
Marine Power on Cancer: Drugs, Lead Compounds, and Mechanisms



Abstract
1. Introduction
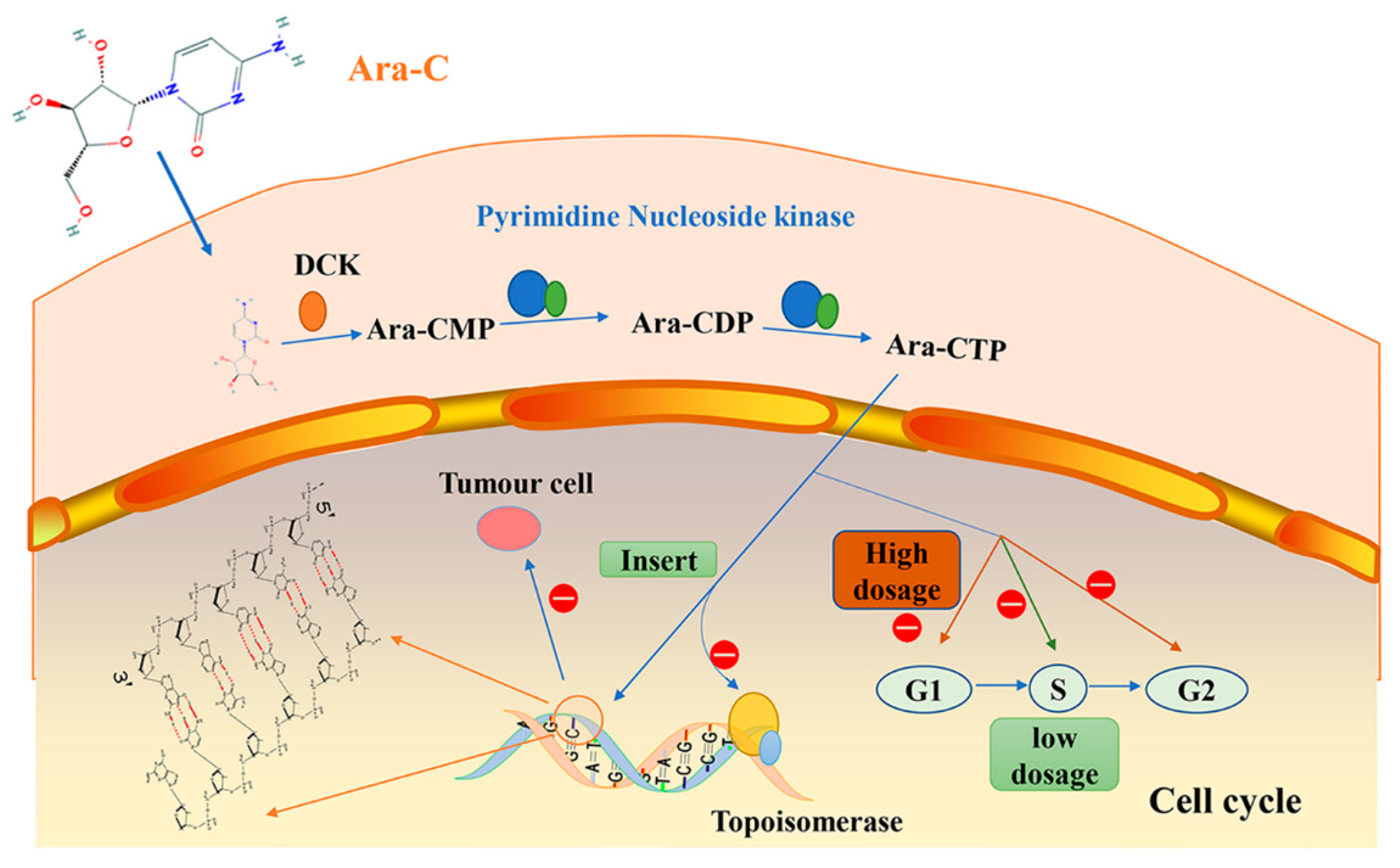
2. Cytarabine/Ara-C
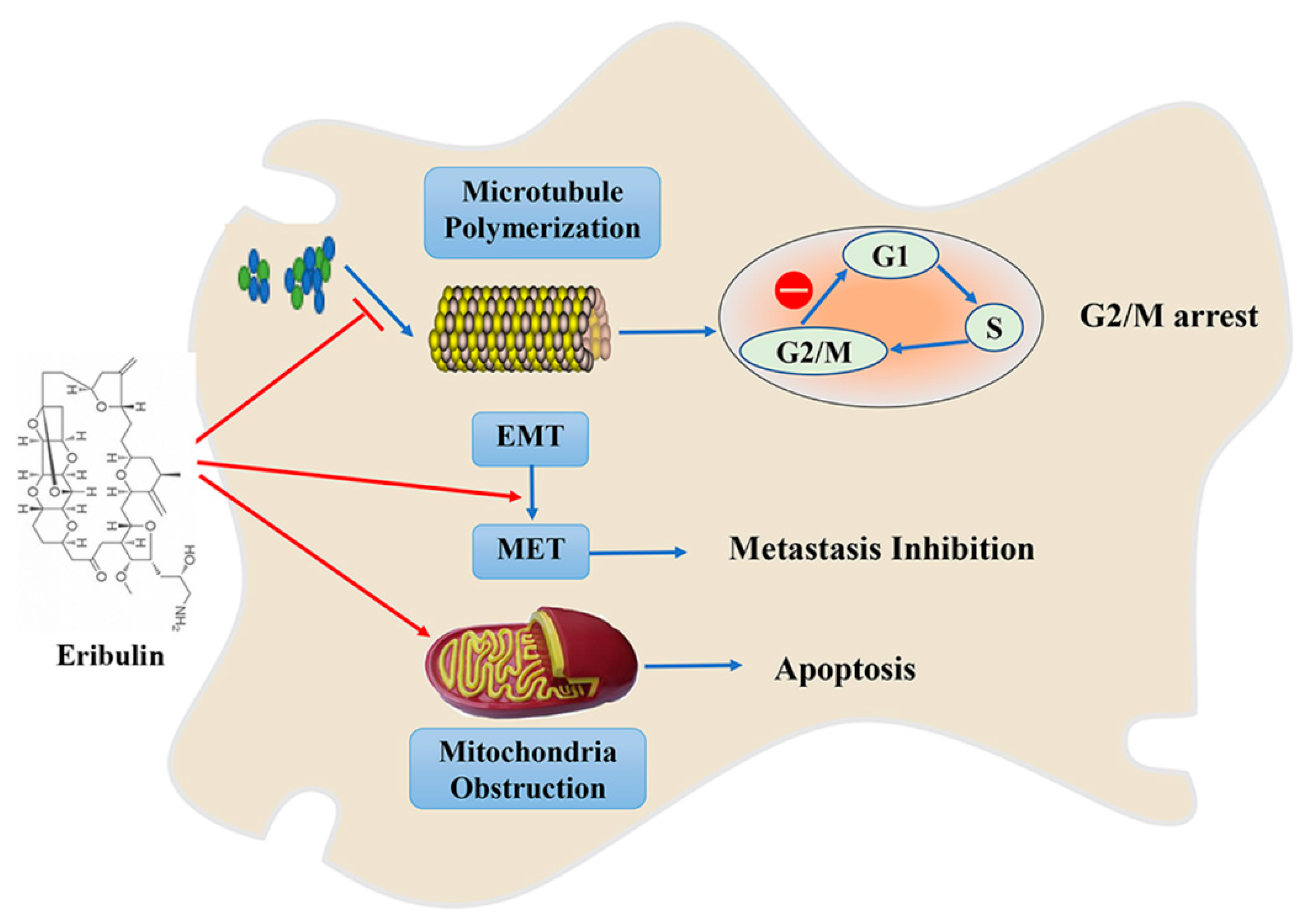
3. Eribulin/E7389
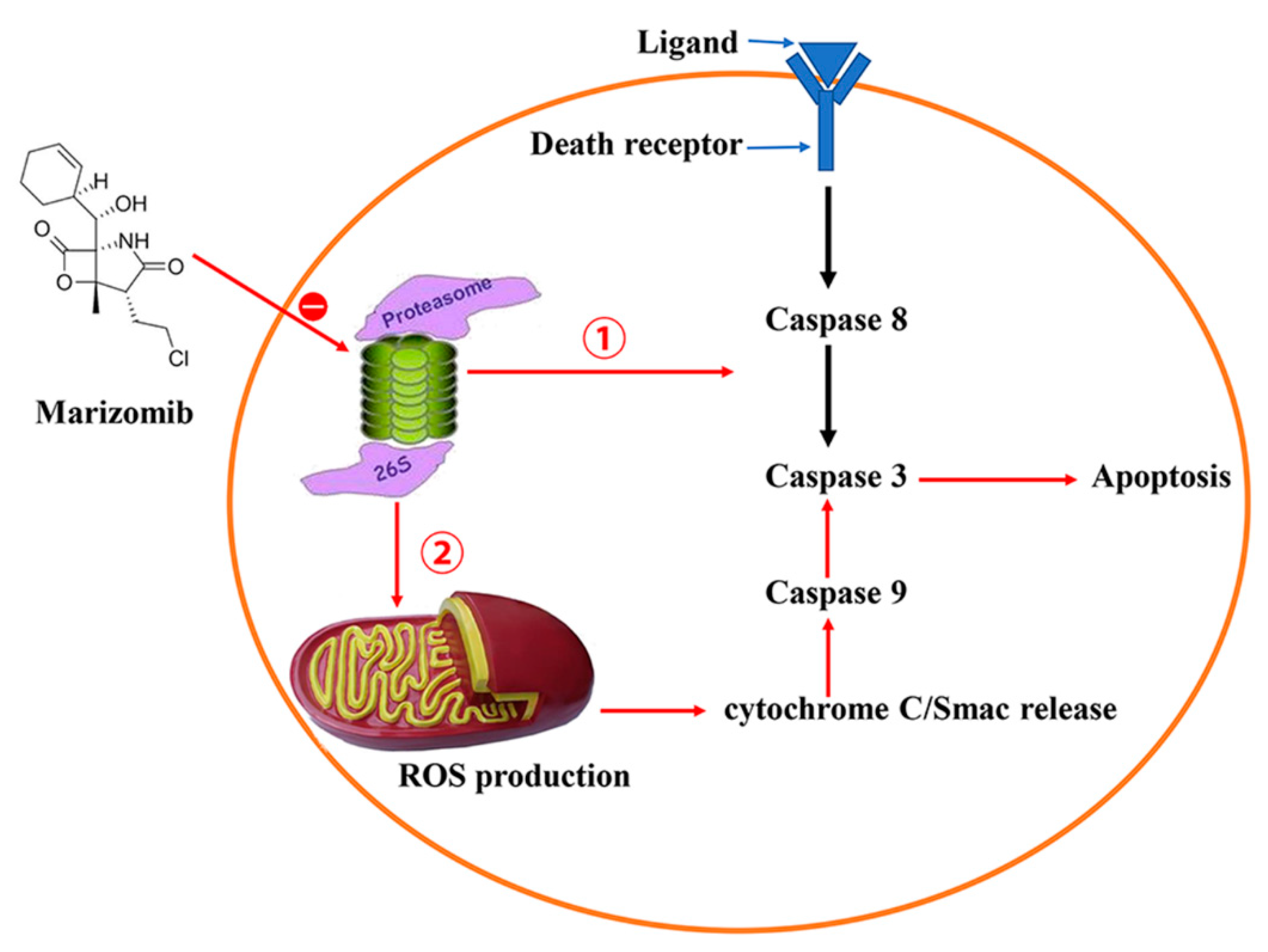
4. Marizomib/NPI-0052
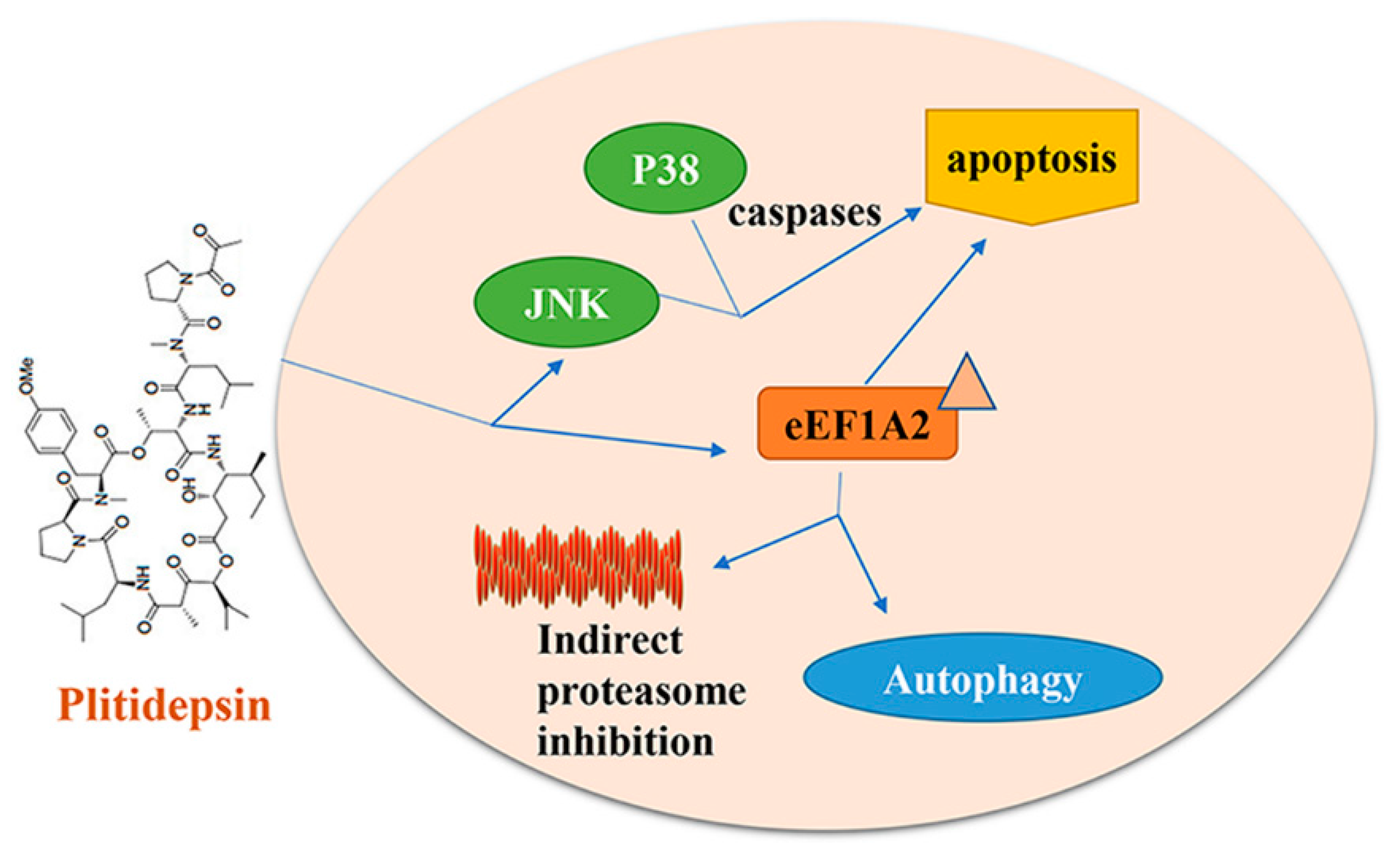
5. Plitidepsin/Aplidine
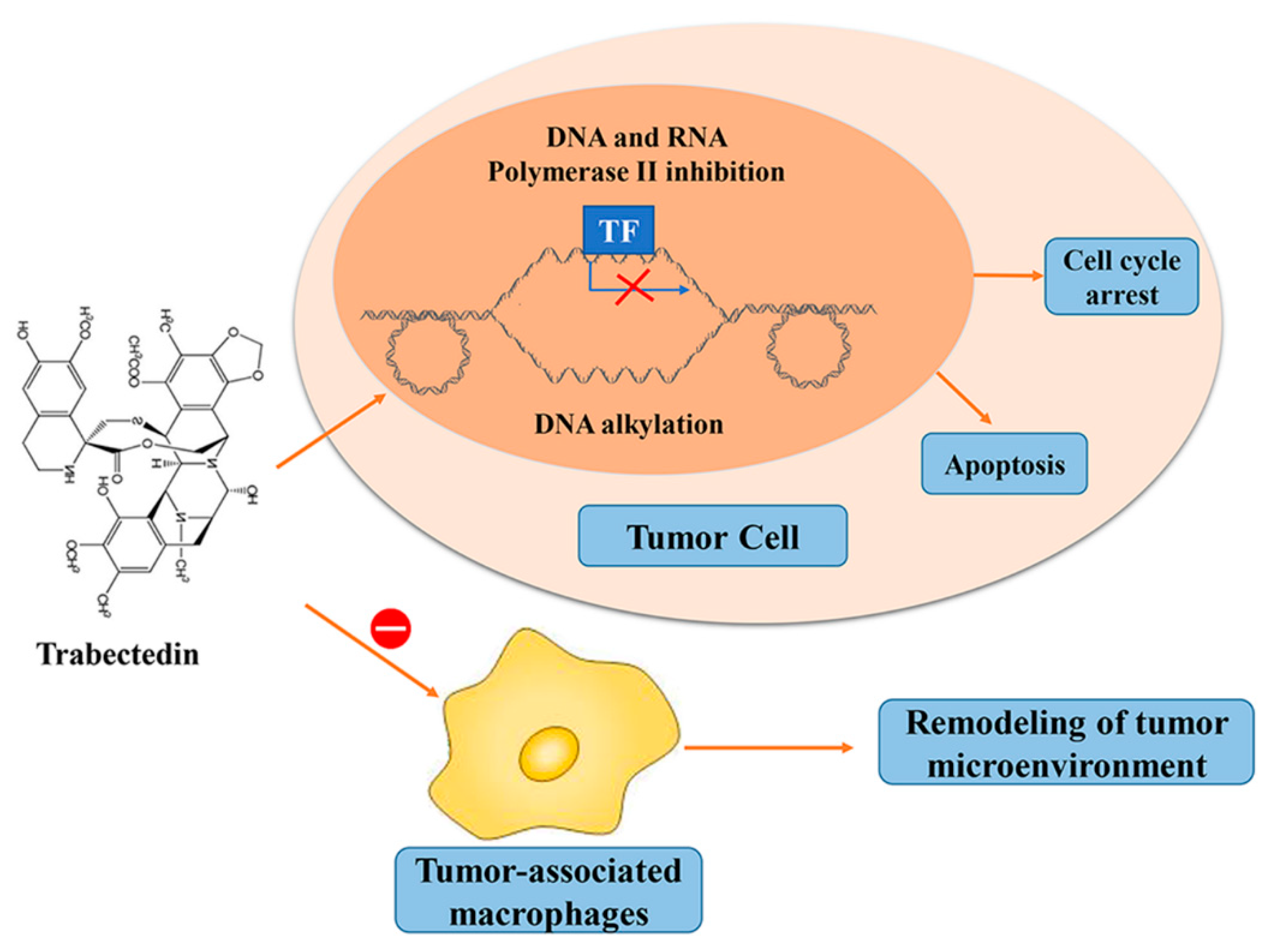
6. Trabectedin/ET-743
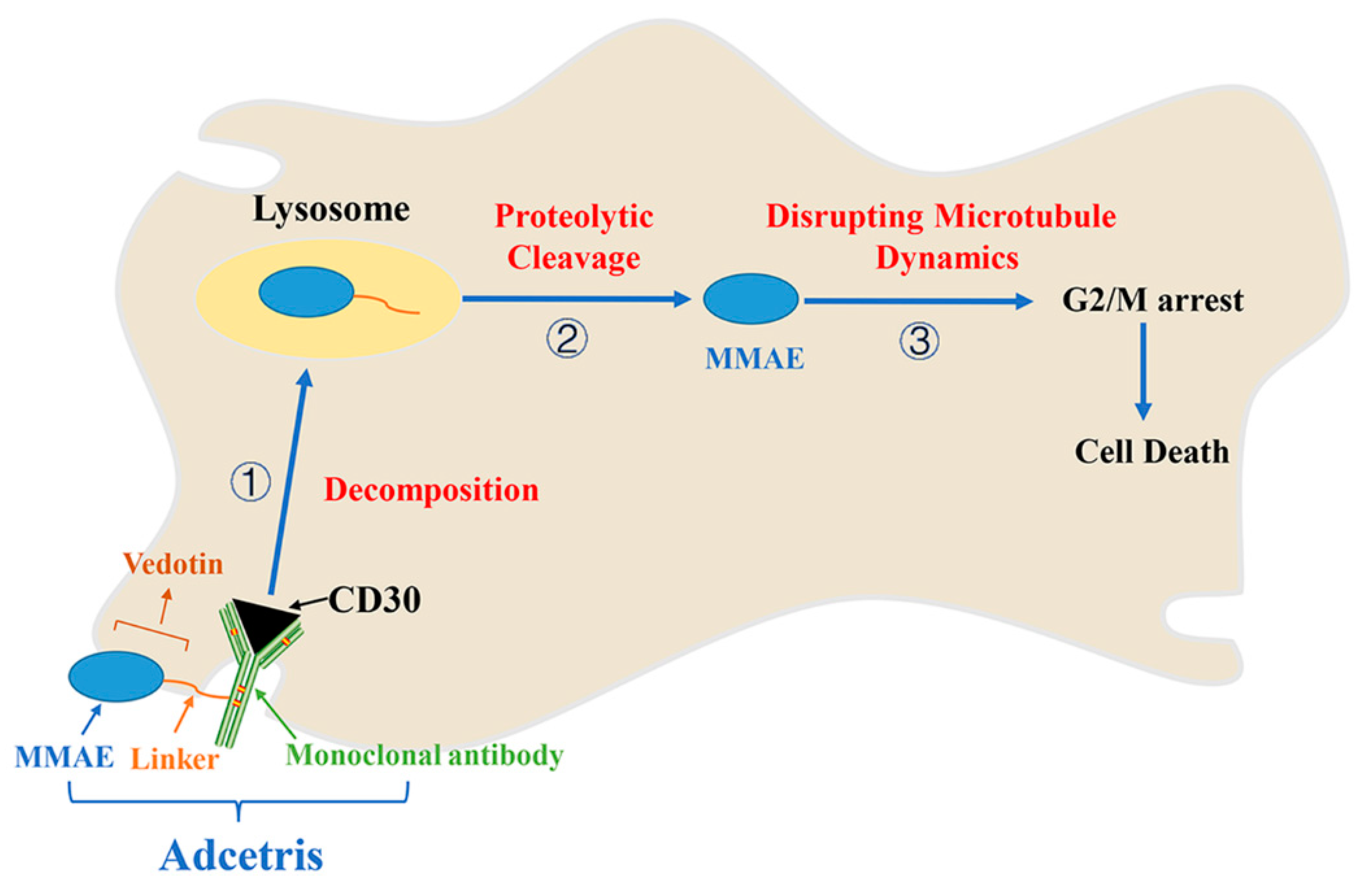
7. Adcetris/Brentuximab Vedotin
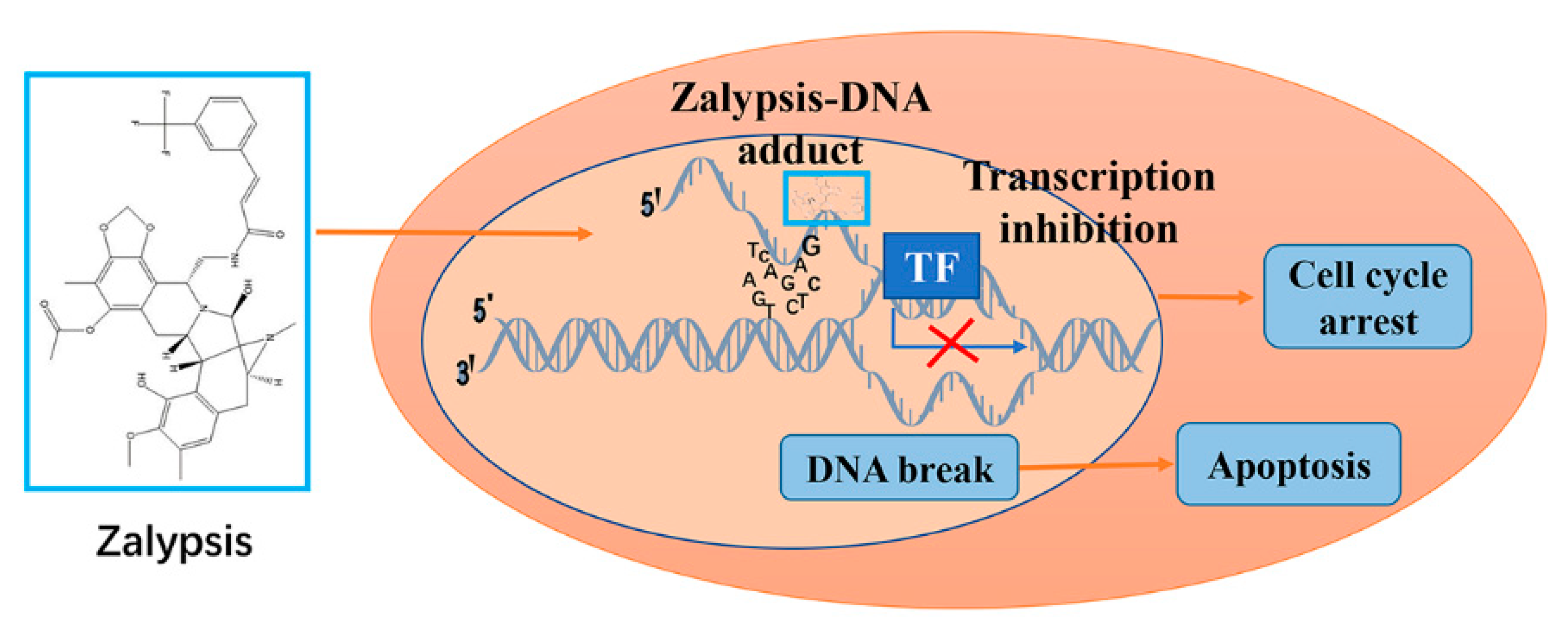
8. Zalypsis/M00104
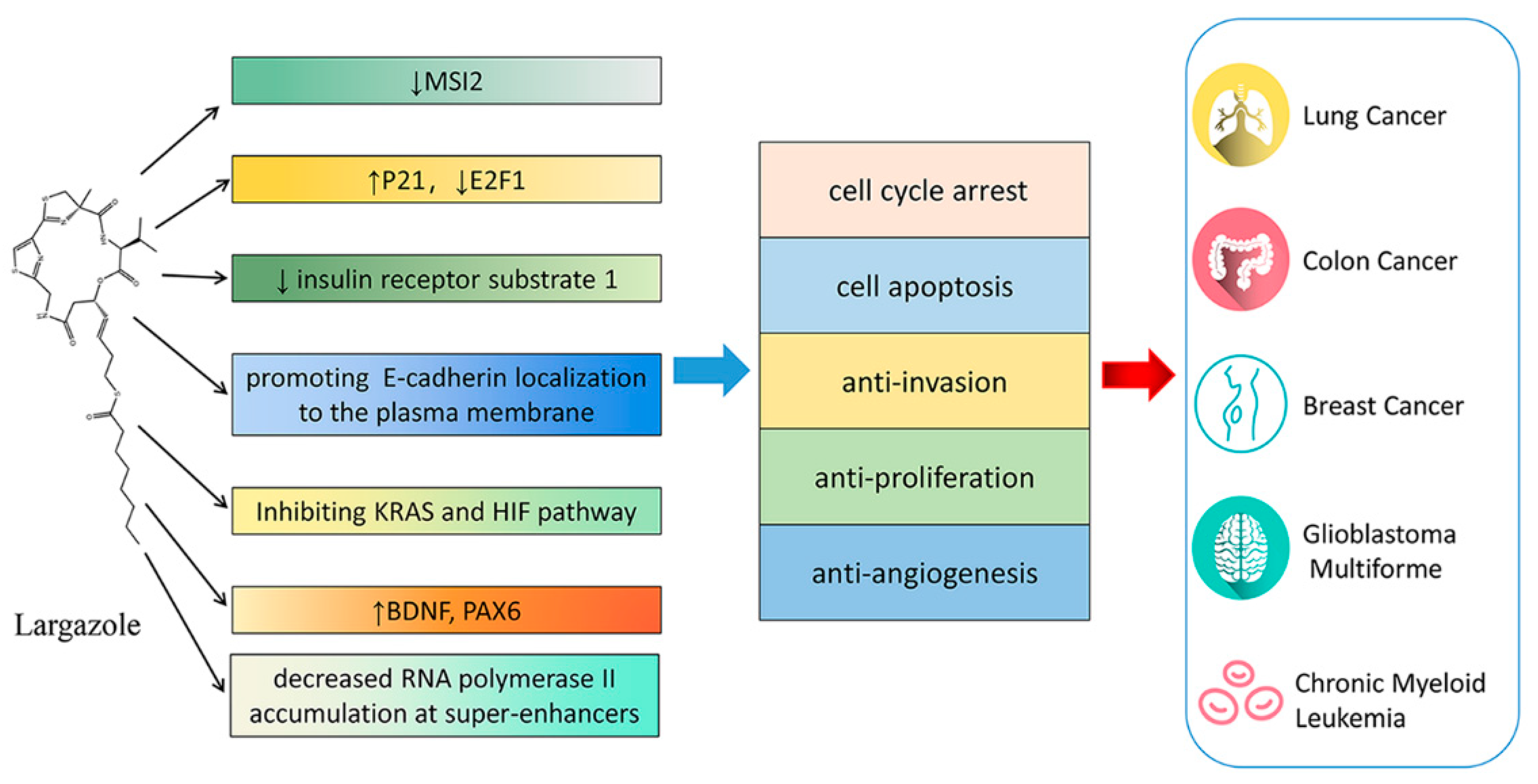
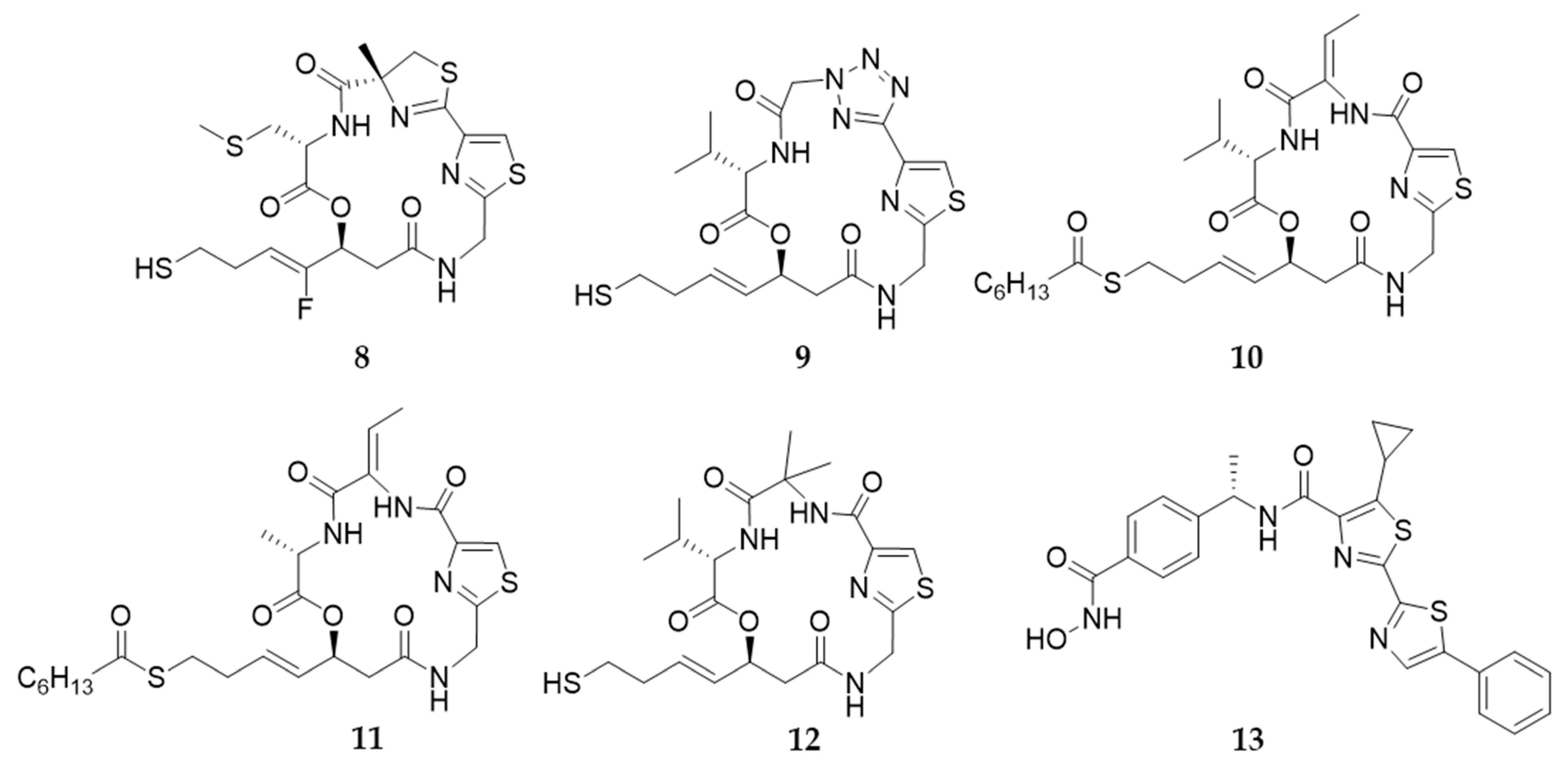
9. Largazole and Its Analogues
10. Conclusions and Prospect
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef]
- Jimenez, C. Marine Natural Products in Medicinal Chemistry. ACS Med. Chem. Lett. 2018, 9, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine Natural Products: A Source of Novel Anticancer Drugs. Mar. Drugs 2019, 17, 491. [Google Scholar] [CrossRef] [PubMed]
- Shinde, P.; Banerjee, P.; Mandhare, A. Marine natural products as source of new drugs: A patent review (2015–2018). Expert. Opin. Pat. 2019, 29, 283–309. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Spano, V.; Venturini, A.; Genovese, M.; Barreca, M.; Raimondi, M.V.; Montalbano, A.; Galietta, L.J.V.; Barraja, P. Current development of CFTR potentiators in the last decade. Eur. J. Med. Chem. 2020, 204, 112631. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M.; Spanò, V.; Montalbano, A.; Cueto, M.; Bertoni, F. Marine Anticancer Agents: An Overview with a Particular Focus on Their Chemical Classes. Mar. Drugs 2020, 18, 619. [Google Scholar] [CrossRef]
- Spanò, V.; Rocca, R.; Barreca, M.; Giallombardo, D.; Barraja, P. Pyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazoles, a New Class of Antimitotic Agents Active against Multiple Malignant Cell Types. J. Med. Chem. 2020, 63, 12023–12042. [Google Scholar] [CrossRef]
- Spanò, V.; Pennati, M.; Parrino, B.; Carbone, A.; Montalbano, A.; Cilibrasi, V.; Zuco, V.; Lopergolo, A.; Cominetti, D.; Diana, P. Preclinical Activity of New [1,2]Oxazolo[5,4-e]isoindole Derivatives in Diffuse Malignant Peritoneal Mesothelioma. J. Med. Chem. 2016, 59, 7223–7238. [Google Scholar] [CrossRef]
- Sippel, T.R.; White, J.; Nag, K.; Tsvankin, V.; Klaassen, M.; Kleinschmidt-DeMasters, B.K.; Waziri, A. Neutrophil Degranulation and Immunosuppression in Patients with GBM: Restoration of Cellular Immune Function by Targeting Arginase I. Clin. Cancer Res. 2011, 17, 6992–7002. [Google Scholar] [CrossRef] [PubMed]
- Dombret, H.; Gardin, C. An update of current treatments for adult acute myeloid leukemia. Blood 2016, 127, 53–61. [Google Scholar] [CrossRef]
- Schwartsmann, G.; da Rocha, A.B.; Berlinck, R.G.S.; Jimeno, J. Marine organisms as a source of new anticancer agents. Lancet Oncol. 2001, 2, 221–225. [Google Scholar] [CrossRef]
- Pourquier, P.; Takebayashi, Y.; Urasaki, Y.; Gioffre, C.; Kohlhagen, G.; Pommier, Y. Induction of topoisomerase I cleavage complexes by 1-beta-D-arabinofuranosylcytosine (ara-C) in vitro and in ara-C-treated cells. Proc. Natl. Acad. Sci. USA 2000, 97, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Sakamoto, S.; Sassa, S.; Suzuki, S.; Kudo, H.; Nagasawa, H. Paradoxical effect of cytosine arabinoside on mouse leukemia cell line L-1210 cells. Anticancer Res. 2005, 25, 157–160. [Google Scholar] [PubMed]
- Herzig, R.H.; Wolff, S.N.; Lazarus, H.M.; Phillips, G.L.; Karanes, C.; Herzig, G.P. High-dose cytosine arabinoside therapy for refractory leukemia. Blood 1983, 62, 361–369. [Google Scholar] [CrossRef]
- Peters, W.G.; Colly, L.P.; Willemze, R. High-dose cytosine arabinoside: Pharmacological and clinical aspects. Blut 1988, 56, 1–11. [Google Scholar] [CrossRef]
- Carden, P.A.; Mitchell, S.L.; Waters, K.D.; Tiedemann, K.; Ekert, H. Prevention of cyclophosphamide/cytarabine-induced emesis with ondansetron in children with leukemia. J. Clin. 1990, 8, 1531–1535. [Google Scholar] [CrossRef]
- Lv, S.Q.; Li, A.H.; Wu, H.J.; Wang, X.L. Observation of clinical efficacy and toxic and side effects of pirarubicin combined with cytarabine on acute myeloid leukemia. Oncol. Lett. 2019, 17, 3411–3417. [Google Scholar] [CrossRef]
- Drenberg, C.D.; Hu, S.; Li, L.; Buelow, D.R.; Orwick, S.J.; Gibson, A.A.; Schuetz, J.D.; Sparreboom, A.; Baker, S.D. ABCC4 Is a Determinant of Cytarabine-Induced Cytotoxicity and Myelosuppression. Clin. Transl. Sci. 2016, 9, 51–59. [Google Scholar] [CrossRef]
- Adema, A.D.; Floor, K.; Smid, K.; Honeywell, R.J.; Scheffer, G.L.; Jansen, G.; Peters, G.J. Overexpression of MRP4 (ABCC4) and MRP5 (ABCC5) confer resistance to the nucleoside analogs cytarabine and troxacitabine, but not gemcitabine. Springerplus 2014, 3, 732. [Google Scholar] [CrossRef] [PubMed]
- Waddick, K.G.; Uckun, F.M. Innovative treatment programs against cancer: II. Nuclear factor-kappaB (NF-kappaB) as a molecular target. Biochem. Pharmacol. 1999, 57, 9–17. [Google Scholar] [CrossRef]
- Wang, C.Y.; Cusack, J.C., Jr.; Liu, R.; Baldwin, A.S., Jr. Control of inducible chemoresistance: Enhanced antitumor therapy through increased apoptosis by inhibition of NF-kappaB. Nat. Med. 1999, 5, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Berenson, J.R.; Ma, H.M.; Vescio, R. The role of nuclear factor-kappaB in the biology and treatment of multiple myeloma. Semin. Oncol. 2001, 28, 626–633. [Google Scholar] [CrossRef]
- Cusack, J.C., Jr.; Liu, R.; Houston, M.; Abendroth, K.; Elliott, P.J.; Adams, J.; Baldwin, A.S., Jr. Enhanced chemosensitivity to CPT-11 with proteasome inhibitor PS-341: Implications for systemic nuclear factor-kappaB inhibition. Cancer Res. 2001, 61, 3535–3540. [Google Scholar]
- Rathe, S.K.; Moriarity, B.S.; Stoltenberg, C.B.; Kurata, M.; Aumann, N.K.; Rahrmann, E.P.; Bailey, N.J.; Melrose, E.G.; Beckmann, D.A.; Liska, C.R.; et al. Using RNA-seq and targeted nucleases to identify mechanisms of drug resistance in acute myeloid leukemia. Sci. Rep. 2014, 4, 6048. [Google Scholar] [CrossRef]
- Hirata, Y.; Uemura, D. Halichondrins—Antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef]
- Seletsky, B.M.; Wang, Y.; Hawkins, L.D.; Palme, M.H.; Habgood, G.J.; DiPietro, L.V.; Towle, M.J.; Salvato, K.A.; Wels, B.F.; Aalfs, K.K.; et al. Structurally simplified macrolactone analogues of halichondrin B. Bioorganic Med. Chem. Lett. 2004, 14, 5547–5550. [Google Scholar] [CrossRef]
- In, G.K.; Hu, J.S.; Tseng, W.W. Treatment of advanced, metastatic soft tissue sarcoma: Latest evidence and clinical considerations. Ther. Adv. Med. Oncol. 2017, 9, 533–550. [Google Scholar] [CrossRef]
- Jimenez, P.C.; Wilke, D.V.; Branco, P.C.; Bauermeister, A.; Rezende-Teixeira, P.; Gaudencio, S.P.; Costa-Lotufo, L.V. Enriching cancer pharmacology with drugs of marine origin. Br. J. Pharmacol. 2020, 177, 3–27. [Google Scholar] [CrossRef]
- Eslamian, G.; Wilson, C.; Young, R.J. Efficacy of eribulin in breast cancer: A short report on the emerging new data. OncoTargets Ther. 2017, 10, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Towle, M.J.; Nomoto, K.; Asano, M.; Kishi, Y.; Yu, M.J.; Littlefield, B.A. Broad spectrum preclinical antitumor activity of eribulin (Halaven(R)): Optimal effectiveness under intermittent dosing conditions. Anticancer Res. 2012, 32, 1611–1619. [Google Scholar]
- Yoshida, T.; Ozawa, Y.; Kimura, T.; Sato, Y.; Kuznetsov, G.; Xu, S.; Uesugi, M.; Agoulnik, S.; Taylor, N.; Funahashi, Y.; et al. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. Br. J. Cancer 2014, 110, 1497–1505. [Google Scholar] [CrossRef]
- Funahashi, Y.; Okamoto, K.; Adachi, Y.; Semba, T.; Uesugi, M.; Ozawa, Y.; Tohyama, O.; Uehara, T.; Kimura, T.; Watanabe, H.; et al. Eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. Cancer Sci. 2014, 105, 1334–1342. [Google Scholar] [CrossRef]
- Oba, T.; Ito, K.I. Combination of two anti-tubulin agents, eribulin and paclitaxel, enhances antitumor effects on triple-negative breast cancer through mesenchymal-epithelial transition. Oncotarget 2018, 9, 22986–23002. [Google Scholar] [CrossRef][Green Version]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Cenci, S.; Mezghrani, A.; Cascio, P.; Bianchi, G.; Cerruti, F.; Fra, A.; Lelouard, H.; Masciarelli, S.; Mattioli, L.; Oliva, L.; et al. Progressively impaired proteasomal capacity during terminal plasma cell differentiation. EMBO J. 2006, 25, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Fenical, W.; Jensen, P.R.; Palladino, M.A.; Lam, K.S.; Lloyd, G.K.; Potts, B.C. Discovery and development of the anticancer agent salinosporamide A (NPI-0052). Bioorganic Med. Chem. 2009, 17, 2175–2180. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Hideshima, T.; Anderson, K.C. A novel proteasome inhibitor NPI-0052 as an anticancer therapy. Br. J. Cancer 2006, 95, 961–965. [Google Scholar] [CrossRef]
- Baritaki, S.; Suzuki, E.; Umezawa, K.; Spandidos, D.; Berenson, J.; Daniels, T.; Penichet, M.; Jazirehi, A.; Palladino, M.; Bonavida, B. Inhibition of Yin Yang 1-dependent repressor activity of DR5 transcription and expression by the novel proteasome inhibitor NPI-0052 contributes to its TRAIL-enhanced apoptosis in cancer cells. J. Immunol. 2008, 180, 6199–6210. [Google Scholar] [CrossRef]
- Chauhan, D.; Catley, L.; Li, G.; Podar, K.; Hideshima, T.; Velankar, M.; Mitsiades, C.; Mitsiades, N.; Yasui, H.; Letai, A.; et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005, 8, 407–419. [Google Scholar] [CrossRef]
- Delic, J.; Masdehors, P.; Omura, S.; Cosset, J.M.; Dumont, J.; Binet, J.L.; Magdelenat, H. The proteasome inhibitor lactacystin induces apoptosis and sensitizes chemo- and radioresistant human chronic lymphocytic leukaemia lymphocytes to TNF-alpha-initiated apoptosis. Br. J. Cancer 1998, 77, 1103–1107. [Google Scholar] [CrossRef]
- Nencioni, A.; Grunebach, F.; Patrone, F.; Ballestrero, A.; Brossart, P. Proteasome inhibitors: Antitumor effects and beyond. Leukemia 2007, 21, 30–36. [Google Scholar] [CrossRef]
- Das, D.S.; Ray, A.; Song, Y.; Richardson, P.; Trikha, M.; Chauhan, D.; Anderson, K.C. Synergistic anti-myeloma activity of the proteasome inhibitor marizomib and the IMiD immunomodulatory drug pomalidomide. Br. J. Haematol. 2015, 171, 798–812. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.; Ocio, E.; San Miguel, J. Novel generation of agents with proven clinical activity in multiple myeloma. Semin. Oncol. 2013, 40, 618–633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, P.; Hu, Y. Clinical and marketed proteasome inhibitors for cancer treatment. Curr. Med. Chem. 2013, 20, 2537–2551. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Alonci, A.; Gerace, D.; Russo, S.; Innao, V.; Calabrò, L.; Musolino, C. New orally active proteasome inhibitors in multiple myeloma. Leuk. Res. 2014, 38, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guan, F.; Chen, D.; Dou, Q.; Yang, H. An analysis of the safety profile of proteasome inhibitors for treating various cancers. Expert Opin. Drug Saf. 2014, 13, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Urdiales, J.L.; Morata, P.; Nunez De Castro, I.; Sanchez-Jimenez, F. Antiproliferative effect of dehydrodidemnin B (DDB), a depsipeptide isolated from Mediterranean tunicates. Cancer Lett. 1996, 102, 31–37. [Google Scholar] [CrossRef]
- Sakai, R.; Rinehart, K.L.; Kishore, V.; Kundu, B.; Faircloth, G.; Gloer, J.B.; Carney, J.R.; Namikoshi, M.; Sun, F.; Hughes, R.G., Jr.; et al. Structure--activity relationships of the didemnins. J. Med. Chem. 1996, 39, 2819–2834. [Google Scholar] [CrossRef]
- Gomes, N.G.M.; Valentao, P.; Andrade, P.B.; Pereira, R.B. Plitidepsin to treat multiple myeloma. Drugs Today 2020, 56, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Santiago, L.; Suarez, Y.; Zarich, N.; Munoz-Alonso, M.J.; Cuadrado, A.; Martinez, T.; Goya, L.; Iradi, A.; Saez-Tormo, G.; Maier, J.V.; et al. Aplidin induces JNK-dependent apoptosis in human breast cancer cells via alteration of glutathione homeostasis, Rac1 GTPase activation, and MKP-1 phosphatase downregulation. Cell Death Differ. 2006, 13, 1968–1981. [Google Scholar] [CrossRef]
- Morande, P.E.; Zanetti, S.R.; Borge, M.; Nannini, P.; Jancic, C.; Bezares, R.F.; Bitsmans, A.; Gonzalez, M.; Rodriguez, A.L.; Galmarini, C.M.; et al. The cytotoxic activity of Aplidin in chronic lymphocytic leukemia (CLL) is mediated by a direct effect on leukemic cells and an indirect effect on monocyte-derived cells. Investig. New Drugs 2012, 30, 1830–1840. [Google Scholar] [CrossRef]
- Garcia-Fernandez, L.F.; Losada, A.; Alcaide, V.; Alvarez, A.M.; Cuadrado, A.; Gonzalez, L.; Nakayama, K.; Nakayama, K.I.; Fernandez-Sousa, J.M.; Munoz, A.; et al. Aplidin induces the mitochondrial apoptotic pathway via oxidative stress-mediated JNK and p38 activation and protein kinase C delta. Oncogene 2002, 21, 7533–7544. [Google Scholar] [CrossRef]
- Losada, A.; Lopez-Oliva, J.M.; Sanchez-Puelles, J.M.; Garcia-Fernandez, L.F. Establishment and characterisation of a human carcinoma cell line with acquired resistance to Aplidin. Br. J. Cancer 2004, 91, 1405–1413. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Losada, A.; Muñoz-Alonso, M.; García, C.; Sánchez-Murcia, P.; Martínez-Leal, J.; Domínguez, J.; Lillo, M.; Gago, F.; Galmarini, C. Translation Elongation Factor eEF1A2 is a Novel Anticancer Target for the Marine Natural Product Plitidepsin. Sci. Rep. 2016, 6, 35100. [Google Scholar] [CrossRef] [PubMed]
- Borjan, B.; Steiner, N.; Karbon, S.; Kern, J.; Francesch, A.; Hermann, M.; Willenbacher, W.; Gunsilius, E.; Untergasser, G. The Aplidin analogs PM01215 and PM02781 inhibit angiogenesis in vitro and in vivo. BMC Cancer 2015, 15, 738. [Google Scholar] [CrossRef] [PubMed]
- Grosso, F.; Jones, R.L.; Demetri, G.D.; Judson, I.R.; Blay, J.-Y.; Le Cesne, A.; Sanfilippo, R.; Casieri, P.; Collini, P.; Dileo, P.; et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: A retrospective study. Lancet Oncol. 2007, 8, 595–602. [Google Scholar] [CrossRef]
- Pautier, P.; Floquet, A.; Chevreau, C.; Penel, N.; Guillemet, C.; Delcambre, C.; Cupissol, D.; Selle, F.; Isambert, N.; Piperno-Neumann, S.; et al. Trabectedin in combination with doxorubicin for first-line treatment of advanced uterine or soft-tissue leiomyosarcoma (LMS-02): A non-randomised, multicentre, phase 2 trial. Lancet Oncol. 2015, 16, 457–464. [Google Scholar] [CrossRef]
- D’Incalci, M.; Galmarini, C.M. A review of trabectedin (ET-743): A unique mechanism of action. Mol. Cancer Ther. 2010, 9, 2157–2163. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, K.L.; Holt, T.G.; Fregeau, N.L.; Stroh, J.G.; Martin, D.G. Ecteinascidins 729, 743, 745, 759A, 759B, and 770: Potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4512–4515. [Google Scholar] [CrossRef]
- Wright, A.; Forleo, D.A.; Gunawardana, G.P.; Gunasekera, S.P.; Koehn, F.E.; Mcconnell, O.J. Antitumor tetrahydroisoquinoline alkaloids from the colonial ascidian Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4508–4512. [Google Scholar] [CrossRef]
- Seaman, F.C.; Hurley, L.H. Molecular Basis for the DNA Sequence Selectivity of Ecteinascidin 736 and 743: Evidence for the Dominant Role of Direct Readout via Hydrogen Bonding. J. Am. Chem. Soc. 1998, 120, 13028–13041. [Google Scholar] [CrossRef]
- Moore, B.M.; Seaman, F.C.; Wheelhouse, R.T.; Hurley, L.H. Mechanism for the Catalytic Activation of Ecteinascidin 743 and Its Subsequent Alkylation of Guanine N2. J. Am. Chem. Soc. 1998, 120, 2490–2491. [Google Scholar] [CrossRef]
- Feuerhahn, S.; Giraudon, C.; Martinez-Diez, M.; Bueren-Calabuig, J.A.; Galmarini, C.M.; Gago, F.; Egly, J.M. XPF-dependent DNA breaks and RNA polymerase II arrest induced by antitumor DNA interstrand crosslinking-mimetic alkaloids. Chem. Biol. 2011, 18, 988–999. [Google Scholar] [CrossRef]
- Aune, G.J.; Takagi, K.; Sordet, O.; Guirouilh-Barbat, J.; Antony, S.; Bohr, V.A.; Pommier, Y. Von Hippel-Lindau-coupled and transcription-coupled nucleotide excision repair-dependent degradation of RNA polymerase II in response to trabectedin. Clin. Cancer Res. 2008, 14, 6449–6455. [Google Scholar] [CrossRef]
- Forni, C.; Minuzzo, M.; Virdis, E.; Tamborini, E.; Simone, M.; Tavecchio, M.; Erba, E.; Grosso, F.; Gronchi, A.; Aman, P.; et al. Trabectedin (ET-743) promotes differentiation in myxoid liposarcoma tumors. Mol. Cancer Ther. 2009, 8, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, G.; Barra, F.; D’Alessandro, G.; Tantari, M.; Stigliani, S.; Corte, L.D.; Bifulco, G.; Ferrero, S. Trabectedin for the therapy of ovarian cancer. Drugs Today 2020, 56, 669–688. [Google Scholar] [CrossRef]
- Jones, R.; Herzog, T.; Patel, S.; von Mehren, M.; Schuetze, S.; Van Tine, B.; Coleman, R.; Knoblauch, R.; Triantos, S.; Hu, P.; et al. Cardiac safety of trabectedin monotherapy or in combination with pegylated liposomal doxorubicin in patients with sarcomas and ovarian cancer. Cancer Med. 2021, 10, 3565–3574. [Google Scholar] [CrossRef] [PubMed]
- Pignata, S.; Scambia, G.; Villanucci, A.; Naglieri, E.; Ibarbia, M.; Brusa, F.; Bourgeois, H.; Sorio, R.; Casado, A.; Reichert, D.; et al. A European, Observational, Prospective Trial of Trabectedin Plus Pegylated Liposomal Doxorubicin in Patients with Platinum-Sensitive Ovarian Cancer. Oncologist 2021, 26, e658–e668. [Google Scholar] [CrossRef]
- Palmerini, E.; Sanfilippo, R.; Grignani, G.; Buonadonna, A.; Romanini, A.; Badalamenti, G.; Ferraresi, V.; Vincenzi, B.; Comandone, A.; Pizzolorusso, A.; et al. Trabectedin for Patients with Advanced Soft Tissue Sarcoma: A Non-Interventional, Retrospective, Multicenter Study of the Italian Sarcoma Group. Cancers 2021, 13, 1053. [Google Scholar] [CrossRef]
- Grosso, F.; D’Ambrosio, L.; Zucchetti, M.; Ibrahim, T.; Tamberi, S.; Matteo, C.; Rulli, E.; Comandini, D.; Palmerini, E.; Baldi, G.; et al. Pharmacokinetics, safety, and activity of trabectedin as first-line treatment in elderly patients who are affected by advanced sarcoma and are unfit to receive standard chemotherapy: A phase 2 study (TR1US study) from the Italian Sarcoma Group. Cancer 2020, 126, 4726–4734. [Google Scholar] [CrossRef]
- Gronchi, A.; Palmerini, E.; Quagliuolo, V.; Martin Broto, J.; Lopez Pousa, A.; Grignani, G.; Brunello, A.; Blay, J.; Tendero, O.; Beveridge, R.D.; et al. Neoadjuvant Chemotherapy in High-Risk Soft Tissue Sarcomas: Final Results of a Randomized Trial From Italian (ISG), Spanish (GEIS), French (FSG), and Polish (PSG) Sarcoma Groups. J. Clin. Oncol. 2020, 38, 2178–2186. [Google Scholar] [CrossRef]
- Carlos, G.; Maurizio, D.; Paola, A. Trabectedin and Plitidepsin: Drugs from the Sea that Strike the Tumor Microenvironment. Mar. Drugs 2014, 12, 719–733. [Google Scholar]
- Martinez-Cruzado, L.; Tornin, J.; Rodriguez, A.; Santos, L.; Allonca, E.; Fernandez-Garcia, M.; Astudillo, A.; Garcia-Pedrero, J.; Rodriguez, R. Trabectedin and Campthotecin Synergistically Eliminate Cancer Stem Cells in Cell-of-Origin Sarcoma Models. Neoplasia 2017, 19, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Acikgoz, E.; Guven, U.; Duzagac, F.; Uslu, R.; Kara, M.; Soner, B.; Oktem, G. Enhanced G2/M Arrest, Caspase Related Apoptosis and Reduced E-Cadherin Dependent Intercellular Adhesion by Trabectedin in Prostate Cancer Stem Cells. PLoS ONE 2015, 10, e0141090. [Google Scholar] [CrossRef] [PubMed]
- Tumini, E.; Herrera-Moyano, E.; San Martín-Alonso, M.; Barroso, S.; Galmarini, C.; Aguilera, A. The Antitumor Drugs Trabectedin and Lurbinectedin Induce Transcription-Dependent Replication Stress and Genome Instability. Mol. Cancer Res. 2019, 17, 773–782. [Google Scholar] [CrossRef]
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and anticancer drug conjugate assemblies: The role of the linkage between components. Toxins 2011, 3, 848–883. [Google Scholar] [CrossRef]
- Martino, M.; Festuccia, M.; Fedele, R.; Console, G.; Cimminiello, M.; Gavarotti, P.; Bruno, B. Salvage treatment for relapsed/refractory Hodgkin lymphoma: Role of allografting, brentuximab vedotin and newer agents. Expert Opin. Biol. Ther. 2016, 16, 347–364. [Google Scholar] [CrossRef]
- Alperovich, A.; Younes, A. Targeting CD30 Using Brentuximab Vedotin in the Treatment of Hodgkin Lymphoma. Cancer J. 2016, 22, 23–26. [Google Scholar] [CrossRef]
- Andrade-Gonzalez, X.; Ansell, S. Novel Therapies in the Treatment of Hodgkin Lymphoma. Curr. Treat. Options Oncol. 2021, 22, 42. [Google Scholar] [CrossRef]
- Wahl, A.F.; Klussman, K.; Thompson, J.D.; Chen, J.H.; Francisco, L.V.; Risdon, G.; Chace, D.F.; Siegall, C.B.; Francisco, J.A. The anti-CD30 monoclonal antibody SGN-30 promotes growth arrest and DNA fragmentation in vitro and affects antitumor activity in models of Hodgkin’s disease. Cancer Res. 2002, 62, 3736–3742. [Google Scholar]
- Ducry, L.; Stump, B. Antibody-drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconjugate Chem. 2010, 21, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Younes, A. CD30-targeted antibody therapy. Curr. Opin. Oncol. 2011, 23, 587–593. [Google Scholar] [CrossRef]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, C.H.; Nademanee, A.; Masszi, T.; Agura, E.; Holowiecki, J.; Abidi, M.H.; Chen, A.I.; Stiff, P.; Gianni, A.M.; Carella, A.; et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 385, 1853–1862. [Google Scholar] [CrossRef]
- Yi, J.H.; Kim, S.J.; Kim, W.S. Brentuximab vedotin: Clinical updates and practical guidance. Blood Res. 2017, 52, 243–253. [Google Scholar] [CrossRef]
- Barreca, M.; Stathis, A.; Barraja, P.; Bertoni, F. An overview on anti-tubulin agents for the treatment of lymphoma patients. Pharmacol. Ther. 2020, 211, 107552. [Google Scholar] [CrossRef]
- Petek, B.J.; Jones, R.L. PM00104 (Zalypsis(R)): A marine derived alkylating agent. Molecules 2014, 19, 12328–12335. [Google Scholar] [CrossRef]
- Leal, J.F.; Garcia-Hernandez, V.; Moneo, V.; Domingo, A.; Bueren-Calabuig, J.A.; Negri, A.; Gago, F.; Guillen-Navarro, M.J.; Aviles, P.; Cuevas, C.; et al. Molecular pharmacology and antitumor activity of Zalypsis in several human cancer cell lines. Biochem. Pharmacol. 2009, 78, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Ocio, E.M.; Maiso, P.; Chen, X.; Garayoa, M.; Pandiella, A. Zalypsis: A novel marine-derived compound with potent antimyeloma activity that reveals high sensitivity of malignant plasma cells to DNA double-strand breaks. Blood 2009, 113, 3781. [Google Scholar] [CrossRef] [PubMed]
- Elices, M.; Sasak, H.; Caylor, T.; Grant, W.; Guillen, M.J.; Martin, J.; Cuevas, C.; Aviles, P.; Faircloth, G. Antitumor activity of the novel investigational compound PM00104. Cancer Res. 2005, 65, 1384. [Google Scholar]
- Lepage, D.; Sasak, H.; Guillen, M.J.; Grant, W.; Cuevas, C.; Aviles, P. Antitumor activity of Zalypsis® (PM00104) in experimental models of bladder, gastric and liver cancer. Mol. Cancer Ther. 2007, 6, C62. [Google Scholar]
- Elices, M.; Grant, W.; Harper, C.; Guillen, M.J.; Cuevas, C.; Faircloth, G.; Aviles, P. The novel compound PM00104 exhibits significant in vivo activity against breast tumors. PM00104 exhibits significant in vivo activity against breast tumors. Cancer Res. 2005, 65, 147. [Google Scholar]
- Greiner, T.; Maier, A.; Bausch, N.; Fiebig, H.; Lepage, D.; Guillen, M.J.; Cuevas, C.; Aviles, P. Preclinical evaluation of PM00104 to support the selection of tumor indications for clinical development. Mol. Cancer Ther. 2007, 6, 3545S–3546S. [Google Scholar]
- Guirouilh-Barbat, J.; Antony, S.; Pommier, Y. Zalypsis (PM00104) is a potent inducer of gamma-H2AX foci and reveals the importance of the C ring of trabectedin for transcription-coupled repair inhibition. Mol. Cancer Ther. 2009, 8, 2007–2014. [Google Scholar] [CrossRef]
- Colado, E.; Paino, T.; Maiso, P.; Ocio, E.M.; Chen, X.; Alvarez-Fernandez, S.; Gutierrez, N.C.; Martin-Sanchez, J.; Flores-Montero, J.; San Segundo, L.; et al. Zalypsis has in vitro activity in acute myeloid blasts and leukemic progenitor cells through the induction of a DNA damage response. Haematologica 2011, 96, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Stojanovic, N.; Hassan, Z.; Wirth, M.; Wenzel, P.; Beyer, M.; Schäfer, C.; Brand, P.; Kroemer, A.; Stauber, R.H.; Schmid, R.M.; et al. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene 2017, 36, 1804–1815. [Google Scholar] [CrossRef]
- McLeod, A.B.; Stice, J.P.; Wardell, S.E.; Alley, H.M.; Chang, C.Y.; McDonnell, D.P. Validation of histone deacetylase 3 as a therapeutic target in castration-resistant prostate cancer. Prostate 2018, 78, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; Qi, Q.; Peng, X.; Yan, L.; Takabe, K.; Hait, N.C. Class I histone deacetylase inhibitor suppresses vasculogenic mimicry by enhancing the expression of tumor suppressor and anti-angiogenesis genes in aggressive human TNBC cells. Int. J. Oncol. 2019, 55, 116–130. [Google Scholar] [CrossRef]
- von Tresckow, B.; Sayehli, C.; Aulitzky, W.E.; Goebeler, M.E.; Schwab, M.; Braz, E.; Krauss, B.; Krauss, R.; Hermann, F.; Bartz, R. Phase I study of domatinostat (4SC-202), a class I histone deacetylase inhibitor in patients with advanced hematological malignancies. Eur. J. Haematol. 2019, 102, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, W. Isoform-selective histone deacetylase inhibitors: The trend and promise of disease treatment. Epigenomics 2015, 7, 5–7. [Google Scholar] [CrossRef]
- Bieliauskas, A.V.; Pflum, M.K. Isoform-selective histone deacetylase inhibitors. Chem. Soc. Rev. 2008, 37, 1402–1413. [Google Scholar] [CrossRef] [PubMed]
- Kuphal, S.; Wallner, S.; Bosserhoff, A.K. Loss of nephronectin promotes tumor progression in malignant melanoma. Cancer Sci. 2008, 99, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Taori, K.; Kim, H.; Hong, J.; Luesch, H. Total synthesis and molecular target of largazole, a histone deacetylase inhibitor. J. Am. Chem. Soc. 2008, 130, 8455–8459. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Liu, Y.; Byeon, S.R.; Kim, H.; Luesch, H.; Hong, J. Synthesis and activity of largazole analogues with linker and macrocycle modification. Org. Lett. 2008, 10, 4021–4024. [Google Scholar] [CrossRef] [PubMed]
- Bowers, A.; West, N.; Taunton, J.; Schreiber, S.L.; Bradner, J.E.; Williams, R.M. Total synthesis and biological mode of action of largazole: A potent class I histone deacetylase inhibitor. J. Am. Chem. Soc. 2008, 130, 11219–11222. [Google Scholar] [CrossRef]
- Zhou, H.; Jiang, S.; Chen, J.; Ren, X.; Jin, J.; Su, S.B. Largazole, an inhibitor of class I histone deacetylases, attenuates inflammatory corneal neovascularization. Eur. J. Pharmacol. 2014, 740, 619–626. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z.; Wang, J.; Lam, W.; Kwong, S.; Li, F.; Friedman, S.L.; Zhou, S.; Ren, Q.; Xu, Z.; et al. A histone deacetylase inhibitor, largazole, decreases liver fibrosis and angiogenesis by inhibiting transforming growth factor-beta and vascular endothelial growth factor signalling. Liver Int. 2013, 33, 504–515. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Perrine, S.P.; Williams, R.M.; Faller, D.V. Histone deacetylase inhibitors are potent inducers of gene expression in latent EBV and sensitize lymphoma cells to nucleoside antiviral agents. Blood 2012, 119, 1008–1017. [Google Scholar] [CrossRef]
- Lee, S.U.; Kwak, H.B.; Pi, S.H.; You, H.K.; Byeon, S.R.; Ying, Y.; Luesch, H.; Hong, J.; Kim, S.H. In Vitro and In Vivo Osteogenic Activity of Largazole. ACS Med. Chem. Lett. 2011, 2, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Giddings, L.-A. Natural products as leads to antitumor drugs. Phytochem. Rev. 2013, 13, 123–137. [Google Scholar] [CrossRef]
- Wu, L.C.; Wen, Z.S.; Qiu, Y.T.; Chen, X.Q.; Chen, H.B.; Wei, M.M.; Liu, Z.; Jiang, S.; Zhou, G.B. Largazole Arrests Cell Cycle at G1 Phase and Triggers Proteasomal Degradation of E2F1 in Lung Cancer Cells. ACS Med. Chem. Lett. 2013, 4, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Salvador, L.A.; Byeon, S.; Ying, Y.; Kwan, J.C.; Law, B.K.; Hong, J.; Luesch, H. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J. Pharmacol. Exp. Ther. 2010, 335, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Law, M.E.; Corsino, P.E.; Jahn, S.C.; Davis, B.J.; Chen, S.; Patel, B.; Pham, K.; Lu, J.; Sheppard, B.; Norgaard, P.; et al. Glucocorticoids and histone deacetylase inhibitors cooperate to block the invasiveness of basal-like breast cancer cells through novel mechanisms. Oncogene 2013, 32, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, M.S.; Ma, J.J.; Ratnayake, R.; Havre, P.A.; Yao, J.; Dang, N.H.; Paul, V.J.; Carney, T.J.; Dang, L.H.; Luesch, H. Multidimensional Screening Platform for Simultaneously Targeting Oncogenic KRAS and Hypoxia-Inducible Factors Pathways in Colorectal Cancer. ACS Chem. Biol. 2016, 11, 1322–1331. [Google Scholar] [CrossRef]
- Sanchez, G.J.; Richmond, P.A.; Bunker, E.N.; Karman, S.S.; Azofeifa, J.; Garnett, A.T.; Xu, Q.; Wheeler, G.E.; Toomey, C.M.; Zhang, Q.; et al. Genome-wide dose-dependent inhibition of histone deacetylases studies reveal their roles in enhancer remodeling and suppression of oncogenic super-enhancers. Nucleic Acids Res. 2018, 46, 1756–1776. [Google Scholar] [CrossRef]
- Al-Awadhi, F.H.; Salvador-Reyes, L.A.; Elsadek, L.A.; Ratnayake, R.; Chen, Q.Y.; Luesch, H. Largazole is a Brain-Penetrant Class I HDAC Inhibitor with Extended Applicability to Glioblastoma and CNS Diseases. ACS Chem. Neurosci. 2020, 11, 1937–1943. [Google Scholar] [CrossRef]
- Min, W.; Xiao-Yan, S.; Yong-Chun, Z.; Kuo-Jun, Z.; Yong-Zhi, L.; Jinsong, L.; Yun-Chao, H.; Gui-Zhen, W.; Sheng, J.; Guang-Biao, Z. Suppression of Musashi-2 by the small compound largazole exerts inhibitory effects on malignant cells. Int. J. Oncol. 2020, 56, 1274–1283. [Google Scholar]
- Clausen, D.J.; Smith, W.B.; Haines, B.E.; Wiest, O.; Bradner, J.E.; Williams, R.M. Modular synthesis and biological activity of pyridyl-based analogs of the potent Class I Histone Deacetylase Inhibitor Largazole. Bioorganic Med. Chem. 2015, 23, 5061–5074. [Google Scholar] [CrossRef]
- Almaliti, J.; Al-Hamashi, A.A.; Negmeldin, A.T.; Hanigan, C.L.; Perera, L.; Pflum, M.K.; Casero, R.A., Jr.; Tillekeratne, L.M. Largazole Analogues Embodying Radical Changes in the Depsipeptide Ring: Development of a More Selective and Highly Potent Analogue. J. Med. Chem. 2016, 59, 10642–10660. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Chai, H.; Su, M.B.; Zhang, Y.M.; Li, J.; Xie, X.; Nan, F.J. Potent and orally efficacious bisthiazole-based histone deacetylase inhibitors. ACS Med. Chem. Lett. 2014, 5, 628–633. [Google Scholar] [CrossRef]
- Cole, K.E.; Dowling, D.P.; Boone, M.A.; Phillips, A.J.; Christianson, D.W. Structural Basis of the Antiproliferative Activity of Largazole, a Depsipeptide Inhibitor of the Histone Deacetylases. J. Am. Chem. Soc. 2011, 133, 12474–12477. [Google Scholar] [CrossRef] [PubMed]
- Seiser, T.; Kamena, F.; Cramer, N. Synthesis and biological activity of largazole and derivatives. Angew. Chem. Int. Ed. 2008, 47, 6483–6485. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Yin, B.; Hu, Z.; Liao, C.; Liu, J.; Li, S.; Li, Z.; Nicklaus, M.C.; Zhou, G.; Jiang, S. Total synthesis and biological evaluation of largazole and derivatives with promising selectivity for cancers cells. Org. Lett. 2010, 12, 1368–1371. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Park, H.; Salvador, L.A.; Serrano, P.E.; Kwan, J.C.; Zeller, S.L.; Chen, Q.-Y.; Ryu, S.; Liu, Y.; Byeon, S.; et al. Evaluation of class I HDAC isoform selectivity of largazole analogues. Bioorganic Med. Chem. Lett. 2014, 24, 3728–3731. [Google Scholar] [CrossRef] [PubMed]
- Bowers, A.A.; West, N.; Newkirk, T.L.; Troutman-Youngman, A.E.; Schreiber, S.L.; Wiest, O.; Bradner, J.E.; Williams, R.M. Synthesis and histone deacetylase inhibitory activity of largazole analogs: Alteration of the zinc-binding domain and macrocyclic scaffold. Org. Lett. 2009, 11, 1301–1304. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Ratnayake, R.; Lee, H.; Shi, G.; Zeller, S.L.; Li, C.; Luesch, H.; Hong, J. Synthesis and biological evaluation of largazole zinc-binding group analogs. Bioorganic Med. Chem. 2017, 25, 3077–3086. [Google Scholar] [CrossRef] [PubMed]
- Bhansali, P.; Hanigan, C.L.; Casero, R.A.; Tillekeratne, L.M. Largazole and analogues with modified metal-binding motifs targeting histone deacetylases: Synthesis and biological evaluation. J. Med. Chem. 2011, 54, 7453–7463. [Google Scholar] [CrossRef]
- Su, J.; Qiu, Y.; Ma, K.; Yao, Y.; Wang, Z.; Li, X.; Zhang, D.; Tu, Z.; Jiang, S. Design, synthesis, and biological evaluation of largazole derivatives: Alteration of the zinc-binding domain. Tetrahedron 2014, 70, 7763–7769. [Google Scholar] [CrossRef]
- Benelkebir, H.; Marie, S.; Hayden, A.L.; Lyle, J.; Loadman, P.M.; Crabb, S.J.; Packham, G.; Ganesan, A. Total synthesis of largazole and analogues: HDAC inhibition, antiproliferative activity and metabolic stability. Bioorganic Med. Chem. 2011, 19, 3650–3658. [Google Scholar] [CrossRef] [PubMed]
- Bhansali, P.; Hanigan, C.L.; Perera, L.; Casero, R.A.; Tillekeratne, L.M.V. Synthesis and biological evaluation of largazole analogues with modified surface recognition cap groups. Eur. J. Med. Chem. 2014, 86, 528–541. [Google Scholar] [CrossRef]
- Zhang, B.; Ruan, Z.W.; Luo, D.; Zhu, Y.; Ding, T.; Sui, Q.; Lei, X. Unexpected Enhancement of HDACs Inhibition by MeS Substitution at C-2 Position of Fluoro Largazole. Mar. Drugs 2020, 18, 344. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Gao, A.-H.; Li, J.; Nan, F.-J. Synthesis and Biological Evaluation of C7-Demethyl Largazole Analogues. ChemMedChem 2009, 4, 1269–1272. [Google Scholar] [CrossRef]
- Souto, J.A.; Vaz, E.; Lepore, I.; Pöppler, A.-C.; Franci, G.; Álvarez, R.; Altucci, L.; de Lera, Á.R. Synthesis and Biological Characterization of the Histone Deacetylase Inhibitor Largazole and C7- Modified Analogues. J. Med. Chem. 2010, 53, 4654–4667. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tu, Z.; Li, H.; Liu, C.; Li, Z.; Sun, Q.; Yao, Y.; Liu, J.; Jiang, S. Biological evaluation of new largazole analogues: Alteration of macrocyclic scaffold with click chemistry. ACS Med. Chem. Lett. 2013, 4, 132–136. [Google Scholar] [CrossRef][Green Version]
- Yao, Y.; Tu, Z.; Liao, C.; Wang, Z.; Li, S.; Yao, H.; Li, Z.; Jiang, S. Discovery of Novel Class I Histone Deacetylase Inhibitors with Promising in Vitro and in Vivo Antitumor Activities. J. Med. Chem. 2015, 58, 7672–7680. [Google Scholar] [CrossRef]
- Zhang, K.; Yao, Y.; Tu, Z.; Liao, C.; Wang, Z.; Qiu, Y.; Chen, D.; Hamilton, D.J.; Li, Z.; Jiang, S. Discovery of class I histone deacetylase inhibitors based on romidpesin with promising selectivity for cancer cells. Futur. Med. Chem. 2020, 12, 311–323. [Google Scholar] [CrossRef]
- Zhang, S.-W.; Gong, C.-J.; Su, M.-B.; Chen, F.; He, T.; Zhang, Y.-M.; Shen, Q.-Q.; Su, Y.; Ding, J.; Li, J.; et al. Synthesis and in Vitro and in Vivo Biological Evaluation of Tissue-Specific Bisthiazole Histone Deacetylase (HDAC) Inhibitors. J. Med. Chem. 2020, 63, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J.R.; Kagihara, J.A.; Liu, X.; Gordon, G.; Heim, A.M.; Winkler, J.; DeMattei, J.A.; Piscopio, A.D.; Eckhardt, S.G. OKI-179 is a novel, oral, class I specific histone deacetylase inhibitor in phase 1 clinical trials. In Proceedings of the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics, Boston, MA, USA, 26–30 October 2019; pp. 26–30. [Google Scholar]
- Diamond, J.; Gordon, G.; Kagihara, J.; Corr, B.; Piscopio, A. Initial results from a phase 1 trial of OKI-179, an oral Class 1-selective depsipeptide HDAC inhibitor, in patients with advanced solid tumors. Eur. J. Cancer 2020, 138, S12. [Google Scholar] [CrossRef]
- Liu, X.; Phillips, A.J.; Ungermannova, D.; Nasveschuk, C.G.; Zhang, G. Macrocyclic Compounds Useful as Inhibitors of Histone Deacetylases. U.S. Patent 9,422,340, 23 August 2016. [Google Scholar]
- Wang, X.; Waschke, B.C.; Woolaver, R.A.; Chen, Z.; Zhang, G.; Piscopio, A.D.; Liu, X.; Wang, J.H. Histone Deacetylase Inhibition Sensitizes PD1 Blockade-Resistant B-cell Lymphomas. Cancer Immunol. Res. 2019, 7, 1318–1331. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Source | Indications | Mechanisms | Clinical Trial |
|---|---|---|---|---|
| Cytarabine | Cryptotethia crypta | AML | DNA synthesis inhibition | Approved |
| Eribulin | Halichondria okadai | Breast cancer, liposarcoma | Prevention the formation of spindles and EMT, apoptosis induction | Approved |
| Marizomib | Salinispora tropica | MM | Selectively inhibiting 20S proteasome | Approved |
| Plitidepsin | Aplidium albican | Solid tumor, MM | Cell cycle arrest, apoptosis induction | Approved |
| Trabectedin | Ecteinascidia turbinata | Liposarcoma, leiomyosarcoma | Binding to guanine residues in the DNA groove to form protein adducts | Approved |
| Adcetris | Dolabella auricularia | Hodgkin’s lymphoma | Inhibiting tubulin formation, apoptosis induction | Approved |
| Zalypsis | Joruna funebris | Urothelial carcinoma, cervical carcinoma, MM | rendering DNA double strand breaks, cell cycle arrest, apoptosis induction | Phase II |
| OKI-179 | Cyanobacterium, Symploca | Advanced solid tumors | Class I HDAC inhibition | Phase I |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, L.; Ye, K.; Jiang, S.; Zhou, G. Marine Power on Cancer: Drugs, Lead Compounds, and Mechanisms. Mar. Drugs 2021, 19, 488. https://doi.org/10.3390/md19090488
Wu L, Ye K, Jiang S, Zhou G. Marine Power on Cancer: Drugs, Lead Compounds, and Mechanisms. Marine Drugs. 2021; 19(9):488. https://doi.org/10.3390/md19090488
Chicago/Turabian StyleWu, Lichuan, Ke Ye, Sheng Jiang, and Guangbiao Zhou. 2021. "Marine Power on Cancer: Drugs, Lead Compounds, and Mechanisms" Marine Drugs 19, no. 9: 488. https://doi.org/10.3390/md19090488
APA StyleWu, L., Ye, K., Jiang, S., & Zhou, G. (2021). Marine Power on Cancer: Drugs, Lead Compounds, and Mechanisms. Marine Drugs, 19(9), 488. https://doi.org/10.3390/md19090488