounds 9 (35.3 mg) and 10 (13.5 mg). D5 was further purified by preparative HPLC with MeOH-H2O (45:55) to yield compounds 8 (1.9 mg) and 11 (6.8 mg). The following are details of the extraction and isolation of the compounds.
Amycolachromone A (1): White, amorphous powder; UV (MeOH) λmax (log ε) 253 (3.28) nm; IR (ZnSe) νmax 3426, 3195, 2844, 1656, 1445, 1008 cm−1; 1H and 13C NMR data, Table 1; HRESIMS m/z 477.1172 [M + Na]+ (calcd for C24H22O9, 477.1162).
Amycolachromone B (2): White, amorphous powder; UV (MeOH) λmax (log ε) 254 (3.46) nm; IR (ZnSe) νmax 3460, 3190, 2894, 1658, 1445, 1008 cm−1; 1H and 13C NMR data, Table 1; HRESIMS m/z 469.1502 [M + H]+ (calcd for C25H24O9, 469.1499), m/z 491.1333 [M + Na]+ (calcd 469.1318).
Amycolachromone C (3): White, amorphous powder; UV (MeOH) λmax (log ε) 250 (3.43), 233 (3.46) nm; IR (ZnSe) νmax 3420, 3199, 2993, 1650, 1310, 1089 cm−1; 1H and 13C NMR data, Table 2; HRESIMS m/z 351.0515 [M + Na]+ (calcd for C14H16O7S, 351.0514).
Amycolachromone D (4): White, amorphous powder; UV (MeOH) λmax (log ε) 250 (3.56), 233 (3.58) nm; IR (ZnSe) νmax 3520, 32,469, 2990, 1750, 1281, 1008 cm−1; 1H and 13C NMR data, Table 2; HRESIMS m/z 321.0406 [M + Na]+ (calcd for C13H14O6S, 321.0409).
Amycolachromone E (5): White, amorphous powder; UV (MeOH) λmax (log ε) 250 (3.61), 240 (3.61) nm; IR (ZnSe) νmax 3470, 3122, 2880, 1603, 1210, 1089 cm−1; 1H and 13C NMR data, Table 2; HRESIMS m/z 305.0462 [M + Na]+ (calcd for C13H14O5S, 305.0460).
Amycolachromone F (6): White, crystal; UV (MeOH) λmax (log ε) 356 (3.20), 277 (3.68) nm; IR (ZnSe) νmax 3477, 2956, 2916, 1748, 1622, 1475, 1349, 1083 cm−1; 1H and 13C NMR data, Table 3; HRESIMS m/z 337.0915 [M + H]+ (calcd for C16H16O8, 337.0923).
6-Ethoxymethyleugenin (7): White, amorphous powder; HR-ESIMS m/z 287.0891 [M + Na]+ (calcd for C14H16O5, 287.0985). 1H-NMR (600 MHz, DMSO-d6): δH 13.19 (s, OH-5), 6.70 (s, 1H, H-8), 6.28 (s, 1H, H-3), 4.41 (s, 2H, CH2OCH3), 3.89 (s, 3H, OCH3), 3.44 (q, J = 7.0 Hz, 2H, 6-CH2OCH2), 2.40 (s, 3H, 2-CH3), 1.08 (t, J = 7.0 Hz, 3H, OCH2CH3). 13C-NMR (150 MHz, DMSO-d6): δC182.6 (C-4), 168.5 (C-2), 164.3 (C-7), 159.9 (C-5), 158.0 (C-8a), 109.1 (C-3), 108.9 (C-6), 104.4 (C-4a), 89.6 (C-8), 65.1 (CH2OCH3), 59.3 (6-CH2OCH2), 56.9 (7-OCH3), 20.4 (2-CH3) and 15.3 (CH2CH3).
6-Methoxymethyleugenin (8): White, amorphous powder; HR-ESIMS m/z 273.0738 [M + Na]+ (calcd for C13H14O, 273.0739). 1H-NMR (600 MHz, CDCl3): δH 13.04 (s, OH-5), 6.36 (s, 1H, H-8), 6.04 (s, 1H, H-3), 4.55 (s, 2H, CH2OCH3), 3.90 (s, 3H, OCH3), 3.40 (s, 3H, CH2OCH3), 2.35 (s, 3H, CH3). 13C-NMR (150 MHz, CDCl3): δC182.4 (C-4), 166.6 (C-2), 164.2 (C-7), 160.6 (C-5), 158.2 (C-8a), 109.1 (C-3), 108.9 (C-6), 105.1 (C-4a), 89.6 (C-8), 61.6 (CH2OCH3), 58.2 (CH2OCH3), 56.2 (OCH3) and 20.4 (2-CH3).
6-Hydroxymethyleugenin (9): White, amorphous powder; HRESIMS m/z 259.0579 [M + Na]+ (calcd for C12H12O5, 259.0582). 1H-NMR (600 MHz, DMSO-d6): δH 13.09 (s, OH-5), 6.65 (s, 1H, H-8), 6.24 (s, 1H, H-3), 4.55 (t, J = 5.3 Hz, CH2OH), 4.43 (d, J = 5.2 Hz, 2H, H-9), 3.87 (s, 3H, OCH3), 2.38 (s, 3H, CH3). 13C-NMR (150 MHz, DMSO-d6): δC182.5 (C-4), 168.4 (C-2), 164.1 (C-7), 159.1 (C-5), 157.7 (C-8a), 112.5 (C-6), 08.8 (C-3), 104.6 (C-4a), 90.7 (C-8), 56.7 (OCH3), 50.9 (CH2OH) and 20.4(CH3).
Emodin (10): White, amorphous powder; HRESIMS m/z 269.0448 [M − H]− (calcd for C15H12O5, 269.0450). 1H-NMR (500 MHz, DMSO-d6): δH 12.07 (d, J = 19.5 Hz, 2H, OH), 7.49 (d, J = 1.1 Hz, 1H, H-5), 7.16 (s, 1H, H-7), 7.10 (d, J = 2.4 Hz, 1H, H-4), 6.57 (d, J = 2.4 Hz, 1H, H-2), 2.41 (s, 3H, CH3). 13C-NMR (150 MHz, DMSO-d6): δC190.0 (C-9), 182.0 (C-10), 166.6 (C-6), 165.0 (C-8), 161.9 (C-1), 148.6 (C-3), 135.6 (C-10a), 133.3 (C-4a), 24.6 (C-2), 120.9 (C-4), 113.9 (C-9a), 109.6 (C-5), 109.2 (C-7), 108.4 (C-8a) and 22.0 (CH3).
Chaetoquadrin D (11): White, amorphous powder; HRESIMS m/z HR-ESIMS m/z 370.0969 [M + H]+ (calcd for C16H19NO7S, 370.0960). 1H-NMR (500 MHz, DMSO-d6): δH 13.43 (s, 5-OH), 8.08 (t, NH), 6.79 (s, H-8), 6.32 (s, H-3), 4.36 (s, H-1′), 3.92 (s, 7-OCH3), 3.45 (dd, J = 13.6, 6.1 Hz, H-4′), 3.20 (t, J = 7.0 Hz, H-3′), 2.41 (s, 2-CH3), 1.81 (s, H-7′). 13C-NMR (125 MHz, DMSO-d6): δC 182.4 (C-4), 70.1 (C-6′), 169.0 (C-2), 163.7 (C-7), 159.9 (C-5), 158.2 (C-8a), 108.9 (C-3), 104.5 (C-4a), 100.8 (C-6), 91.4 (C-8), 57.3 (7-OCH3), 52.9 (C-3′), 48.5 (C-1′), 32.9 (C-4′), 22.9 (C-7′) and 20.42 (2-CH3).
X-ray Crystallographic Analysis of Compound 6. Crystals of 6 were obtained in the mixed solvent comprising MeOH and H2O, and crystallographic data were deposited at the Cambridge Crystallographic Data Centre (CCDC) under the reference number CCDC 1873441. The X-ray diffraction data were collected with Mo Kα radiation (λ = 0.71073 Å). The structure was solved by direct methods using the SHELXS-97 program. Orthorhombic C16H16O8, CH4O, H2O, a = 7.7760(3) Å, b = 8.6993(4) Å, c = 26.8196(11) Å, α = 90°, β = 90°, γ = 90°, V = 1814.23(13) Å3, Z = 4, ρCalcd = 1.414 g/cm3, μ = 0.118 mm−1, and F (0 0 0) = 816. Measurements were in the range 3.038° ≤ θ ≤ 26.368°, with 3697 independent reflections, of which 3133 unique reflections with I > 2σ (I) were collected for the analysis, Rint = 0.0332. The final R indices: R1 = 0.0455, wR2 = 0.1177 [I > 2σ(I)], R indices (all data): R1 = 0.0566, wR2 = 0.1256, and largest difference peak and hole: 0.560 and-0.231 e Å-3.
The ABH2 family DNA repair enzyme assay. Effects of compounds 1–11 on the ABH2 family demethylase activity reactions on m3c-ss-DNA were evaluated. All reactions were performed at 37 °C in reaction buffer [5 μM Fe(NH4)2(SO4)2, 0.93 mMα-ketoglutarate, 1.86 mM ascorbic acid, and 46.56 mM HEPES (pH 8.0)] for 1 h. Varying concentrations of compounds 1–11 (0, 5.0, 7.5, 20, 30, 40, 50, 75 and 100 μM) were used for tests. The m3c-ss-DNA was pre-mixed with reaction buffer in a concentration of 5.0 μM. The reactions were initiated by adding 2.0 μM ABH2. The reactions were stopped by adding 10.0 mM EDTA followed by heating to 95 °C for 5 min. All the results of reaction were analyzed by HPLC. All reaction samples were quantified by DNApac PA-100 column (4 mm × 250 mm, Thermo Scientific, (Waltham, MA, USA) with isocratic 60 % mobile B, 1.5 M ammonium acetate, under a constant flow rate of 1.0 mL/min. Mobile A was water. The UV detection wavelength was 260 nm.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/md20030162/s1. Figures S1–S35: 1D and 2D NMR, HRESI mass spectra, and crystal data for compounds 1–6. Table S1. Crystallographic data for Amycochromone F (6).
Author Contributions
J.C. (Jianwei Chen), J.C. (Jun Chen), S.W., X.B., S.L., B.W., H.Z. and H.W. performed the experiments and analyzed the data; S.L. and B.W. contributed materials and analysis tools; J.C. (Jianwei Chen) and H.W. wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Key Research and Development Program of Zhejiang Province (2021C03084), Natural Foundation of Zhejiang Province (LGF21H300003), National Key R&D Program of China (2018YFC0311003, 2017YFE0103100), High-level Talent Special Support Plan of Zhejiang Province (2019R52009).
Institutional Review Board Statement
No applicable.
Informed Consent Statement
No applicable.
Acknowledgments
We thank Hang Ma, Hilary Ranson, Navindra P. Seeram, Deyu Li, David C. Rowley and Al Bach at the College of Pharmacy, University of Rhode Island for their kind assistance.
Conflicts of Interest
The authors declare no competing financial interest.
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2017, 34, 235–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2016, 33, 382–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2015, 32, 116–211. [Google Scholar] [CrossRef] [Green Version]
- Nutzmann, H.W.; Reyes-Dominguez, Y.; Scherlach, K.; Schroeckh, V.; Horn, F.; Gacek, A.; Schumann, J.; Hertweck, C.; Strauss, J.; Brakhage, A.A. Bacteria-induced Natural Product Formation in the Fungus Aspergillus nidulans Requires Saga/Ada-mediated Histone Acetylation. Proc. Natl. Acad. Sci. USA 2011, 108, 14282–14287. [Google Scholar] [CrossRef] [Green Version]
- Bentley, S.D.; Chater, K.F.; Cerdeno-Tarraga, A.M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.A.; Kieser, H.; Harper, D.; et al. Complete Genome Sequence of the Model Actinomycete Streptomyces coelicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar] [CrossRef]
- Nett, M.; Ikeda, H.; Moore, B.S. Genomic Basis for Natural Product Biosynthetic Diversity in the Actinomycetes. Nat. Prod. Rep. 2009, 26, 1362–1384. [Google Scholar] [CrossRef]
- Bode, H.B.; Bethe, B.; Hofs, R.; Zeeck, A. Big Effects from Small Changes: Possible Ways to Explore Nature’s Chemical Diversity. Chembiochem 2002, 3, 619–627. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, H.; Ye, X.; Wei, B.; Emam, M.; Zhang, H.; Wang, H. The Structural Diversity of Marine Microbial Secondary Metabolites Based on Co-Culture Strategy: 2009–2019. Mar. Drugs 2020, 18, 449. [Google Scholar] [CrossRef]
- Henrikson, J.C.; Hoover, A.R.; Joyner, P.M.; Cichewicz, R.H. A Chemical Epigenetics Approach for Engineering the in Situ Biosynthesis of a Cryptic Natural Product from Aspergillus niger. Org. Biomol. Chem. 2009, 7, 435–438. [Google Scholar] [CrossRef]
- Ochi, K.; Hosaka, T. New strategies for drug discovery: Activation of silent or weakly expressed microbial gene clusters. Appl. Microbiol. Biotechnol. 2013, 97, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Ochi, K.; Tanaka, Y.; Tojo, S. Activating the Expression of Bacterial Cryptic Genes by rpoB Mutations in RNA Polymerase or by Rare Earth Elements. J. Ind. Microbiol. Biotechnol. 2014, 41, 403–414. [Google Scholar] [CrossRef]
- Shima, J.; Hesketh, A.; Okamoto, S.; Kawamoto, S.; Ochi, K. Induction of Actinorhodin Production by rpsL (Encoding Ribosomal Protein S12) Mutations that Confer Streptomycin Resistance in Streptomyces lividans and Streptomyces coelicolor A3(2). J. Bacteriol. 1996, 178, 7276–7284. [Google Scholar] [CrossRef] [Green Version]
- Fdhila, F.; Vazquez, V.; Sanchez, J.L.; Riguera, R. DD-diketopiperazines: Antibiotics Active against Vibrio anguillarum Isolated from Marine Bacteria Associated with Cultures of Pecten maximus. J. Nat. Prod. 2003, 66, 1299–1301. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, H.; Xu, S.; Wang, B.; Ju, J.; Tan, H.; Li, W. Activation and Enhancement of Fredericamycin A production in Deepsea-derived Streptomyces somaliensis SCSIO ZH66 by Using Ribosome Engineering and Response Surface Methodology. Microb. Cell Fact. 2015, 14, 64. [Google Scholar] [CrossRef] [Green Version]
- Fu, P.; Jamison, M.; La, S.; MacMillan, J.B. Inducamides A-C, Chlorinated Alkaloids from an RNA Polymerase Mutant Strain of Streptomyces sp. Org. Lett. 2014, 16, 5656–5659. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, A.; Matos, M.J.; Garrido, J.; Uriarte, E.; Borges, F. Chromone: A Valid Scaffold in Medicinal Chemistry. Chem. Rev. 2014, 114, 4960–4992. [Google Scholar] [CrossRef] [Green Version]
- Reis, J.; Gaspar, A.; Milhazes, N.; Borges, F. Chromone as a Privileged Scaffold in Drug Discovery: Recent Advances. J. Med. Chem. 2017, 60, 7941–7957. [Google Scholar] [CrossRef]
- Sumarah, M.W.; Puniani, E.; Blackwell, B.A.; Miller, J.D. Characterization of Polyketide Metabolites from Foliar Endophytes of Picea glauca. J. Nat. Prod. 2008, 71, 1393–1398. [Google Scholar] [CrossRef]
- Zhang, F.; Li, L.; Niu, S.; Si, Y.; Guo, L.; Jiang, X.; Che, Y. A Thiopyranchromenone and Other Chromone Cerivatives from an Endolichenic fungus, Preussia africana. J. Nat. Prod. 2012, 75, 230–237. [Google Scholar] [CrossRef]
- Tuntiwachwuttikul, P.; Phansa, P.; Pootaeng-On, Y.; Taylor, W.C. Chromones from the Branches of Harrisonia perforata. Chem. Pharm. Bull. 2006, 54, 44–47. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.J.; Blunt, J.W.; Cole, A.L.J.; Munro, M.H.G. The Isolation of Two New Chromone Derivatives from the New Zealand fungus Tolypocladium extinguens. J. Nat. Prod. 2002, 65, 1681–1682. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.; Kaleem, S.; Yi, W.W.; Wu, B.; Zhang, Z.Z. New Polyhydroxanthones from the Marine-Associated Fungus Penicillium sp. ZZ1750. Tetrahedron Lett. 2021, 81, 153354. [Google Scholar] [CrossRef]
- El-Elimat, T.; Raja, H.A.; Day, C.S.; McFeeters, H.; McFeeters, R.L.; Oberlies, N.H. α-Pyrone derivatives, Tetra/Hexahydroxanthones, and Cyclodpsipeptides from Two Freshwater Fungi. Bioorg. Med. Chem. 2017, 25, 795–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, T.; Porco, J.A., Jr. Total Syntheses of Secalonic Acids A and D. Angew. Chem. Int. Ed. Engl. 2014, 53, 3107–3110. [Google Scholar] [CrossRef] [Green Version]
- Qin, T.; Iwata, T.; Ransom, T.T.; Beutler, J.A.; Porco, J.A., Jr. Syntheses of Dimeric Tetrahydroxanthones with Varied Linkages: Investigation of “Shapeshifting” Properties. J. Am. Chem. Soc. 2015, 137, 15225–15233. [Google Scholar] [CrossRef] [Green Version]
- Franck, B.; Stöckigt, J.; Zeidler, U.; Franckowiak, G. Stereospezifische Synthese der 10-Methyl-10-desmethoxycarbonyl-hemisecalonsäure A. Chem. Ber. 1973, 106, 1198–1220. [Google Scholar] [CrossRef]
- Chen, F.; Tang, Q.; Bian, K.; Humulock, Z.T.; Yang, X.; Jost, M.; Drennan, C.L.; Essigmann, J.M.; Li, D. Adaptive Response Enzyme AlkB Preferentially Repairs 1-Methylguanine and 3-Methylthymine Adducts in Double-Stranded DNA. Chem. Res. Toxicol. 2016, 29, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Bian, K.; Chen, F.; Humulock, Z.T.; Tang, Q.; Li, D. Copper Inhibits the alkb Family DNA Repair Enzymes under Wilson’s Disease Condition. Chem. Res. Toxicol. 2017, 30, 1794–1796. [Google Scholar] [CrossRef]
- Pilzys, T.; Marcinkowski, M.; Kukwa, W.; Garbicz, D.; Dylewska, M.; Ferenc, K.; Mieczkowski, A.; Kukwa, A.; Migacz, E.; Wolosz, D. ALKBH Overexpression in Head and Neck Cancer: Potential Target for Novel Anticancer Therapy. Sci. Rep. 2019, 9, 13249. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).